Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes



Abstract
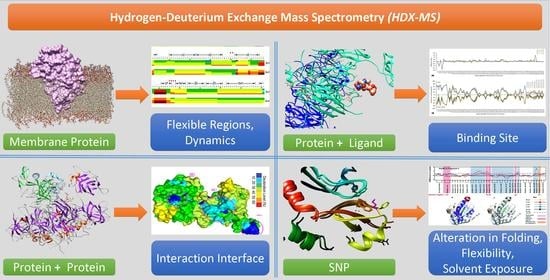
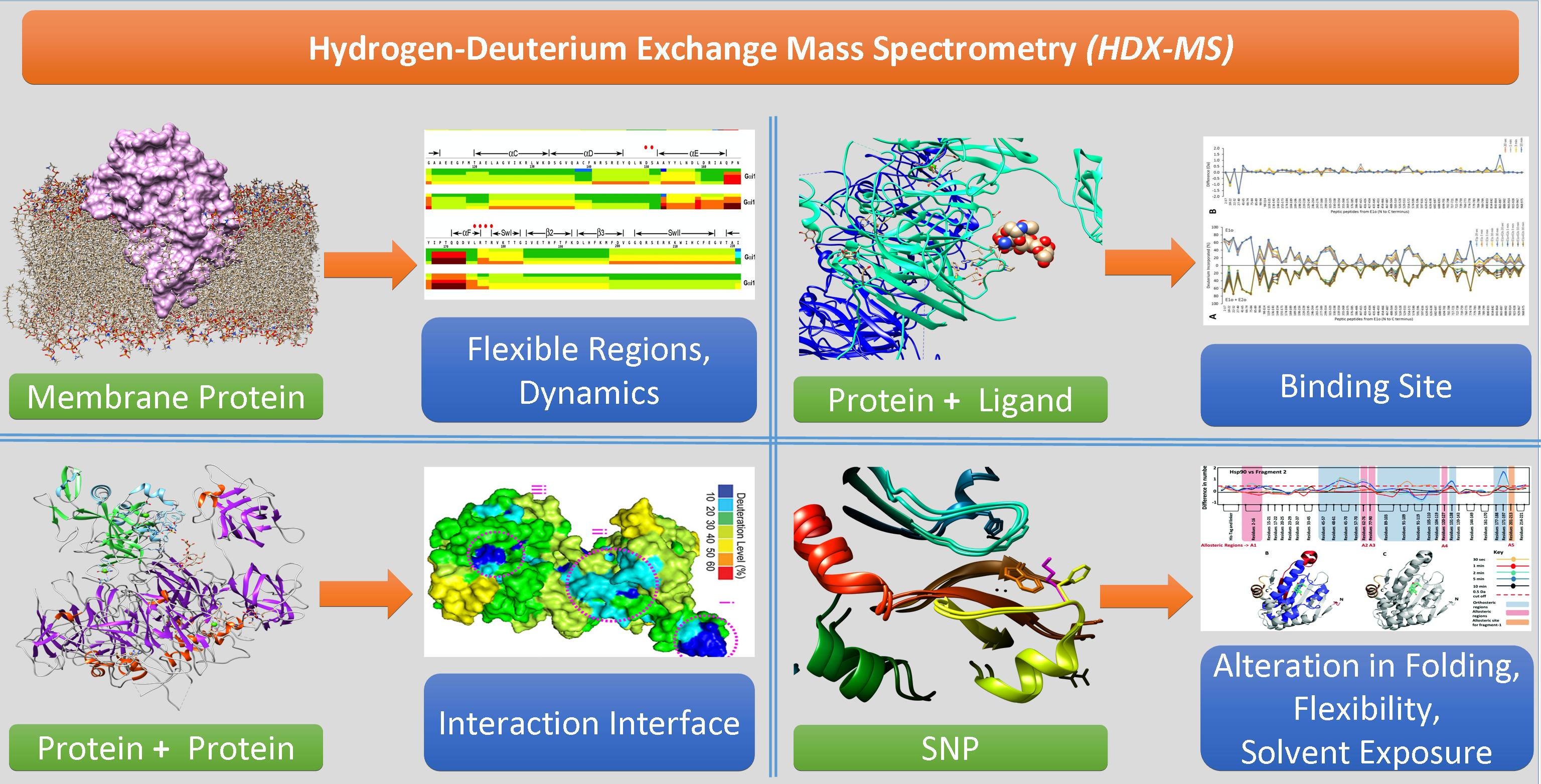{kind=link}
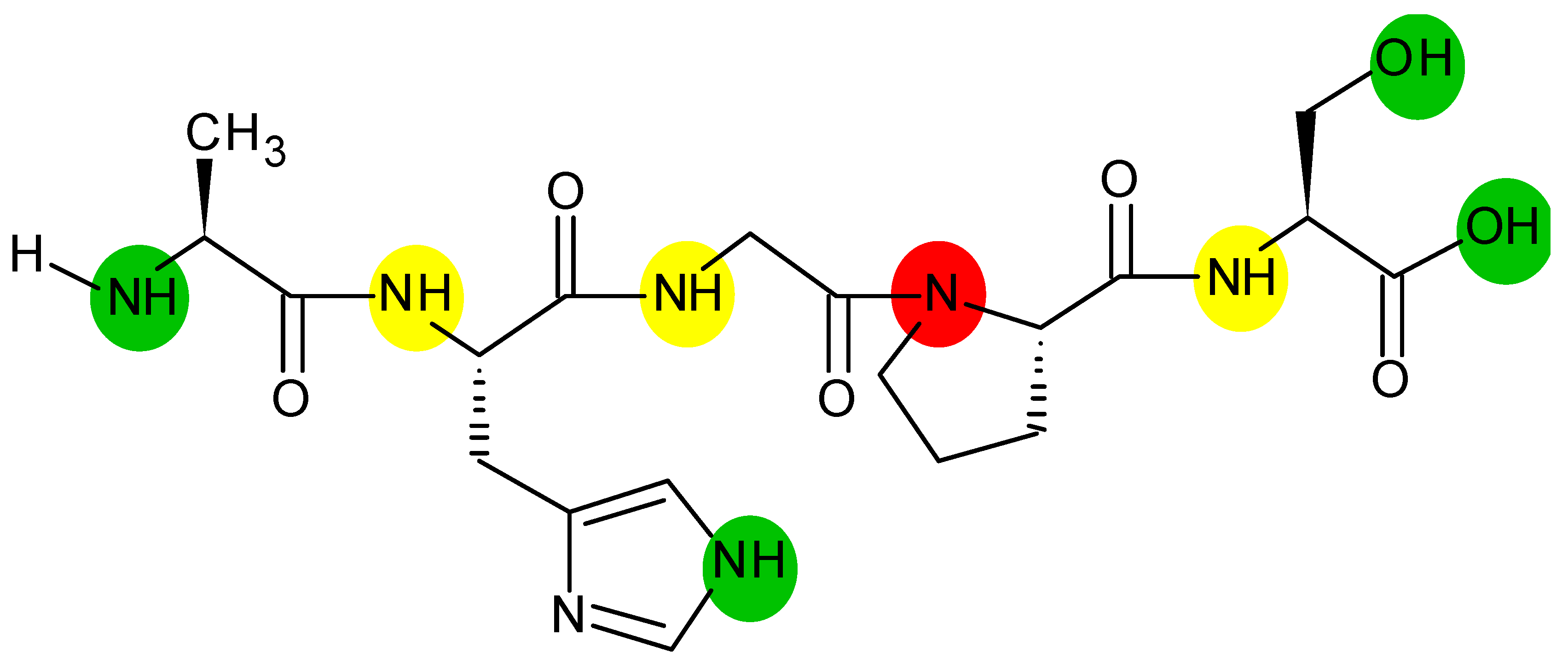{kind=link}
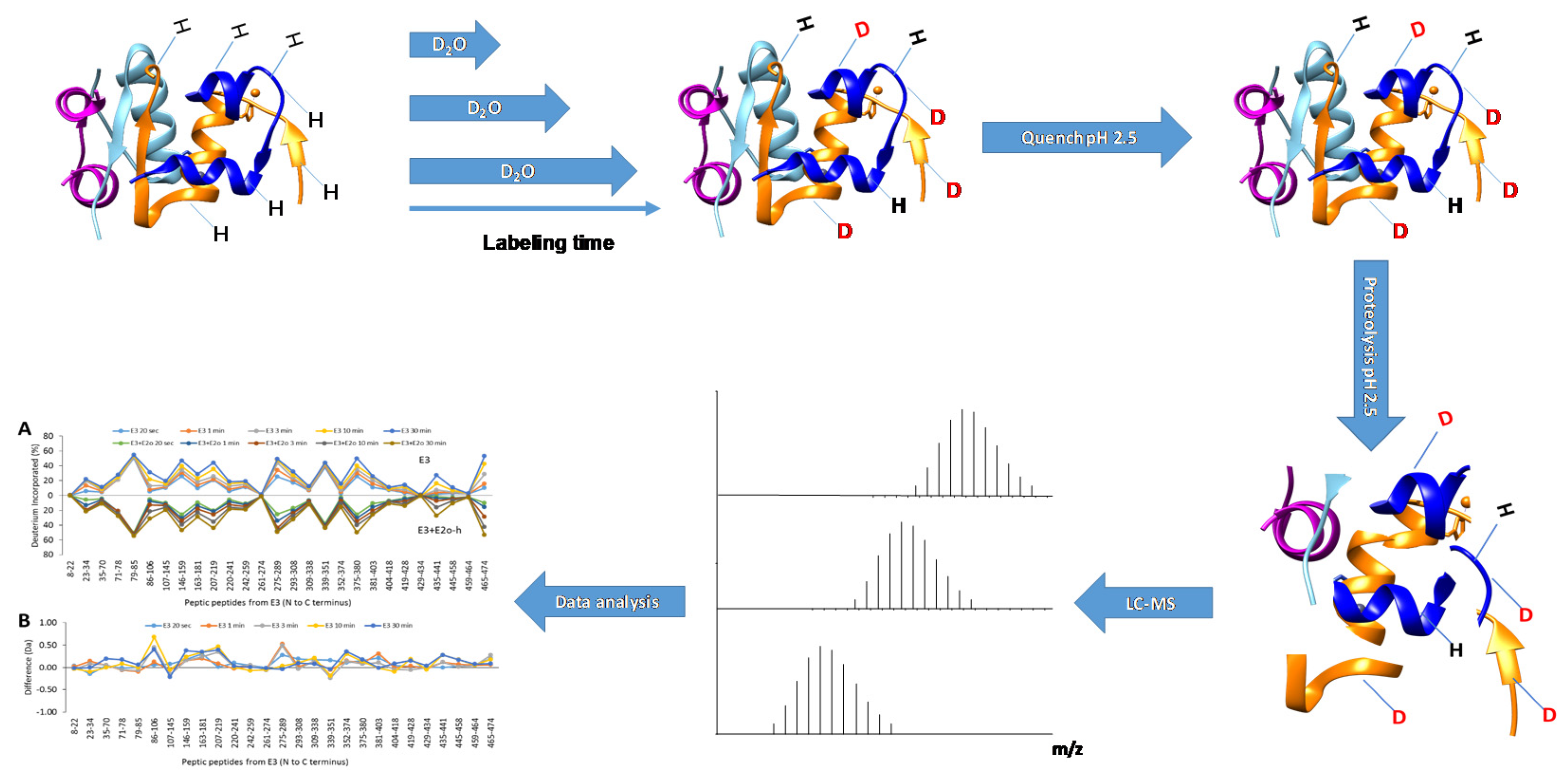{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theory of HDX-MS
2.1. Workflow and Equipment
2.2. Strengths and Weaknesses—Comparison to NMR
3. Applications
3.1. Analysis of Protein Conformation and Folding
3.2. Analysis of Protein Interactions
3.3. Proteins with Glycosylation and Disulfide Bonding
3.4. Membrane Proteins
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Englander, S.W.; Mayne, L.; Bai, Y.; Sosnick, T.R. Hydrogen exchange: The modern legacy of Linderstrøm-Lang. Protein Sci. Publ. Protein Soc. 1997, 6, 1101–1109. [Google Scholar] [CrossRef]
- Englander, S.W.; Sosnick, T.R.; Englander, J.J.; Mayne, L. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 1996, 6, 18–23. [Google Scholar] [CrossRef]
- Weis, D.D. (Ed.) Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications; John Wiley & Sons Ltd.: Chichester, UK, 2016. [Google Scholar]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef]
- Karch, K.R.; Coradin, M.; Zandarashvili, L.; Kan, Z.Y.; Gerace, M.; Englander, S.W.; Black, B.E.; Garcia, B. Hydrogen-Deuterium Exchange Coupled to Top- and Middle-Down Mass Spectrometry Reveals Histone Tail Dynamics before and after Nucleosome Assembly. Structure 2018, 26, 1651–1663.e3. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Sarpe, V.; Burns, K.M.; Buse, J.; Baker, C.A.; van Dijk, M.; Wordeman, L.; Bonvin, A.M.J.J.; Schriemer, D.C. Mass spec studio for integrative structural biology. Structure 2014, 22, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Rostislavleva, K.; Soler, N.; Ohashi, Y.; Zhang, L.; Pardon, E.; Burke, J.E.; Masson, G.R.; Johnson, C.; Steyaert, J.; Ktistakis, N.T.; et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science 2015, 350, aac7365. [Google Scholar] [CrossRef] [PubMed]
- Sheff, J.G.; Hepburn, M.; Yu, Y.; Lees-Miller, S.P.; Schriemer, D.C. Nanospray HX-MS configuration for structural interrogation of large protein systems. Analyst 2017, 142, 904–910. [Google Scholar] [CrossRef]
- Zhang, Z.; Vachet, R.W. Kinetics of Protein Complex Dissociation Studied by Hydrogen/Deuterium Exchange and Mass Spectrometry. Anal. Chem. 2015, 87, 11777–11783. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, L.; Ozohanics, O.; Zhang, X.; Wang, J.; Ambrus, A.; Arjunan, P.; Brukh, R.; Nemeria, N.S.; Furey, W.; et al. A multipronged approach unravels unprecedented protein-protein interactions in the human 2-oxoglutarate dehydrogenase multienzyme complex. J. Biol. Chem. 2018, 293, 19213–19227. [Google Scholar] [CrossRef]
- Chandramohan, A.; Krishnamurthy, S.; Larsson, A.; Nordlund, P.; Jansson, A.; Anand, G.S. Predicting Allosteric Effects from Orthosteric Binding in Hsp90-Ligand Interactions: Implications for Fragment-Based Drug Design. PloS Comput. Biol. 2016, 12, e1004840. [Google Scholar] [CrossRef]
- De Vera, I.M.S.; Zheng, J.; Novick, S.; Shang, J.; Hughes, T.S.; Brust, R.; Munoz-Tello, P.; Gardner, W.J.; Marciano, D.P.; Kong, X.; et al. Synergistic Regulation of Coregulator/Nuclear Receptor Interaction by Ligand and DNA. Structure 2017, 25, 1506–1518.e4. [Google Scholar] [CrossRef] [PubMed]
- Espada, A.; Haro, R.; Castañon, J.; Sayago, C.; Perez-Cozar, F.; Cano, L.; Redero, P.; Molina-Martin, M.; Broughton, H.; Stites, R.E.; et al. A Decoupled Automation Platform for Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. J. Am. Soc. Mass Spectrom. 2019, 30, 2580–2583. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.S.; Hudgens, J.W. Mapping Protein-Ligand Interactions with Proteolytic Fragmentation, Hydrogen/Deuterium Exchange-Mass Spectrometry. In Isotope Labeling of Biomolecules—Applications; Kelman, Z., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2016; Volume 566, pp. 357–404. [Google Scholar]
- Giladi, M.; Khananshvili, D. Hydrogen-Deuterium Exchange Mass-Spectrometry of Secondary Active Transporters: From Structural Dynamics to Molecular Mechanisms. Front. Pharmacol. 2020, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.; Marciano, D.P.; Pascal, B.D.; Chalmers, M.J.; Griffin, P.R. Application of Differential Hydrogen Exchange Mass Spectrometry in Small Molecule Drug Discovery. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 209–223. [Google Scholar]
- Zhang, Y.; Rempel, D.L.; Gross, M.L. Hydrogen Exchange Mass Spectrometry for the Analysis of Ligand Binding and Protein Aggregation. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 185–207. [Google Scholar]
- Huang, R.Y.; Kuhne, M.; Deshpande, S.; Rangan, V.; Srinivasan, M.; Wang, Y.; Chen, G. Mapping binding epitopes of monoclonal antibodies targeting major histocompatibility complex class I chain-related A (MICA) with hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Husain, N.; Tulsian, N.K.; Chien, W.L.; Suresh, S.; Anand, G.S.; Sivaraman, J. Ligand-mediated changes in conformational dynamics of NpmA: Implications for ribosomal interactions. Sci. Rep. 2016, 6, 37061. [Google Scholar] [CrossRef]
- Masson, G.R.; Jenkins, M.L.; Burke, J.E. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert. Opin. Drug Discov. 2017, 12, 981–994. [Google Scholar] [CrossRef]
- Pacholarz, K.J.; Burnley, R.J.; Jowitt, T.A.; Ordsmith, V.; Pisco, J.P.; Porrini, M.; Larrouy-Maumus, G.; Garlish, R.A.; Taylor, R.J.; De Carvalho, L.P.S.; et al. Hybrid Mass Spectrometry Approaches to Determine How L-Histidine Feedback Regulates the Enzyzme MtATP-Phosphoribosyltransferase. Structure 2017, 25, 730–738.e4. [Google Scholar] [CrossRef]
- Redhair, M.; Hackett, J.C.; Pelletier, R.D.; Atkins, W.M. Dynamics and Location of the Allosteric Midazolam Site in Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 2020, 59, 766–779. [Google Scholar] [CrossRef]
- Sowole, M.A.; Konermann, L. Effects of protein-ligand interactions on hydrogen/deuterium exchange kinetics: Canonical and noncanonical scenarios. Anal. Chem. 2014, 86, 6715–6722. [Google Scholar] [CrossRef]
- Zhang, X.; Chien, E.Y.T.; Chalmers, M.J.; Pascal, B.D.; Gatchalian, J.; Stevens, R.C.; Griffin, P.R. Dynamics of the β2-Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen−Deuterium Exchange. Anal. Chem. 2010, 82, 1100–1108. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; DeColli, A.; Freel Meyers, C.; Nemeria, N.S.; Jordan, F. Conformational dynamics of 1-deoxy-d-xylulose 5-phosphate synthase on ligand binding revealed by H/D exchange MS. Proc. Natl. Acad. Sci. USA 2017, 114, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Trabjerg, E.; Nazari, Z.E.; Rand, K.D. Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): Challenges and emerging solutions. TrAC Trends Anal. Chem. 2018, 106, 125–138. [Google Scholar] [CrossRef]
- Englander, J.J.; Del Mar, C.; Li, W.; Englander, S.W.; Kim, J.S.; Stranz, D.D.; Hamuro, Y.; Woods, V.L. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. USA 2003, 100, 7057–7062. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Krishnamurthy, S.; Anand, G.S. Hydrogen Exchange Mass Spectrometry Experimental Design. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 19–35. [Google Scholar]
- Guo, C.; Steinberg, L.K.; Cheng, M.; Song, J.H.; Henderson, J.P.; Gross, M.L. Site-Specific Siderocalin Binding to Ferric and Ferric-Free Enterobactin As Revealed by Mass Spectrometry. ACS Chem. Biol. 2020, 15, 1154–1160. [Google Scholar] [CrossRef]
- Majumdar, R.C.; Middaugh, R.; Weis, D.D.; Volkin, D.B. Hydrogen Exchange Mass Spectrometry as an Emerging Analytical Tool for Stabilization and Formulation Development of Therapeutic Monoclonal Antibodies. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 323–342. [Google Scholar]
- Alexovič, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116. [Google Scholar] [CrossRef]
- Huang, R.Y.C.; Tymiak, A.A.; Chen, G. Utility of Hydrogen Exchange Mass Spectrometry in Epitope Mapping. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 247–263. [Google Scholar]
- Skorupska, A.; Bystranowska, D.; Dąbrowska, K.; Ożyhar, A. Calcium ions modulate the structure of the intrinsically disordered Nucleobindin-2 protein. Int. J. Biol. Macromol. 2020, 154, 1091–1104. [Google Scholar] [CrossRef]
- Harrison, R.A.; Engen, J.R. Conformational insight into multi-protein signaling assemblies by hydrogen-deuterium exchange mass spectrometry. Curr. Opin. Struct. Biol. 2016, 41, 187–193. [Google Scholar] [CrossRef]
- Kim, S.J.; Fernandez-Martinez, J.; Nudelman, I.; Shi, Y.; Zhang, W.; Raveh, B.; Herricks, T.; Slaughter, B.D.; Hogan, J.A.; Upla, P.; et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature 2018, 555, 475–482. [Google Scholar] [CrossRef]
- Masson, G.R.; Maslen, S.L.; Williams, R.L. Analysis of phosphoinositide 3-kinase inhibitors by bottom-up electron-transfer dissociation hydrogen/deuterium exchange mass spectrometry. Biochem. J. 2017, 474, 1867–1877. [Google Scholar] [CrossRef]
- Kaltashov, I.A.; Abzalimov, R.R.; Wang, G.; Bobst, C.E. Top-Down Hydrogen Exchange Mass Spectrometry. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 149–164. [Google Scholar]
- Sanguantrakun, N.; Chanthamontri, C.; Gross, M.L. Top-Down Analysis of In-Source HDX of Native Protein Ions. J. Am. Soc. Mass Spectrom. 2020, 31, 1151–1154. [Google Scholar] [CrossRef]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Rand, K.D.; Zehl, M.; Jorgensen, T.J.D. Measuring the Hydrogen/Deuterium Exchange of Proteins at High Spatial Resolution by Mass Spectrometry: Overcoming Gas-Phase Hydrogen/Deuterium Scrambling. Acc. Chem. Res. 2014, 47, 3018–3027. [Google Scholar] [CrossRef]
- Moroco, J.A.; Engen, J.R. Replication in bioanalytical studies with HDX MS: Aim as high as possible. Bioanalysis 2015, 7, 1065–1067. [Google Scholar] [CrossRef]
- Oganesyan, I.; Lento, C.; Wilson, D.J. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 2018, 144, 27–42. [Google Scholar] [CrossRef]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Bioinform. 1993, 17, 75–86. [Google Scholar] [CrossRef]
- Nguyen, D.; Mayne, L.; Phillips, M.C.; Englander, S.W. Reference Parameters for Protein Hydrogen Exchange Rates. J. Am. Soc. Mass Spectrom. 2018, 29, 1936–1939. [Google Scholar] [CrossRef]
- Covington, A.K.; Paabo, M.; Robinson, R.A.; Bates, R.G. Use of the glass electrode in deuterium oxide and the relation between the standardized pD (paD) scale and the operational pH in heavy water. Anal. Chem. 1968, 40, 700–706. [Google Scholar] [CrossRef]
- Hudgens, J.W.; Gallagher, E.S.; Karageorgos, I.; Anderson, K.W.; Filliben, J.J.; Huang, R.Y.C.; Chen, G.; Bou-Assaf, G.M.; Espada, A.; Chalmers, M.J.; et al. Interlaboratory Comparison of Hydrogen–Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Anal. Chem. 2019, 91, 7336–7345. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 16, 521–655. [Google Scholar] [CrossRef]
- Hudgens, J.W.; Gallagher, E.S.; Karageorgos, I.; Anderson, K.W.; Huang, R.Y.C.; Chen, G.; Bou-Assaf, G.M.; Espada, A.; Chalmers, M.J.; Harguindey, E.; et al. Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) Centroid Data Measured between 3.6 °C and 25.4 °C for the Fab Fragment of NISTmAb. J. Res. Natl. Inst. Stand. Technol. 2019, 124, 1–7. [Google Scholar] [CrossRef]
- Wales, T.E.; Fadgen, K.E.; Eggertson, M.J.; Engen, J.R. Subzero Celsius separations in three-zone temperature controlled hydrogen deuterium exchange mass spectrometry. J. Chromatogr. A 2017, 1523, 275–282. [Google Scholar] [CrossRef]
- Venable, J.D.; Okach, L.; Agarwalla, S.; Brock, A. Subzero temperature chromatography for reduced back-exchange and improved dynamic range in amide hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2012, 84, 9601–9608. [Google Scholar] [CrossRef]
- Hudgens, J.W. Construction of a Dual Protease Column, Subzero (−30 °C) Chromatography System and Multi-channel Precision Temperature Controller for Hydrogen-Deuterium Exchange Mass Spectrometry. J. Res. Natl. Inst. Stand. Technol. 2020, 125, 7. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Z.; Smith, D.L. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J. Am. Soc. Mass Spectrom. 1999, 10, 675–684. [Google Scholar] [CrossRef]
- Slysz, G.W.; Percy, A.J.; Schriemer, D.C. Restraining expansion of the peak envelope in H/D exchange-MS and its application in detecting perturbations of protein structure/dynamics. Anal. Chem. 2008, 80, 7004–7011. [Google Scholar] [CrossRef]
- Arora, J.; Hickey, J.M.; Majumdar, R.; Esfandiary, R.; Bishop, S.M.; Samra, H.S.; Middaugh, C.R.; Weis, D.D.; Volkin, D.B. Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. mAbs 2015, 7, 525–539. [Google Scholar] [CrossRef]
- Konermann, L.; Tong, X.; Pan, Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J. Mass Spectrom. 2008, 43, 1021–1036. [Google Scholar] [CrossRef]
- Kaltashov, I.A. Probing protein dynamics and function under native and mildly denaturing conditions with hydrogen exchange and mass spectrometry. Int. J. Mass Spectrom. 2005, 240, 249–259. [Google Scholar] [CrossRef]
- Abzalimov, R.R.; Kaltashov, I.A. Extraction of local hydrogen exchange data from HDX CAD MS measurements by deconvolution of isotopic distributions of fragment ions. J. Am. Soc. Mass Spectrom. 2006, 17, 1543–1551. [Google Scholar] [CrossRef][Green Version]
- Xiao, H.; Hoerner, J.K.; Eyles, S.J.; Dobo, A.; Voigtman, E.; Mel’čuk, A.I.; Kaltashov, I.A. Mapping protein energy landscapes with amide hydrogen exchange and mass spectrometry: I. A generalized model for a two-state protein and comparison with experiment. Protein Sci. 2005, 14, 543–557. [Google Scholar] [CrossRef]
- Ambrus, A.; Wang, J.; Mizsei, R.; Zambo, Z.; Torocsik, B.; Jordan, F.; Adam-Vizi, V. Structural alterations induced by ten disease-causing mutations of human dihydrolipoamide dehydrogenase analyzed by hydrogen/deuterium-exchange mass spectrometry: Implications for the structural basis of E3 deficiency. Biochim. Biophys. Acta 2016, 1862, 2098–2109. [Google Scholar] [CrossRef]
- Zhang, H.-M.; McLoughlin, S.M.; Frausto, S.D.; Tang, H.; Emmett, M.R.; Marshall, A.G. Simultaneous Reduction and Digestion of Proteins with Disulfide Bonds for Hydrogen/Deuterium Exchange Monitored by Mass Spectrometry. Anal. Chem. 2010, 82, 1450–1454. [Google Scholar] [CrossRef]
- Trabjerg, E.; Jakobsen, R.U.; Mysling, S.; Christensen, S.; Jørgensen, T.J.; Rand, K.D. Conformational analysis of large and highly disulfide-stabilized proteins by integrating online electrochemical reduction into an optimized H/D exchange mass spectrometry workflow. Anal. Chem. 2015, 87, 8880–8888. [Google Scholar] [CrossRef]
- Comamala, G.; Wagner, C.; de la Torre, P.S.; Jakobsen, R.U.; Hilger, M.; Brouwer, H.-J.; Rand, K.D. Hydrogen/deuterium exchange mass spectrometry with improved electrochemical reduction enables comprehensive epitope mapping of a therapeutic antibody to the cysteine-knot containing vascular endothelial growth factor. Anal. Chim. Acta 2020, 1115, 41–51. [Google Scholar] [CrossRef]
- Cravello, L.; Lascoux, D.; Forest, E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 2003, 17, 2387–2393. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Akeroyd, M.; Olsthoorn, M.; Heck, A.J.R. Aspergillus niger Prolyl Endoprotease for Hydrogen–Deuterium Exchange Mass Spectrometry and Protein Structural Studies. Anal. Chem. 2017, 89, 7966–7973. [Google Scholar] [CrossRef]
- Nirudodhi, S.N.; Sperry, J.B.; Rouse, J.C.; Carroll, J.A. Application of Dual Protease Column for HDX-MS Analysis of Monoclonal Antibodies. J. Pharm. Sci. 2017, 106, 530–536. [Google Scholar] [CrossRef]
- Black, W.A.; Stocks, B.B.; Mellors, J.S.; Engen, J.R.; Ramsey, J.M. Utilizing Microchip Capillary Electrophoresis Electrospray Ionization for Hydrogen Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 6280–6287. [Google Scholar] [CrossRef]
- Wales, T.E.; Fadgen, K.E.; Gerhardt, G.C.; Engen, J.R. High-Speed and High-Resolution UPLC Separation at Zero Degrees Celsius. Anal. Chem. 2008, 80, 6815–6820. [Google Scholar] [CrossRef]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Wollenberg, D.T.W.; Pengelley, S.; Mouritsen, J.C.; Suckau, D.; Jørgensen, C.I.; Jørgensen, T.J.D. Avoiding H/D Scrambling with Minimal Ion Transmission Loss for HDX-MS/MS-ETD Analysis on a High-Resolution Q-TOF Mass Spectrometer. Anal. Chem. 2020, 92, 7453–7461. [Google Scholar] [CrossRef]
- Zehl, M.; Rand, K.D.; Jensen, O.N.; Jørgensen, T.J.D. Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc. 2008, 130, 17453–17459. [Google Scholar] [CrossRef]
- Kabaria, S.R.; Mangion, I.; Makarov, A.A.; Pirrone, G.F. Use of MALDI-MS with solid-state hydrogen deuterium exchange for semi-automated assessment of peptide and protein physical stability in lyophilized solids. Anal. Chim. Acta 2019, 1054, 114–121. [Google Scholar] [CrossRef]
- Pascal, B.D.; Willis, S.; Lauer, J.L.; Landgraf, R.R.; West, G.M.; Marciano, D.; Novick, S.; Goswami, D.; Chalmers, M.J.; Griffin, P.R. HDX Workbench: Software for the Analysis of H/D Exchange MS Data. J. Am. Soc. Mass Spectrom. 2012, 23, 1512–1521. [Google Scholar] [CrossRef]
- Lumpkin, R.J.; Komives, E.A. DECA, A Comprehensive, Automatic Post-processing Program for HDX-MS Data. Mol. Cell. Proteom. 2019, 18, 2516–2523. [Google Scholar] [CrossRef]
- Slysz, G.W.; Baker, C.A.H.; Bozsa, B.M.; Dang, A.; Percy, A.J.; Bennett, M.; Schriemer, D.C. Hydra: Software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinform. 2009, 10, 162. [Google Scholar] [CrossRef]
- Kan, Z.Y.; Mayne, L.; Chetty, P.S.; Englander, S.W. ExMS: Data analysis for HX-MS experiments. J. Am. Soc. Mass Spectrom. 2011, 22, 1906–1915. [Google Scholar] [CrossRef]
- Liu, S.M.; Liu, L.T.; Uzuner, U.; Zhou, X.; Gu, M.X.; Shi, W.B.; Zhang, Y.; Uzuner, U.; Yuan, J.S. HDX-Analyzer: A novel package for statistical analysis of protein structure dynamics. BMC Bioinform. 2011, 12, 10. [Google Scholar] [CrossRef]
- Lindner, R.; Lou, X.H.; Reinstein, J.; Shoeman, R.L.; Hamprecht, F.A.; Winkler, A. Hexicon 2: Automated Processing of Hydrogen-Deuterium Exchange Mass Spectrometry Data with Improved Deuteration Distribution Estimation. J. Am. Soc. Mass Spectrom. 2014, 25, 1018–1028. [Google Scholar] [CrossRef]
- Bouyssié, D.; Lesne, J.; Locard-Paulet, M.; Albigot, R.; Burlet-Schiltz, O.; Marcoux, J. HDX-Viewer: Interactive 3D visualization of hydrogen–deuterium exchange data. Bioinformatics 2019, 35, 5331–5333. [Google Scholar] [CrossRef]
- Claesen, J.; Burzykowski, T. Computational methods and challenges in hydrogen/deuterium exchange mass spectrometry. Mass Spectrom Rev. 2017, 36, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Weis, D.D. Comment on Houde, D.; Berkowitz, S.A.; Engen, J.R. The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J. Pharm. Sci. 2011, 100, 2071–2086. J. Pharm. Sci. 2019, 108, 807–810. [Google Scholar] [CrossRef]
- Wales, T.E.; Eggertson, M.J.; Engen, J.R. Considerations in the Analysis of Hydrogen Exchange Mass Spectrometry Data. In Mass Spectrometry Data Analysis in Proteomics; Matthiesen, R., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 263–288. [Google Scholar]
- Sarpe, V.; Schriemer, D.C. Data Processing in Bottom-Up Hydrogen Exchange Mass Spectrometry. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 37–53. [Google Scholar]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, B.S.; Ghomi, H.T.; Lill, M.A.; Topp, E.M. Structural transitions and interactions in the early stages of human glucagon amyloid fibrillation. Biophys. J. 2015, 108, 937–948. [Google Scholar] [CrossRef]
- Moorthy, B.S.; Schultz, S.G.; Kim, S.G.; Topp, E.M. Predicting protein aggregation during storage in lyophilized solids using solid state amide hydrogen/deuterium exchange with mass spectrometric analysis (ssHDX-MS). Mol. Pharm. 2014, 11, 1869–1879. [Google Scholar] [CrossRef]
- Wang, H.; Shu, Q.; Frieden, C.; Gross, M.L. Deamidation Slows Curli Amyloid-Protein Aggregation. Biochemistry 2017, 56, 2865–2872. [Google Scholar] [CrossRef]
- Liu, G.X.; Tu, Z.C.; Wang, H.; Zhang, L.; Huang, T.; Ma, D. Monitoring of the functional properties and unfolding change of Ovalbumin after DHPM treatment by HDX and FTICR MS: Functionality and unfolding of Oval after DHPM by HDX and FTICR MS. Food Chem. 2017, 227, 413–421. [Google Scholar] [CrossRef]
- Geromanos, S.J.; Hughes, C.; Ciavarini, S.; Vissers, J.P.; Langridge, J.I. Using ion purity scores for enhancing quantitative accuracy and precision in complex proteomics samples. Anal. Bioanal. Chem. 2012, 404, 1127–1139. [Google Scholar] [CrossRef]
- Jensen, P.F.; Rand, K.D. Gas-Phase Fragmentation of Peptides to Increase the Spatial Resolution of the Hydrogen Exchange Mass Spectrometry Experiment. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 127–147. [Google Scholar]
- Huang, R.Y.; Garai, K.; Frieden, C.; Gross, M.L. Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry 2011, 50, 9273–9282. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, J. Extracting Information from Hydrogen Exchange Mass Spectrometry Data. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 107–125. [Google Scholar]
- Rand, K.D.; Zehl, M.; Jensen, O.N.; Jorgensen, T.J.D. Loss of Ammonia during Electron-Transfer Dissociation of Deuterated Peptides as an Inherent Gauge of Gas-Phase Hydrogen Scrambling. Anal. Chem. 2010, 82, 9755–9762. [Google Scholar] [CrossRef]
- Artigues, A.; Nadeau, O.W.; Rimmer, M.A.; Villar, M.T.; Du, X.; Fenton, A.W.; Carlson, G.M. Protein Structural Analysis via Mass Spectrometry-Based Proteomics. Adv. Exp. Med. Biol. 2016, 919, 397–431. [Google Scholar] [PubMed]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. Publ. Protein Soc. 1993, 2, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Weis, D.D.; Engen, J.R.; Lee, K.K. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J. Am. Soc. Mass Spectrom. 2013, 24, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Weis, D.D.; Engen, J.R.; Kass, I.J. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom 2006, 17, 1700–1703. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.Y.; Walters, B.T.; Mayne, L.; Englander, S.W. Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis. Proc. Natl. Acad. Sci. USA 2013, 110, 16438–16443. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ahn, J.; Yu, Y.Q.; Tymiak, A.; Engen, J.R.; Chen, G. Using hydrogen/deuterium exchange mass spectrometry to study conformational changes in granulocyte colony stimulating factor upon PEGylation. J. Am. Soc. Mass Spectrom. 2012, 23, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Berkowitz, S.A.; Engen, J.R. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011, 100, 2071–2086. [Google Scholar] [CrossRef]
- Skinner, J.J.; Lim, W.K.; Bédard, S.; Black, B.E.; Englander, S.W. Protein hydrogen exchange: Testing current models. Protein Sci. Publ. Protein Soc. 2012, 21, 987–995. [Google Scholar] [CrossRef]
- Brown, K.A.; Wilson, D.J. Bottom-up hydrogen deuterium exchange mass spectrometry: Data analysis and interpretation. Analyst 2017, 142, 2874–2886. [Google Scholar] [CrossRef]
- McKenzie-Coe, A.; Shortt, R.; Jones, L.M. The Making of a Footprint in Protein Footprinting: A Review in Honor of Michael L. Gross. Mass Spectrom. Rev. 2020, 24. [Google Scholar] [CrossRef]
- Engen, J.R.; Komives, E.A. Complementarity of Hydrogen/Deuterium Exchange Mass Spectrometry and Cryo-Electron Microscopy. Trends Biochem. Sci. 2020, 45, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Deredge, D.J.; Nagarajan, A.; Forrest, L.R.; Wintrode, P.L.; Singh, S.K. Conformational dynamics of a neurotransmitter: Sodium symporter in a lipid bilayer. Proc. Natl. Acad. Sci. USA 2017, 114, E1786–E1795. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.X.; Chandramohan, A.; Lim, X.E.; Crowe, J.E., Jr.; Lok, S.M.; Anand, G.S. Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 2017, 25, 1391–1402.e3. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.A.; Bondensgaard, K.; Nørskov-Lauritsen, L.; Christensen, L.; Becker, P.; Andersen, M.D.; Maltesen, M.J.; Rand, K.D.; Breinholt, J. Crystal structure of a prolactin receptor antagonist bound to the extracellular domain of the prolactin receptor. J. Biol. Chem. 2008, 283, 19085–19094. [Google Scholar] [CrossRef] [PubMed]
- Forest, E.; Rey, M. Hydrogen Exchange Mass Spectrometry of Membrane Proteins. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 279–294. [Google Scholar]
- Hodge, E.A.; Benhaim, M.A.; Lee, K.K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 2020, 29, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, R.; Migas, L.G.; Das, R.K.; Pappu, R.V.; Kriwacki, R.W.; Barran, P.E. Ion Mobility Mass Spectrometry Uncovers the Impact of the Patterning of Oppositely Charged Residues on the Conformational Distributions of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2019, 141, 4908–4918. [Google Scholar] [CrossRef]
- Rob, T.; Gill, P.K.; Golemi-Kotra, D.; Wilson, D.J. An electrospray ms-coupled microfluidic device for sub-second hydrogen/deuterium exchange pulse-labelling reveals allosteric effects in enzyme inhibition. Lab Chip 2013, 13, 2528–2532. [Google Scholar] [CrossRef]
- Wilson, D.J.; Konermann, L. A Capillary Mixer with Adjustable Reaction Chamber Volume for Millisecond Time-Resolved Studies by Electrospray Mass Spectrometry. Anal. Chem. 2003, 75, 6408–6414. [Google Scholar] [CrossRef]
- Rob, T.; Wilson, D.J. Time-Resolved Mass Spectrometry for Monitoring Millisecond Time-Scale Solution-Phase Processes. Eur. J. Mass Spectrom. 2012, 18, 205–214. [Google Scholar] [CrossRef]
- Zhu, S.; Khatun, R.; Lento, C.; Sheng, Y.; Wilson, D.J. Enhanced Binding Affinity via Destabilization of the Unbound State: A Millisecond Hydrogen-Deuterium Exchange Study of the Interaction between p53 and a Pleckstrin Homology Domain. Biochemistry 2017, 56, 4127–4133. [Google Scholar] [CrossRef]
- Wilson, D.J. Millisecond Hydrogen Exchange. In Hydrogen Exchange Mass Spectrometry of Proteins; Weis, D.D., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 73–91. [Google Scholar]
- Bowman, G.R.; Voelz, V.A.; Pande, V.S. Taming the complexity of protein folding. Curr. Opin. Struct. Biol. 2011, 21, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Trabjerg, E.; Kartberg, F.; Christensen, S.; Rand, K.D. Conformational characterization of nerve growth factor-β reveals that its regulatory pro-part domain stabilizes three loop regions in its mature part. J. Biol. Chem. 2017, 292, 16665–16676. [Google Scholar] [CrossRef] [PubMed]
- Iacob, R.E.; Pene-Dumitrescu, T.; Zhang, J.; Gray, N.S.; Smithgall, T.E.; Engen, J.R. Conformational disturbance in Abl kinase upon mutation and deregulation. Proc. Natl. Acad. Sci. USA 2009, 106, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Udgaonkar, J.B. Dissection of conformational conversion events during prion amyloid fibril formation using hydrogen exchange and mass spectrometry. J. Mol. Biol. 2013, 425, 3510–3521. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zheng, Q. Applications of Mass Spectrometry in the Onset of Amyloid Fibril Formation: Focus on the Analysis of Early-Stage Oligomers. Front. Chem. 2020, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Friedrich, K.; Somogyi, A. Oligomerization of nitrophorins. Anal. Biochem. 2006, 352, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A. An Updated View on the Molecular Pathomechanisms of Human Dihydrolipoamide Dehydrogenase Deficiency in Light of Novel Crystallographic Evidence. Neurochem. Res. 2019, 44, 2307–2313. [Google Scholar] [CrossRef]
- Ambrus, A.; Adam-Vizi, V. Human dihydrolipoamide dehydrogenase (E3) deficiency: Novel insights into the structural basis and molecular pathomechanism. Neurochem. Int. 2018, 117, 5–14. [Google Scholar] [CrossRef]
- Ambrus, A.; Adam-Vizi, V. Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch. Biochem. Biophys. 2013, 538, 145–155. [Google Scholar] [CrossRef]
- Szabo, E.; Mizsei, R.; Wilk, P.; Zambo, Z.; Torocsik, B.; Weiss, M.S.; Adam-Vizi, V.; Ambrus, A. Crystal structures of the disease-causing D444V mutant and the relevant wild type human dihydrolipoamide dehydrogenase. Free Radic. Biol. Med. 2018, 124, 214–220. [Google Scholar] [CrossRef]
- Szabo, E.; Wilk, P.; Nagy, B.; Zambo, Z.; Bui, D.; Weichsel, A.; Arjunan, P.; Torocsik, B.; Hubert, A.; Furey, W.; et al. Underlying molecular alterations in human dihydrolipoamide dehydrogenase deficiency revealed by structural analyses of disease-causing enzyme variants. Hum. Mol. Genet. 2019, 28, 3339–3354. [Google Scholar] [CrossRef] [PubMed]
- Underbakke, E.S.; Iavarone, A.T.; Chalmers, M.J.; Pascal, B.D.; Novick, S.; Griffin, P.R.; Marletta, M.A. Nitric Oxide-Induced Conformational Changes in Soluble Guanylate Cyclase. Structure 2014, 22, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Tal, I.; Khananshvili, D. Structural Features of Ion Transport and Allosteric Regulation in Sodium-Calcium Exchanger (NCX) Proteins. Front. Physiol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Nguyen, T.; Moroz, O.V.; Turkenburg, J.P.; Nielsen, J.E.; Wilson, K.S.; Rand, K.D.; Teilum, K. Conformational heterogeneity of Savinase from NMR, HDX-MS and X-ray diffraction analysis. PeerJ 2020, 8, e9408. [Google Scholar] [CrossRef] [PubMed]
- Nyíri, K.; Mertens, H.D.T.; Tihanyi, B.; Nagy, G.N.; Kőhegyi, B.; Matejka, J.; Harris, M.J.; Szabó, J.E.; Papp-Kádár, V.; Németh-Pongrácz, V.; et al. Structural model of human dUTPase in complex with a novel proteinaceous inhibitor. Sci. Rep. 2018, 8, 4326. [Google Scholar]
- Singh, H.R.; Nardozza, A.P.; Möller, I.R.; Knobloch, G.; Kistemaker, H.A.V.; Hassler, M.; Harrer, N.; Blessing, C.; Eustermann, S.; Kotthoff, C.; et al. A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Mol. Cell 2017, 68, 860–871.e7. [Google Scholar] [CrossRef]
- Masson, G.R.; Perisic, O.; Burke, J.E.; Williams, R.L. The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem. J. 2016, 473, 135–144. [Google Scholar] [CrossRef]
- Jensen, P.F.; Larraillet, V.; Schlothauer, T.; Kettenberger, H.; Hilger, M.; Rand, K.D. Investigating the Interaction between the Neonatal Fc Receptor and Monoclonal Antibody Variants by Hydrogen/Deuterium Exchange Mass Spectrometry. Mol. Cell. Proteom. 2015, 14, 148–161. [Google Scholar] [CrossRef]
- Lim, X.-X.; Chandramohan, A.; Lim, X.Y.E.; Bag, N.; Sharma, K.K.; Wirawan, M.; Wohland, T.; Lok, S.-M.; Anand, G.S. Conformational changes in intact dengue virus reveal serotype-specific expansion. Nat. Commun. 2017, 8, 14339. [Google Scholar] [CrossRef]
- Cryar, A.; Groves, K.; Quaglia, M. Online Hydrogen-Deuterium Exchange Traveling Wave Ion Mobility Mass Spectrometry (HDX-IM-MS): A Systematic Evaluation. J. Am. Soc. Mass Spectrom. 2017, 28, 1192–1202. [Google Scholar] [CrossRef]
- Khakinejad, M.; Kondalaji, S.G.; Maleki, H.; Arndt, J.R.; Donohoe, G.C.; Valentine, S.J. Combining ion mobility spectrometry with hydrogen-deuterium exchange and top-down MS for peptide ion structure analysis. J. Am. Soc. Mass Spectrom. 2014, 25, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Vahidi, S.; Bi, Y.; Dunn, S.D.; Konermann, L. Load-dependent destabilization of the γ-rotor shaft in FOF1 ATP synthase revealed by hydrogen/deuterium-exchange mass spectrometry. Proc. Natl. Acad. Sci. USA 2016, 113, 2412–2417. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.F.; Comamala, G.; Trelle, M.B.; Madsen, J.B.; Jørgensen, T.J.D.; Rand, K.D. Removal of N-Linked Glycosylations at Acidic pH by PNGase A Facilitates Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of N-Linked Glycoproteins. Anal. Chem. 2016, 88, 12479–12488. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Scian, M.; Lee, K.K. Tracking Hydrogen/Deuterium Exchange at Glycan Sites in Glycoproteins by Mass Spectrometry. Anal. Chem. 2011, 83, 7492–7499. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.C.; Hudgens, J.W. Effects of Desialylation on Human α1-Acid Glycoprotein–Ligand Interactions. Biochemistry 2013, 52, 7127–7136. [Google Scholar] [CrossRef] [PubMed]
- Benhaim, M.; Lee, K.K.; Guttman, M. Tracking Higher Order Protein Structure by Hydrogen-Deuterium Exchange Mass Spectrometry. Protein Pept. Lett. 2019, 26, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Gallagher, E.S.; Hudgens, J.W. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem. 2018, 90, 6409–6412. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozohanics, O.; Ambrus, A. Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10, 286. https://doi.org/10.3390/life10110286
Ozohanics O, Ambrus A. Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life. 2020; 10(11):286. https://doi.org/10.3390/life10110286
Chicago/Turabian StyleOzohanics, Oliver, and Attila Ambrus. 2020. "Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes" Life 10, no. 11: 286. https://doi.org/10.3390/life10110286
APA StyleOzohanics, O., & Ambrus, A. (2020). Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life, 10(11), 286. https://doi.org/10.3390/life10110286