Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas


 ,
,
Abstract
1. Introduction
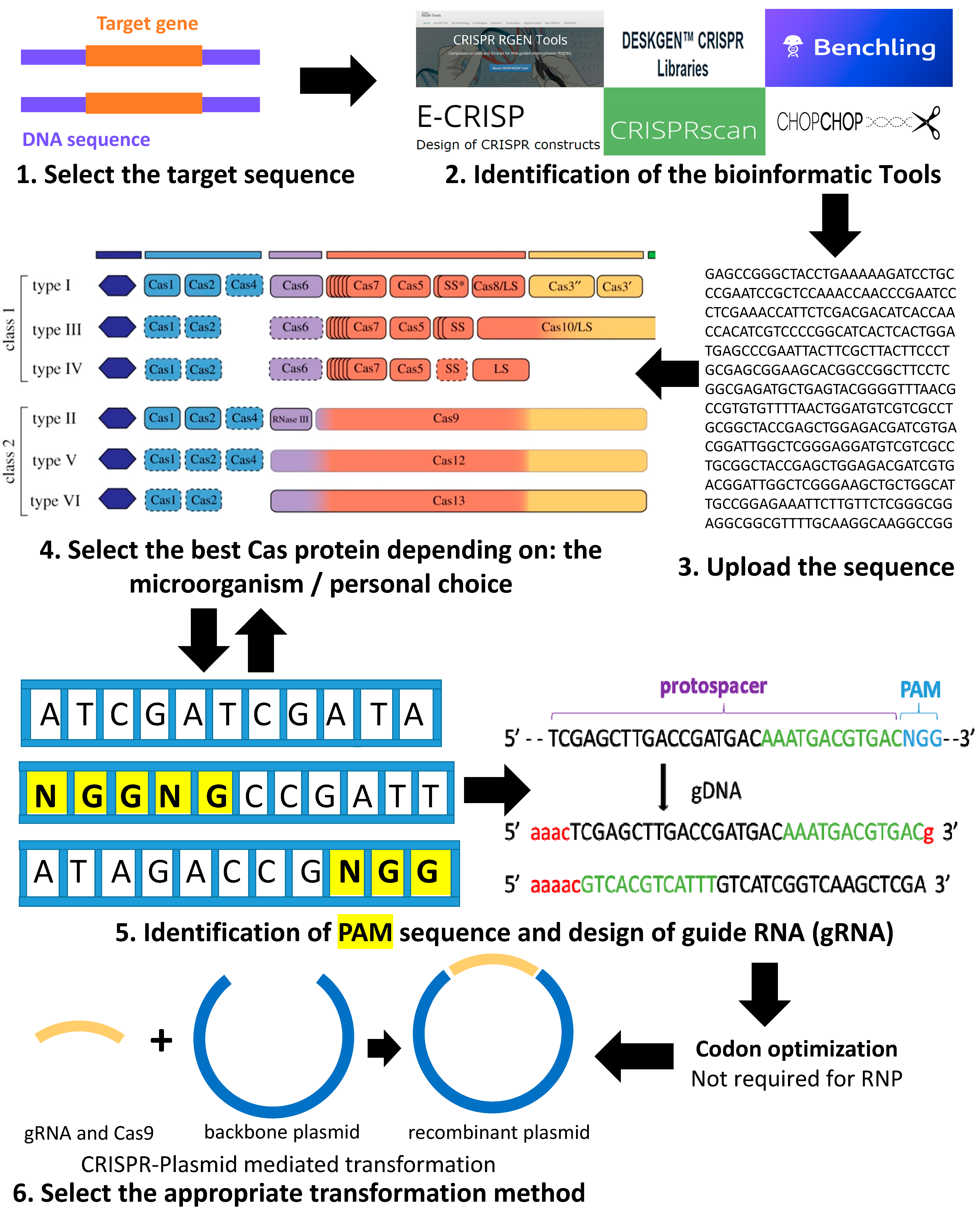
2. Summary of the Pipeline for Using the CRISPR-Cas System for Genome Editing in Green Microalgae C. reinhardtii
Codon Optimization
3. CRISPR-Cas System in C. reinhardtii
3.1. Cas/Cas9 Protein and Guide RNA
3.1.1. Definition
3.1.2. Regarding the Off Target and On Target Efficiency
3.1.3. The Case of the Cpf1 Protein
3.1.4. Dead Cas9
3.1.5. Nickase Cas9
3.1.6. The Size of sgRNA
3.2. Current CRISPR-Cas Systems in C. reinhardtii
3.2.1. Integrative CRISPR System
3.2.2. RNP DNA Free CRISPR System
3.2.3. Episomal CRISPR System
4. Repair System in C. reinhardtii
4.1. NHEJ
- (i)
- DNA end-binding and bridging,
- (ii)
- Terminal-end processing,
- (iii)
- Ligation.
4.1.1. (i) DNA End-Binding and Bridging
4.1.2. (ii) Terminal End Processing and (iii) Ligation
4.2. HDR
5. Transformation Methods
5.1. Electroporation
5.2. Glass-Beads
5.3. Biolistic
5.4. Agrobacterium tumefaciens
6. Applications of CRISPR Systems in C. reinhardtii
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khan, M.I.; Shin, J.H.; Kim, J.D. The promising future of microalgae: Current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb. Cell Factories 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.H. Chlamydomonas as a Model Organism. 2001. Available online: www.annualreviews.org (accessed on 10 May 2020).
- Rochaix, J.D. The three genomes of Chlamydomonas. Photosynth. Res. 2002, 73, 285–293. [Google Scholar] [CrossRef] [PubMed]
- D’Halluin, K.; Ruiter, R. Directed genome engineering for genome optimization. Int. J. Dev. Biol. 2013, 57, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Zorin, B.; Hegemann, P.; Sizova, I. Nuclear-gene targeting by using single-stranded DNA avoids illegitimate DNA integration in Chlamydomonas reinhardtii. Eukaryot. Cell 2005, 4, 1264–1272. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Cyranoski, D. The CRISPR-baby scandal: What’s next for human gene-editing. Nature 2019, 566, 440–442. [Google Scholar] [CrossRef]
- Poliner, E.; Takeuchi, T.; Du, Z.Y.; Benning, C.; Farré, E.M. Nontransgenic Marker-Free Gene Disruption by an Episomal CRISPR System in the Oleaginous Microalga, Nannochloropsis oceanica CCMP1779. ACS Synth. Biol. 2018, 7, 962–968. [Google Scholar] [CrossRef]
- Stukenberg, D.; Zauner, S.; Dell’Aquila, G.; Maier, U.G. Optimizing CRISPR/cas9 for the diatom Phaeodactylum tricornutum. Front. Plant Sci. 2018, 9, 740. [Google Scholar] [CrossRef]
- Hopes, A.; Nekrasov, V.; Kamoun, S.; Mock, T. Editing of the urease gene by CRISPR-Cas in the diatom Thalassiosira pseudonana. Plant Methods 2016, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-R.; Ng, I.-S. Development of CRISPR/Cas9 system in Chlorella vulgaris FSP-E to enhance lipid accumulation. Enzym. Microb. Technol. 2020, 133, 109458. [Google Scholar] [CrossRef] [PubMed]
- Greiner, A.; Kelterborn, S.; Evers, H.; Kreimer, G.; Sizova, I.; Hegemann, P. Targeting of Photoreceptor Genes in Chlamydomonas reinhardtii via Zinc-Finger Nucleases and CRISPR/Cas9. Plant Cell 2017, 29, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Kim, D.H.; Jeong, J.; Sim, S.J.; Melis, A.; Kim, J.-S.; Jin, E.; Bae, S. DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci. Rep. 2016, 6, 30620. [Google Scholar] [CrossRef]
- Poliner, E.; Cummings, C.; Newton, L.; Farré, E.M. Identification of circadian rhythms in Nannochloropsis species using bioluminescence reporter lines. Plant J. 2019, 99, 112–127. [Google Scholar] [CrossRef]
- European Nucleoteide Archive. Chlamydomonas Genome Search. 2020. Available online: https://www.ebi.ac.uk/ena/browser/text-search?query=chlamydomonas (accessed on 14 October 2020).
- Jiang, W.; Brueggeman, A.J.; Horken, K.M.; Plucinak, T.M.; Weeks, D.P. Successful Transient Expression of Cas9 and Single Guide RNA Genes in Chlamydomonas reinhardtii. Eukaryot. Cell 2014, 13, 1465–1469. [Google Scholar] [CrossRef]
- Jiang, W.Z.; Weeks, D.P. A gene-within-a-gene Cas9/sgRNA hybrid construct enables gene editing and gene replacement strategies in Chlamydomonas reinhardtii. Algal Res. 2017, 26, 474–480. [Google Scholar] [CrossRef]
- Guzmán-Zapata, D.; Sandoval-Vargas, J.M.; Macedo-Osorio, K.S.; Salgado-Manjarrez, E.; Castrejón-Flores, J.L.; Oliver-Salvador, M.D.C.; Durán-Figueroa, N.V.; Nogue, F.; Badillo-Corona, J.A. Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module. Int. J. Mol. Sci. 2019, 20, 1247. [Google Scholar] [CrossRef]
- Ferenczi, A.; Pyott, D.E.; Xipnitou, A.; Molnar, A. Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 13567–13572. [Google Scholar] [CrossRef]
- Shin, S.-E.; Lim, J.-M.; Koh, H.G.; Kim, E.K.; Kang, N.K.; Jeon, S.; Kwon, S.; Shin, W.-S.; Lee, B.; Hwangbo, K.; et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 2016, 6, 27810. [Google Scholar] [CrossRef]
- Baek, K.; Yu, J.; Jeong, J.; Sim, S.J.; Bae, S.; Jin, E. Photoautotrophic production of macular pigment in a Chlamydomonas reinhardtii strain generated by using DNA-free CRISPR-Cas9 RNP-mediated mutagenesis. Biotechnol. Bioeng. 2018, 115, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. The basic building blocks and evolution of CRISPR-Cas systems. Biochem. Soc. Trans. 2013, 41, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Neupert, J.; Karcher, D.; Bock, R. Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant J. 2009, 57, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Barahimipour, R.; Strenkert, D.; Neupert, J.; Schroda, M.; Merchant, S.S.; Bock, R. Dissecting the contributions of GC content and codon usage to gene expression in the model alga Chlamydomonas reinhardtii. Plant J. 2015, 84, 704–717. [Google Scholar] [CrossRef]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- Codon Usage Database. Available online: http://www.kazusa.or.jp/codon/ (accessed on 14 May 2020).
- Baier, T.; Wichmann, J.; Kruse, O.; Lauersen, K.J. Intron-containing algal transgenes mediate efficient recombinant gene expression in the green microalga Chlamydomonas reinhardtii. Nucleic Acids Res. 2018, 46, 6909–6919. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Annotation and classification of CRISPR-Cas systems. Methods Mol. Biol. 2015, 1311, 47–75. [Google Scholar] [CrossRef]
- Cas9-an Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/cas9 (accessed on 10 May 2020).
- Mohr, S.E.; Hu, Y.; Ewen-Campen, B.; Housden, B.E.; Viswanatha, R.; Perrimon, N. CRISPR guide RNA design for research applications. FEBS J. 2016, 283, 3232–3238. [Google Scholar] [CrossRef]
- Zeng, Y.; Cui, Y.; Zhang, Y.; Zhang, Y.; Liang, M.; Chen, H.; Lan, J.; Song, G.; Lou, J. The initiation, propagation and dynamics of CRISPR-SpyCas9 R-loop complex. Nucleic Acids Res. 2018, 46, 350–361. [Google Scholar] [CrossRef]
- Huai, C.; Li, G.; Yao, R.; Zhang, Y.; Cao, M.; Kong, L.; Jia, C.; Yuan, H.; Chen, H.; Lu, D.; et al. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Walsh, R.M.; Hochedlinger, K. A variant CRISPR-Cas9 system adds versatility to genome engineering. Proc. Natl. Acad. Sci. USA 2013, 110, 15514–15515. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Zhang, Y.; Propson, N.E.; Howden, S.E.; Chu, L.-F.; Sontheimer, E.J.; Thomson, J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 2013, 110, 15644–15649. [Google Scholar] [CrossRef] [PubMed]
- Esvelt, K.M.; Mali, P.; Braff, J.L.; Moosburner, M.; Yaung, S.J.; Church, G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, J.A.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Cas-OFFinder: A Fast and Versatile Algorithm that Searches for Potential Off-Target Sites of Cas9 RNA-Guided Endonucleases. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4016707/ (accessed on 10 May 2020).
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef]
- Pliatsika, V.; Rigoutsos, I. ‘Off-Spotter’: Very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct 2015, 10. [Google Scholar] [CrossRef]
- Xie, X.; Ma, X.; Zhu, Q.; Zeng, D.; Li, G.; Liu, Y.G. CRISPR-GE: A Convenient Software Toolkit for CRISPR-Based Genome Editing. Mol. Plant 2017, 10, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Hough, S.H.; Ajetunmobi, A.; Brody, L.; Humphryes-Kirilov, N.; Perello, E. Desktop genetics. Pers. Med. 2016, 13, 517–521. [Google Scholar] [CrossRef] [PubMed]
- CHOPCHOP: A CRISPR/Cas9 and TALEN Web Tool for Genome Editing|Nucleic Acids Research | Oxford Academic. Available online: https://academic.oup.com/nar/article/42/W1/W401/2437392 (accessed on 10 May 2020).
- Prykhozhij, S.V.; Rajan, V.; Gaston, D.; Berman, J.N. CRISPR multitargeter: A web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS ONE 2015, 10, e0119372. [Google Scholar] [CrossRef]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Cebrian-Serrano, A.; Davies, B. CRISPR-Cas orthologues and variants: Optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm. Genome 2017, 28, 247–261. [Google Scholar] [CrossRef]
- Tang, L. PAM-less is more. Nat. Methods 2020, 17, 559. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Sharma, S. SSFinder: High throughput CRISPR-Cas target sites prediction tool. BioMed Res. Int. 2014, 2014, 742482. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther.-Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Yang, Z.; Edwards, H.; Xu, P. CRISPR-Cas12a/Cpf1-assisted precise, efficient and multiplexed genome-editing in Yarrowia lipolytica. Metab. Eng. Commun. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Whinn, K.S.; Kaur, G.; Lewis, J.S.; Schauer, G.D.; Mueller, S.H.; Jergic, S.; Maynard, H.; Gan, Z.Y.; Naganbabu, M.; Bruchez, M.P.; et al. Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.F.; Sturme, M.H.J.; D’Adamo, S.; Weusthuis, R.A.; Wijffels, R.H. Stable transformation of the green algae Acutodesmus obliquus and Neochloris oleoabundans based on E. coli conjugation. Algal Res. 2019, 39. [Google Scholar] [CrossRef]
- Karas, B.J.; Diner, R.E.; Lefebvre, S.C.; McQuaid, J.; Phillips, A.P.; Noddings, C.M.; Brunson, J.K.; Valas, R.E.; Deerinck, T.J.; Jablanovic, J.; et al. Designer diatom episomes delivered by bacterial conjugation. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Diner, R.E.; Bielinski, V.A.; Dupont, C.L.; Allen, A.E.; Weyman, P.D. Refinement of the diatom episome maintenance sequence and improvement of conjugation-based DNA delivery methods. Front. Bioeng. Biotechnol. 2016, 4, 65. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Pastwa, E.; Błasiak, J. Non-homologous DNA end joining. Acta Biochim. Pol. 2003, 50, 891–908. [Google Scholar] [CrossRef]
- Daley, J.M.; Laan, R.L.V.; Suresh, A.; Wilson, T.E. DNA joint dependence of Pol X family polymerase action in nonhomologous end joining. J. Biol. Chem. 2005, 280, 29030–29037. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. DNA end resection: Many nucleases make light work. DNA Repair 2009, 8, 983–995. [Google Scholar] [CrossRef]
- CRISPR 101: Homology Directed Repair. Available online: https://blog.addgene.org/crispr-101-homology-directed-repair (accessed on 13 May 2020).
- Pal, A.; Levy, Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 2019, 15, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Triana, J.A.; Tavhelidse, T.; Thumberger, T.; Thomas, I.; Wittbrodt, B.; Kellner, T.; Anlas, K.; Tsingos, E.; Wittbrodt, J. Efficient single-copy HDR by 5’ modified long dsDNA donors. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- León-Bañares, R.; González-Ballester, D.; Galván, A.; Fernández, E. Transgenic microalgae as green cell-factories. Trends Biotechnol. 2004, 22, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Shimogawara, K.; Fujiwara, S.; Grossman, A.; Usuda, H. High-efficiency transformation of Chlamydomonas reinhardtii by electroporation. Genetics 1998, 148, 1821–1828. [Google Scholar]
- Nelson, J.A.E.; Lefebvre, P.A. Chapter 73 Transformation of Chlamydomonas reinhardtii. Methods Cell Biol. 1995, 47, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Remacle, C.; Cardol, P.; Coosemans, N.; Gaisne, M.; Bonnefoy, N. High-efficiency biolistic transformation of Chlamydomonas mitochondria can be used to insert mutations in complex I genes. Proc. Natl. Acad. Sci. USA 2006, 103, 4771–4776. [Google Scholar] [CrossRef] [PubMed]
- Mini, P.; Demurtas, O.C.; Valentini, S.; Pallara, P.; Aprea, G.; Ferrante, P.; Giuliano, G. Agrobacterium-mediated and electroporation-mediated transformation of Chlamydomonas reinhardtii: A comparative study. BMC Biotechnol. 2018, 18, 11. [Google Scholar] [CrossRef]
- Brown, L.E.; Sprecher, S.L.; Keller, L.R. Introduction of exogenous DNA into Chlamydomonas reinhardtii by electroporation. Mol. Cell. Biol. 1991, 11, 2328–2332. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wen, X.; Chen, Z.; Liang, Q.; Li, J.; Wang, W. Rapid and high efficiency transformation of Chlamydomonas reinhardtii by square-wave electroporation. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Kindle, K.L. High-Frequency Nuclear Transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef]
- Boynton, J.E.; Gillham, N.W. Chloroplast Transformation in Chlamydomonas. Methods Enzymol. 1993, 217, 510–536. [Google Scholar] [CrossRef] [PubMed]
- Boynton, J.E.; Gillham, N.W.; Harris, E.H.; Hosler, J.P.; Johnson, A.M.; Jones, A.R.; Randolph-Anderson, B.L.; Robertson, D.; Klein, T.M.; Shark, K.B.; et al. Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 1988, 240, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Pratheesh, P.T.; Vineetha, M.; Kurup, G.M. An efficient protocol for the Agrobacterium-mediated genetic transformation of microalga Chlamydomonas reinhardtii. Mol. Biotechnol. 2014, 56, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Park, R.V.; Asbury, H.; Miller, S.M. Modification of a Chlamydomonas reinhardtii CRISPR/Cas9 transformation protocol for use with widely available electroporation equipment. MethodsX 2020, 7, 100855. [Google Scholar] [CrossRef]
- Purton, S.; Rochaix, J.D. Complementation of a Chlamydomonas reinhardtii mutant using a genomic cosmid library. Plant Mol. Biol. 1994, 24, 533–537. [Google Scholar] [CrossRef]
- Findinier, J.; Delevoye, C.; Cohen, M.M. The dynamin-like protein fzl promotes thylakoid fusion and resistance to light stress in Chlamydomonas reinhardtii. PLoS Genet. 2019, 15, e1008047. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Baek, K.; Jin, E. Site-Specific Gene Knock-Out and On-Site Heterologous Gene Overexpression in Chlamydomonas reinhardtii via a CRISPR-Cas9-Mediated Knock-in Method. Front. Plant Sci. 2020, 11, 306. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cas Protein | Bacterial Species | PAM Sequences | Used in C. reinhardtii | References |
|---|---|---|---|---|
| spCas9 native | Streptococcus pyrogenes | NGG | Yes | [35] |
| StCas9 | Streptococcus thermophilus | NNAGAAW | Yes | [36] |
| saCas9 | Staphylococcus aureus | NNGRRT or NNGRR(N) | Yes | [37] |
| nmCas9 | Neisseria meningitidis | NNNNGATT | Not yet | [36] |
| Cpf1 (Cas12) | Prevoltella and francisella 1 | NTT | Yes | [38] |
| dCas9 (dead) | Engineered S. pyrogenes | NGG | Not yet | [39] |
| nCas9 (nikase) | Engineered S. pyrogenes | NGG | Not yet | [40] |
| Tool Name | Species | Website | References |
|---|---|---|---|
| CRISPRko | Human and mouse. | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design | [41] |
| Cas-OFFinder | Vertebrates. Insects, plants, fungi, prokaryotes, C. reinhardtii | http://www.rgenome.net/cas-offinder/ | [42] |
| CRISPOR | Vertebrates, insects, plants, fungi, prokaryotes. | http://crispor.tefor.net/ | [43] |
| E-CRISP | Vertebrates, invertebrates. Plants, fungi, protists, and C. reinhardtii. | http://www.e-crisp.org/E-CRISP/aboutpage.html | [44] |
| Off–spotter | Human, mouse, fungi | https://cm.jefferson.edu/Off-Spotter/ | [45] |
| CRISPR-GE | Vertebrates, invertebrates, plants | http://skl.scau.edu.cn/targetdesign/ | [46] |
| Benchling | Various: vertebrates. Insects, plants, fungi, prokaryotes, and C. reinhardtii | https://benchling.com/ | - |
| Deskgen | Various: vertebrates. Insects, plants, fungi, prokaryotes | https://www.deskgen.com/ | [47] |
| CHOPCHOP | Various: vertebrates. Insects, plants, fungi, prokaryotes and Chlamydomonas | https://chopchop.rc.fas.harvard.edu/ | [48] |
| CRISPR MultiTargeter | Various: vertebrates. Insects, plants, fungi, prokaryotes | http://www.multicrispr.net/ | [49] |
| sgRNA Scorer | Various: vertebrates. Insects, plants, fungi, prokaryotes. | https://crispr.med.harvard.edu/sgRNAScorerV1/ | [50] |
| CRISPRscan | Various: vertebrates. Insects, plants, fungi, prokaryotes. | http://www.crisprscan.org/ | [51] |
| Methods of Transformation | DNA Type | DNA Quantity | Cells Quantity | Cells Type | Plasmid Size | Efficiency of Transformation | References |
|---|---|---|---|---|---|---|---|
| Electroporation | Not CRISPR | 100 ng | 1.0–2.0 × 107 cells ml−1 | WT | >10 Kb | 0.4–3 × 103 per μg | [76] |
| CRISPR | 2–4 µg | 100× 106 | Wall-less strain | Not mentioned | 31–367 | [81] | |
| Glass beads | Not CRISPR | 1–3 µg | 5 × 105 or OD750 nm: 0.5–0.8 | Preferably cell wall-less cells but also WT | Up to 10 Kb | Low (103–104) | [82] |
| CRISPR | 3 µg | 2 × 108 cells | 0.5 × 108 | [20] | |||
| Biolistic | Not CRISPR | 1–3 µg/µL | 106 to 107 cells/mL | Cell wall deficient strain | Up to 10 Kb | Low 300–600/3 µg of DNA | [73] |
| CRISPR | 1 μg/µL | 1 × 108 cells | Wild type | 0.5 × 108 | [20] | ||
| A. tumefaciens | Not CRISPR | Bacterial culture 40–200 µL | 108 cells/mL | Cell wall deficient strains | >10 Kb, specific plasmid | 108 | [80] |
| Author and Year | System | Targeted Gene | Codon Optimization | Cas | Proof of Concept | C. reinhardtii Strain | Efficiency of Mutations | Transformation Method | Off Target | Antibiotics |
|---|---|---|---|---|---|---|---|---|---|---|
| Jiang et al., 2014 [18] | knock-out to disrupt FKB12 gene | FKB12: C. reinhardtii cell lacking FKB12 are fully resistant to rapamycin antibiotics | yes | Cas9 and dCas9 as control of toxicity | Cells becoming resistant to rapamycin | CC-503 | Only one colony and failure to recover more transformants | electroporation | not studied | rapamycin |
| Baek et al., 2016 [15] | knock-out using DNA-free CRISPR-Cas9 method, disrupting CpFTSY and ZEP and generating a strain producing zeaxanthin | The knockout of the CpFTSY gene confers smaller, or truncated, chlorophyll (Chl) antenna size of the photosystems. The knockout of zeaxanthin epoxidase (ZEP) leads to constitutive accumulation of zeaxanthin | yes | Cas9 | CpFTSY: pale green color ZEP: zeaxanthin accumulation CpFTSY/ZEP: zeaxanthin and golden color | CC-4349 cw15 mt- | CpFTSY:0.56% ZEP: 0.46% | electroporation | CpFTSY: far up to 4 nucleotides from the on-target ZEP: no off-target | no antibiotics used |
| Sung shin et al., 2016 [22] | Complex RNP for knock-out, plasmids CpSRP43 and pCr202 | MAA7 encodes for the beta subunit of tryptophan synthase | yes | Cas9 | 5-fluoro-indole | CC-124 | 40% | electroporation | no off-target events | no antibiotics used |
| CpSRP43: antenna assembly genes encode for chloroplast | Light colony color and low chlorophyll dosage | 1.4% | ||||||||
| ChLM: when disrupted affects chlorophyll biosynthesis and reduces the accumulation of major thylakoid-associated proteins | 1.7% | |||||||||
| Greiner et al., 2017 [14] | RNP and plasmid knock-out of PSY1 | PSY1: Phytoene synthase 1 catalyze carotenoids synthesis and photosynthesis | yes | Cas9 | white colony | CC-125 | not mentioned | electroporation | no off-target events | paromomycin antibiotics |
| Baek et al., 2017 [23] | Knock-out of zeaxanthin epoxidase with RNP CRISPR | ZEP: zeaxanthin epoxidase, when disrupted the microalgae accumulates zeaxanthin | - | Cas9 | zeaxanthin dosage | CC-4349, CC-124, CC-620, CC-4051, CC-4533 | not mentioned | glass beads | no off-target (Cas-OFFinder, reported in [15]) | no antibiotics used |
| Ferenczi et al., 2017 [21] | RNP CRISPR for the gene Fkb12 with a repair plasmid ssODN | Fkb12: mediates the interaction between the antibiotic rapamycin and the cell cycle regulator target of rapamycin, which leads to cell death. | yes | LbCPpf1 | high tolerance to rapamycin | CC-1883, CC-2931 | 29% | electroporation | no predicted off-target sites with Cas-ORFinder | Rapamycin |
| Findinier et al., 2019 [83] | RNP CRISPR for the gene CrFzI | CrFzi: phenotypic analyses revealed a specific requirement of CrFzl for survival upon light stress. Consistent with this, strong irradiance leads to increased photoinhibition of photosynthesis in mutant cells | not mentioned | Cas9 | when Fzi is disrupted, it promotes thylakoid fusion and resistance to light stress | cc-4533, cw15.J3 | not mentioned | electroporation | not mentioned | Hygromycin resistant gene |
| Guzmán-Zapata et al., 2019 [20] | plasmid CRISPR | Apt: sensitivity to a toxin 2-fluoroadenine (2-FA) | Yes | Cas9 | growth on the 2-FA toxin | CC-125, CC-3403, SAG73.72, CC-3403-uvr8-2 | not mentioned | glass beads/particle bombardment | not mentioned | no antibiotic used |
| Kim et al., 2020 [84] | knock- out RNP CRISPR Cas9 and knock-in antibiotics resistance gene and YFP | CrFTSY: confers smaller, or truncated, chlorophyll (Chl) antenna size of the photosystems ([15]) | not mentioned | Cas9 | pale green color and measurement of luciferase activity | CC-4349, CC-124, and CC-503 | up to 30% | electroporation | not mentioned | hygromycin, paromomycin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghribi, M.; Nouemssi, S.B.; Meddeb-Mouelhi, F.; Desgagné-Penix, I. Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas. Life 2020, 10, 295. https://doi.org/10.3390/life10110295
Ghribi M, Nouemssi SB, Meddeb-Mouelhi F, Desgagné-Penix I. Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas. Life. 2020; 10(11):295. https://doi.org/10.3390/life10110295
Chicago/Turabian StyleGhribi, Manel, Serge Basile Nouemssi, Fatma Meddeb-Mouelhi, and Isabel Desgagné-Penix. 2020. "Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas" Life 10, no. 11: 295. https://doi.org/10.3390/life10110295
APA StyleGhribi, M., Nouemssi, S. B., Meddeb-Mouelhi, F., & Desgagné-Penix, I. (2020). Genome Editing by CRISPR-Cas: A Game Change in the Genetic Manipulation of Chlamydomonas. Life, 10(11), 295. https://doi.org/10.3390/life10110295