Whole Genome Sequencing and Comparative Genome Analysis of the Halotolerant Deep Sea Black Yeast Hortaea werneckii

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolates and Genomic DNA Isolation
2.2. Library Preparation, Sequencing and Genome Assembly
2.3. Evaluation of the Quality and Completeness of the Genome Assembly and Determination of the Level of Heterozygosity
2.4. Gene Prediction, Functional Genome Annotation and Phylogenomic Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Gostinčar, C.; Muggia, L.; Grube, M. Polyextremotolerant black fungi: Oligotrophism, adaptive potential, and a link to lichen symbioses. Front. Microbiol. 2012, 3, 390. [Google Scholar] [CrossRef]
- Moreno, L.F.; Vicente, V.A.; de Hoog, S. Black yeasts in the omics era: Achievements and challenges. Med. Mycol. 2018, 56, S32–S41. [Google Scholar] [CrossRef]
- Butinar, L.; Sonjak, S.; Zalar, P.; Plemenitaš, A.; Gunde-Cimerman, N. Melanized halophilic fungi are eukaryotic members of microbial communities in hypersaline waters of solar salterns. Bot. Mar. 2005, 48, 73–79. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Zalar, P.; de Hoog, G.S.; Plemenitaš, A. Hypersaline waters in salterns- Natural ecological niches for halophilic black yeasts. FEMS Microbiol. Ecol. 2000, 32, 235–240. [Google Scholar] [CrossRef]
- Petrovič, U.; Gunde-Cimerman, N.; Plemenitaš, A. Cellular responses to environmental salinity in the halophilic black yeast Hortaea werneckii. Mol. Microbiol. 2002, 45, 665–672. [Google Scholar] [CrossRef]
- Kogej, T.; Stein, M.; Volkmann, M.; Gorbushina, A.A.; Galinski, E.A.; Gunde-Cimerman, N. Osmotic adaptation of the halophilic fungus Hortaea werneckii: Role of osmolytes and melanization. Microbiology 2007, 153, 4261–4273. [Google Scholar] [CrossRef]
- Plemenitaš, A.; Lenassi, M.; Konte, T.; Kejžar, A.; Zajc, J.; Gostinčar, C.; Gunde-Cimerman, N. Adaptation to high concentrations in halotolerant/halophilic fungi: A molecular prospective. Front. Microbiol. 2014, 5, 199. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Plemenitaš, A. Ecology and molecular adaptations of the halophilic black yeast Hortaeawerneckii. Rev. Environ. Sci. Biotechnol. 2006, 5, 323–331. [Google Scholar] [CrossRef]
- Nishimura, K.; Miyaji, M. Hortaea, a new genus to accommodate Cladosporium werneckii. Jpn. J. Med. Mycol. 1984, 25, 139–146. [Google Scholar] [CrossRef]
- Crous, P.W.; Schoch, C.L.; Hyde, K.D.; Wood, A.R.; Gueidan, C.; de Hoog, G.S.; Groenewald, J.Z. Phylogenetic lineages in the Capnodiales. Stud. Mycol. 2009, 64, 17–47. [Google Scholar] [CrossRef]
- Zalar, P.; Zupančič, J.; Gostinčar, C.; Zajc, J.; de Hoog, G.S.; De Leo, F.; Azua-Bustos, A.; Gunde-Cimerman, N. The extremely halotolerant black yeast Hortaea werneckii–a model for intraspecific hybridization in clonal fungi. IMA Fungus 2019, 10, 10. [Google Scholar] [CrossRef]
- Marchetta, A.; van den Ende, G.B.; Al-Hatmi, A.M.S.; Hagen, F.; Zalar, P.; Sudhadham, M.; Gunde-Cimerman, N.; Urzì, C.; de Hoog, S.; De Leo, F. Global molecular diversity of the halotolerant fungus Hortaea werneckii. Life 2018, 8, 31. [Google Scholar] [CrossRef]
- Göttlich, E.; de Hoog, G.S.; Yoshida, S.; Takeo, K.; Nishimura, K.; Miyaji, M. Cell-surface hydrophobicity and lipolysis as essential factors in human tinea nigra. Mycoses 1995, 38, 489–494. [Google Scholar] [CrossRef]
- Perez, C.; Colella, M.T.; Olaizola, C.; de Capriles, C.H.; Magaldi, S.; Mata-Essayag, S. Tineanigra: Report of twelvecases in Venezuela. Mycopathologia 2005, 160, 235–238, ds11046–s005. [Google Scholar] [CrossRef]
- Bonifaz, A.; Badali, H.; de Hoog, G.S.; Cruz, M.; Araiza, J.; Cruz, M.A.; Fierro, L.; Ponce, R.M. Tinea nigra by Hortaea werneckii, a report of 22 cases from Mexico. Stud. Mycol. 2008, 61, 77–82. [Google Scholar] [CrossRef]
- Giordano, M.C.; De la Fuente, A.; Lorca, M.B.; Kramer, D. Tinea nigra: Report of three pediatrics cases. Rev. Chil. Pediatr. 2018, 89, 506–510. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Zalar, P. Extremely halotolerant and halophilic fungi inhabit brine in solar salterns around the globe. Food Technol. Biotech. 2014, 52, 170–179. [Google Scholar]
- Xu, W.; Pang, K.-L.; Luo, Z.-H. High fungal diversity and abundance recovered in the deep-sea sediments of the Pacific Ocean. Microb. Ecol. 2014, 68, 688–698. [Google Scholar] [CrossRef]
- Formoso, A.; Heidrich, D.; Felix, C.R.; Tenório, A.C.; Leite, B.R.; Pagani, D.M.; Ortiz-Monsalve, S.; Ramírez-Castrillón, M.; FontesLandell, M.; Scroferneker, M.L.; et al. Enzymatic activity and susceptibility to antifungal agents of brazilian environmental isolates of Hortaeawerneckii. Mycopathologia 2015, 180, 345–352, ds11046–s015. [Google Scholar] [CrossRef]
- Jones, E.B.G.; Suetrong, S.; Bahkali, A.H.; Abdel-Wahab, M.A.; Boekhout, T.; Pang, K.-L. Classification of marine Ascomycota, Basidiomycota, Blastocladiomycota and Chytridiomycota. Fungal Divers. 2015, 3, 1–72. [Google Scholar] [CrossRef]
- Le Calvez, T.; Burgaud, G.; Mahé, S.; Barbier, G.; Vandenkoornhuyse, P. Fungal diversity in deep-sea hydrothermal ecosystems. Appl. Environ. Microbiol. 2009, 75, 6415–6421. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Raghukumar, C.; Meena, R.M.; Verma, P.; Shouche, Y. Fungal diversity in deep-sea sediments revealed by culture-dependent and culture-independent approaches. Fungal Ecol. 2012, 5, 543–555. [Google Scholar] [CrossRef]
- Pang, K.-L.; Guo, S.-Y.; Chen, I.-A.; Burgaud, G.; Luo, Z.-H.; Dahms, H.U.; Hwang, J.-S.; Lin, Y.-L.; Huang, J.-S.; Ho, T.-W.; et al. Insights into fungal diversity of a shallow-water hydrothermal vent field at Kueishan Island, Taiwan by culture-based and metabarcoding analyses. PLoS ONE 2019, 14, e0226616. [Google Scholar] [CrossRef]
- De Leo, F.; Lo Giudice, A.; Alaimo, C.; De Carlo, G.; Rappazzo, A.C.; Graziano, M.; De Domenico, E.; Urzì, C. Occurrence of the blackyeast Hortaeawerneckii in the Mediterranean Sea. Extremophiles 2019, 23, 9–17. [Google Scholar] [CrossRef]
- Gostinčar, C.; Stajich, J.E.; Zupančič, J.; Zalar, P.; Gunde-Cimerman, N. Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genom. 2018, 19, 364. [Google Scholar] [CrossRef]
- Lenassi, M.; Gostinčar, C.; Jackman, S.; Turk, M.; Sadowski, I.; Nislow, C.; Jones, S.; Birol, I.; Gunde-Cimerman, N.; Plemenitaš, A. Whole genome duplication and enrichment of metal cation transporters revealed by de-novo genome sequencing of extremely halotolerant black yeast Hortaeawerneckii. PLoS ONE 2013, 8, e71328. [Google Scholar] [CrossRef]
- Sinha, S.; Flibotte, S.; Neira, M.; Formby, S.; Plemenitaš, A.; Gunde-Cimerman, N.; Lenassi, M.; Gostinčar, C.; Stajich, J.E.; Nislow, C. Insight into the recent genome duplication of the halophilic yeast Hortaea werneckii: Combining an improved genome with gene expression and chromatin structure. G3 2017, 7, 2015–2022. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 4554–4577. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 2272–2345. [Google Scholar] [CrossRef]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4–11. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 11–14. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 15401–15542. [Google Scholar] [CrossRef] [PubMed]
- Denton, J.F.; Lugo-Martinez, J.; Tucker, A.E.; Schrider, D.R.; Warren, W.C.; Hahn, M.W. Extensive error in the number of genes inferred from draft genome assemblies. Ploscomput. Biol. 2014, 10, e1003998. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef]
- Coleine, C.; Masonjones, S.; Sterflinger, K.; Onofri, S.; Selbmann, L.; Stajich, J.E. Peculiar genomic traits in the stress-adapted cryptoendolithic Antarctic fungus Friedmanniomyces Endolithicus. Fungal Biol. 2020, 124, 458–467. [Google Scholar] [CrossRef]
- Ma, L.-J.; Ibrahim, A.S.; Skory, C.; Grabherr, M.G.; Burger, G.; Butler, M.; Elias, M.; Idnurm, A.; Lang, B.F.; Sone, T.; et al. Genomic analysis of the basal lineage fungus Rhizopus oryzae reveals a whole-genome duplication. PLoS Genet. 2009, 5, e1000549. [Google Scholar] [CrossRef]
- Corrochano, L.M.; Kuo, A.; Marcet-Houben, M.; Polaino, S.; Salamov, A.; Villalobos-Escobedo, J.M.; Grimwood, J.; Álvarez, M.I.; Avalos, J.; Bauer, D.; et al. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016, 26, 1577–1584. [Google Scholar] [CrossRef]
- Lidzbarsky, G.A.; Shkolnik, T.; Nevo, E. Adaptive response to DNA-damaging agents in natural Saccharomyces cerevisiae populations from Evolution Canyon, Mt. Carmel, Israel. PLoS ONE 2009, 4, e5914. [Google Scholar] [CrossRef]
- Dhar, R.; Sägesser, R.; Weikert, C.; Yuan, J.; Wagner, A. Adaptation of Saccharomyces cerevisiae to saline stress through laboratory evolution. J. Evol. Biol. 2011, 24, 1135–1153. [Google Scholar] [CrossRef]



{kind=link}
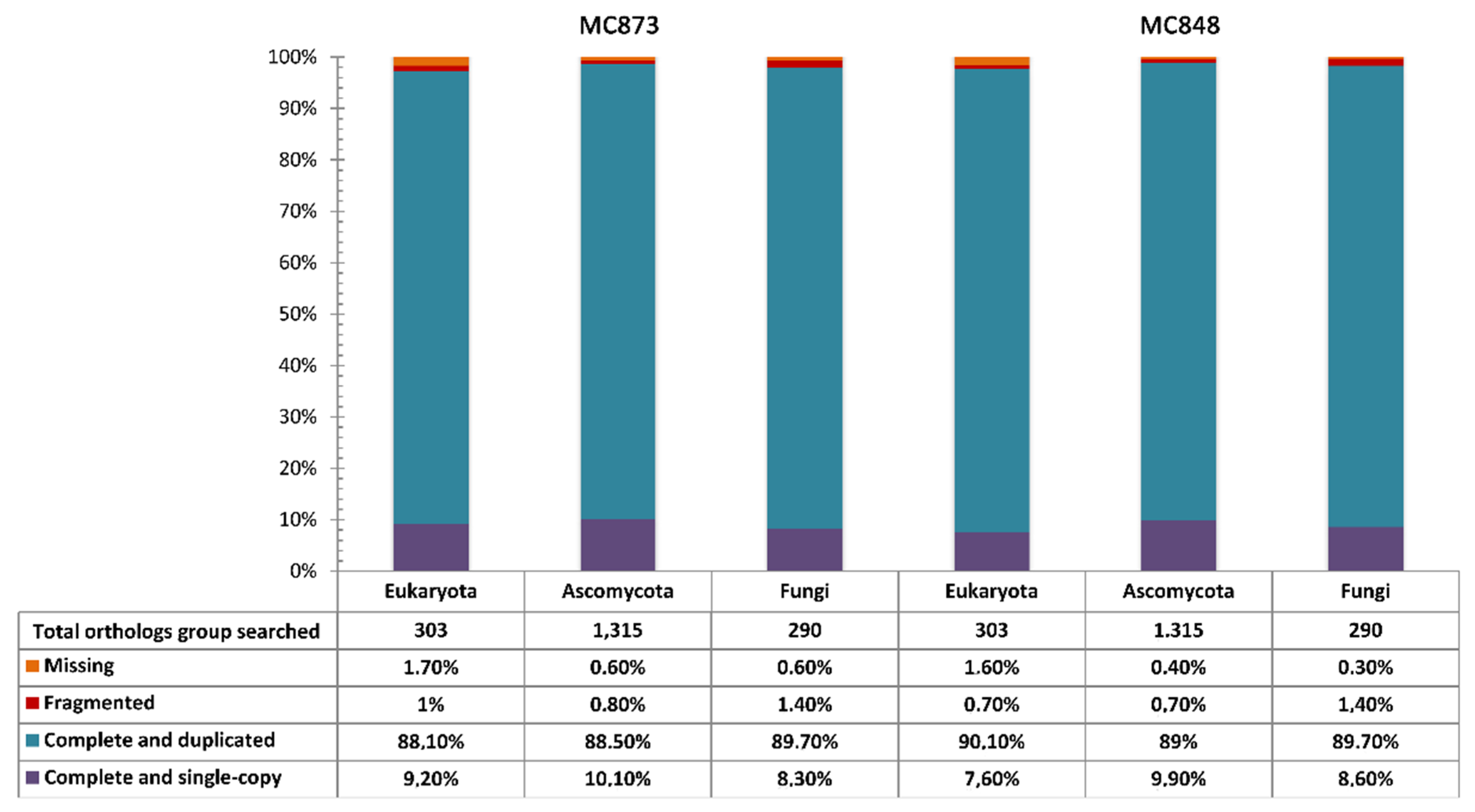{kind=link}
| Strain | Other Collection | Source | Country | Reference |
|---|---|---|---|---|
| EXF-2000 | CBS 100457 | Hypersaline water of salt-pans | Slovenia | [28] |
| EXF-2788 | Hypersaline water of salt-pans | Slovenia | [26] | |
| EXF-120 | Hypersaline water of salt-pans | Spain | [26] | |
| EXF-562 | Soil on the sea cost | Namibia | [26] | |
| EXF-171 | CBS 111.31 | Human, Keratomycosis | Brazil | [26] |
| EXF-2682 | CBS 126.35 | Human, Trichomycosis nigra | Italy | [26] |
| EXF-151 | CBS 107.67 | Human, Tinea nigra | Portugal | [26] |
| EXF-6651 | Spider web, Atacama desert | Chile | [26] | |
| EXF-6654 | Spider web, Atacama desert | Chile | [26] | |
| EXF-6669 | Spider web, Atacama desert | Chile | [26] | |
| EXF-6656 | Rock in a cave, Atacama desert | Chile | [26] | |
| EXF-10513 | MC848 V2500 b | Seawater of Mediterranean Sea; 2500 m depth, Vector station | Italy | [26]; This study |
| MC873 | Geo f 100 c | Seawater, Mediterranean Sea; 3400 m depth, Geostar station | Italy | This study |
| H. werneckii Genome Features | MC873 Strain | MC848 Strain |
|---|---|---|
| Total number of sequenced bases | 10,481,489,013 | 14,036,248,558 |
| Total number of reads with Q-score ≥25 | 70,784,397 | 98,305,008 |
| Total number of mapped reads (%) | 70,711,190 (99.9%) | 95,135,691 (96.8%) |
| Unmapped reads | 73,207 (0.1%) | 3,169,317 (3.2%) |
| Number of total contigs | 925 | 1218 |
| Number of contigs >1 Kbp | 761 | 612 |
| Largest contig (bp) | 604,832 | 576,474 |
| Genome size (bp) | 50,705,820 | 51,030,830 |
| Average coverage depth | ~207× | ~276× |
| GC content (%) | 53.4 | 53.4 |
| Total number of predicted genes | 15,542 | 15,565 |
| Protein-coding genes | 15,397 | 15,410 |
| tRNAs | 126 | 132 |
| snRNAs | 5 | 5 |
| Total gene length (bp) | 26,021,394 | 26,184,209 |
| Longest gene (bp) | 15,052 | 19,877 |
| Number of exons | 32,681 | 32,716 |
| Longest exon (bp) | 12,966 | 12,966 |
| Exon average length (bp) | ~742 | ~746 |
| Gene average length (bp) | ~1690 | ~1699 |
| Tandem Repeat Number | 13,188 | 13,315 |
| Simple repeats | 11,183 | 11,282 |
| Low-complexity repeats | 2005 | 2033 |
| TE Class | MC848 | MC873 | TE Class | MC848 | MC873 |
|---|---|---|---|---|---|
| DNA (notclassified) | 2 | 3 | LINE I-Jockey | 1 | 1 |
| DNA CMC-EnSpm | 2 | 3 | LINE L1 | 4 | 3 |
| DNA Dada | 10 | 8 | LINE L1-Tx1 | 4 | 3 |
| DNA Ginger-1 | 2 | 2 | LINE L2 | 1 | 0 |
| DNA hAT | 1 | 2 | LINE RTE | 1 | 0 |
| DNA hAT-Ac | 18 | 17 | LINE RTE-BovB | 3 | 5 |
| DNA hAT-Charlie | 2 | 2 | LTR (notclassified) | 3 | 3 |
| DNA Kolobok-T2 | 1 | 0 | LTR Copia | 8 | 1 |
| DNA Merlin | 1 | 0 | LTR ERV1 | 7 | 6 |
| DNA-Maverick | 0 | 1 | LTR ERVK | 6 | 8 |
| DNA Mule-MuDR | 1 | 1 | LTR Gypsy | 165 | 151 |
| DNA P | 1 | 1 | LTR Ngaro | 12 | 12 |
| DNA TcMar-ISRm11 | 1 | 1 | LTR Pao | 18 | 17 |
| DNA-TcMar-Tc1 | 0 | 1 | RC Helitron | 1 | 2 |
| LINE CR1 | 1 | 0 | SINE B2 | 1 | 1 |
| LINE I | 1 | 0 | SINE-tRNA-RTE | 0 | 1 |
| Total | 279 | 256 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, O.; Marchetta, A.; Giosa, D.; Giuffrè, L.; Urzì, C.; De Leo, F. Whole Genome Sequencing and Comparative Genome Analysis of the Halotolerant Deep Sea Black Yeast Hortaea werneckii. Life 2020, 10, 229. https://doi.org/10.3390/life10100229
Romeo O, Marchetta A, Giosa D, Giuffrè L, Urzì C, De Leo F. Whole Genome Sequencing and Comparative Genome Analysis of the Halotolerant Deep Sea Black Yeast Hortaea werneckii. Life. 2020; 10(10):229. https://doi.org/10.3390/life10100229
Chicago/Turabian StyleRomeo, Orazio, Alessia Marchetta, Domenico Giosa, Letterio Giuffrè, Clara Urzì, and Filomena De Leo. 2020. "Whole Genome Sequencing and Comparative Genome Analysis of the Halotolerant Deep Sea Black Yeast Hortaea werneckii" Life 10, no. 10: 229. https://doi.org/10.3390/life10100229
APA StyleRomeo, O., Marchetta, A., Giosa, D., Giuffrè, L., Urzì, C., & De Leo, F. (2020). Whole Genome Sequencing and Comparative Genome Analysis of the Halotolerant Deep Sea Black Yeast Hortaea werneckii. Life, 10(10), 229. https://doi.org/10.3390/life10100229