Abstract
The site preference for each cation and site in beryl based on bond-length calculations was determined and compared with analytical data. Tetrahedral SiO4 six-membered rings normally have no substitutions which results from very compact Si4+–O bonds in tetrahedra. Any substitution except Be would require significant tetrahedral ring distortion. The Be tetrahedron should also be negligibly substituted based on the bond-valence calculation; the tetrahedral Li–O bond length is almost 20% larger than Be2+–O. Similar or smaller bond lengths were calculated for Cr3+, V3+, Fe3+, Fe2+, Mn3+, Mg2+, and Al3+, which can substitute for Be but also can occupy a neighboring tetrahedrally coordinated site which is completely vacant in the full Be occupancy. The octahedral site is also very compressed due to dominant Al with short bond lengths; any substitution results in octahedron expansion. There are two channel sites in beryl: the smaller 2b site can be occupied by Na+, Ca2+, Li+, and REE3+ (Rare Earth Elements); Fe2+ and Fe3+ are too small; K+, Cs+, Rb+, and Ba2+ are too large. The channel 2a-site average bond length is 3.38 Å which allows the presence of simple molecules such as H2O, CO2, or NH4 and the large-sized cations-preferring Cs+.
1. Introduction
The hexagonal structure of beryl, Be3Al2Si6O18, has a 3D framework of AlO6 octahedra, SiO4 and BeO4 tetrahedra, and channels delimited by SiO4 tetrahedral rings [1]. Compared to similar minerals or mineral groups, i.e., the tourmaline supergroup and axinite group, the beryl group is less complex in composition, and substitutions are usually limited. The site occupancy can be relatively exactly determined by electron microanalysis (EMPA), laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), and structural refinement (SREF) using single-crystal X-ray and neutron diffraction [1,2,3,4,5,6,7,8]. However, the accuracy of SREF is limited for minor or trace elements in the beryl structure. Their position in the beryl structure can be determined by additional spectroscopic methods. The transition elements, including Fe, Mn, Cr, and V, can be analyzed with optical absorption spectroscopy [9,10,11,12,13,14]. Its accuracy is limited in quantitative determination, but it is very useful for revealing properties such as cationic charge, coordination, and subtle phenomena such as Jahn–Teller distortion of octahedral Mn3+ [12]. A more accurate determination of Fe charge and coordination is possible with Mössbauer spectroscopy [8,15,16,17,18], but it requires substantial Fe content which is not usual in beryl. Channel constituents can be studied by Raman and infrared spectroscopy [14,19,20,21,22,23,24,25,26,27,28], but exact quantification is usually hard or even impossible to provide. Less often used are electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) spectroscopy [21,29,30,31,32,33,34,35].
This work provides the site-occupancy assessment in beryl based on bond-length constraints. It builds mostly on the comprehensive crystal-chemical study by [1], which is reviewed and extended here. The main extension involves the theoretical bond-length calculations from ideal bond valences for each ion and coordination, which allows to predict ion site preference in the structure.
2. Beryl Structure
The beryl hexagonal structure consists of AlO6 octahedra and SiO4 and BeO4 tetrahedra (Figure 1). Rings of SiO4 tetrahedra are connected to AlO6 octahedra and BeO4 tetrahedra forming spatial framework. The most frequent substituting cations include Fe2+, Mn2+, Mg2+, Fe3+, Cr3+, V3+, and Ti4+ for Al3+ in the octahedron and Li+ for Be2+ in the tetrahedron. As most of these substitutions are heterovalent, it is necessary to balance the charge by entering alkali cations, such as Na+, K+, Rb+, and Cs+, to the channel [1].
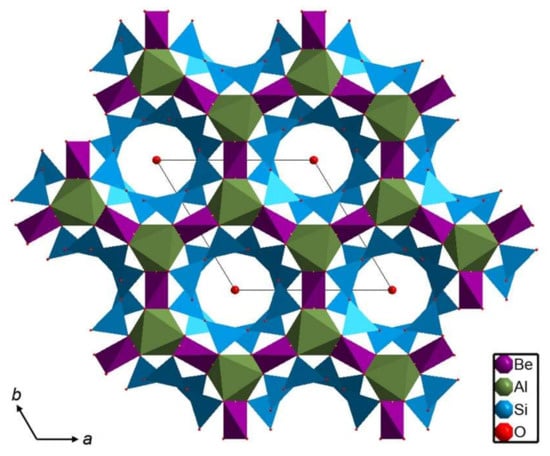
Figure 1.
The beryl structure (based on [12]) with the framework structural sites pictured as coordination polyhedral and channel sites located at the center of each six-membered tetrahedral ring indicated by a red ball.
Aluminum in distorted octahedron can be substituted by ions of comparable size. The substitution of Al by Mg and/or Fe has two main effects on the structure: the volume of the polyhedron must increase to receive ions with greater radii, therefore, an elongation of Me–O distances (Me—octahedral substituent) would be expected accompanied by an increase in the polyhedron distortion, since the length of the edges shared with BeO4 adjacent groups is constrained by the Be tetrahedron dimensions. There are positive correlations among the divalent ions content, the cation–oxygen distance, the volume of the site, and the octahedral bond-angle variance. To attain the electric neutrality, the substitution of trivalent cations by divalent ones is compensated by alkali ions in the channels. These ions usually occupy the 2a site and are weakly linked to ring O. They induce a distortion in the Si tetrahedra, as seen by the increase of bond-angle variance with total alkali content in the channels (correlation coefficient = 0.86). Since the 2a site has twofold multiplicity in the hexagonal cell of the beryl, there is an upper limit of one alkali ion per six Si atoms, i.e., an upper limit of one divalent ion in the two octahedral sites of the formula [1].
In the BeO4 tetrahedron, Li+ is the main substituent [1,6]. The entry of Li+ into the BeO4 tetrahedron causes a bond-strength deficiency to O. Heterovalent substitution of Li+ for Be2+ is charge-balanced by an entry of alkali cations into the channel and therefore, there is an upper limit in the Be-Li substitution; only one-third of the tetrahedral positions can be occupied by Li+. The substitution of Li for Be increasing the cation–oxygen distances and the volume of the Be tetrahedron partially releases the strain on the adjacent octahedron [1]. For the Si tetrahedron, all refined beryls have an invariable mean Si–O bond length (1.609 + 0.002 Å), which indicates uniform Si occupation at these ring sites [1]. There is a direct correlation between the a unit-cell parameter and the divalent-ion content and between c and the Li content. The divalent-cations presence increases the cation-oxygen bond length in the octahedron; the octahedron, constrained by the short edges shared with Be tetrahedra, is flattened in the c direction, and therefore, the increase in length influences directly the a value. In contrast, an increase in the tetrahedral cation–oxygen distance due to the Li–Be substitution is manifested as an increase in c. The dominant substitutions directly influence the length of unit-cell parameters; consequently, beryls were divided into three series. “Octahedral” beryls are characterized by c/a values in the range of 0.991–0.996. “Tetrahedral” beryls have c/a values in the range of 0.999–1.003. “Normal” beryls are typical with a c/a ratio in the range of 0.997–0.998 [1]. The different behavior of the lattice parameters for C+Al3+□−1Me2+−1 and C+Li+□−1Be2+−1 (Me—octahedral cation; C—channel cation) substitutions reflects the opposite distortion arising in the beryl structure with special regard to the octahedron and Be tetrahedron [1].
Lithium is the only alkali ion which can substitute for Be2+ in the tetrahedron because of a small ionic radius. Ionic radii of other alkali cations are significantly higher and, therefore, they enter the channel [14]. Channel interstices parallel to c are surrounded by the stacked hexagonal rings of SiO4 tetrahedra. Channel interstice can be divided in two distinct sites partially filled with alkali cations, molecules (mainly H2O, CO2), and other impurities. Lithium, except the tetrahedron, can be also located in the channel [7]. Alkali cations in the interstice cause a charge-balancing deficiency which results from heterovalent substitutions at octahedral or tetrahedral sites [1,14]. Sodium can be incorporated at the center of each six-membered Si ring (2b interstitial site), and larger Cs, Rb, and K enter the 2a interstitial site between the stacked ring [20,22,25,31]. Water is present only at the 2a site in two orientations: (1) twofold axis of water type I is oriented perpendicular to the sixfold beryl c axis; (2) water type II occurs with alkali, and its twofold axis is oriented parallel to the sixfold beryl c axis [14,19,22,23,25,26].
3. Methods
Bond-valence and bond-length calculations are based on the following equation:
where dij is the bond length (in Å) between the two given atoms, the bond valence (νij) measures bond strength (in vu—valence units), R0 is the length of a single bond (for which νij = 1 vu), and b is the universal parameter for each bond [36]. Many smaller-scale studies have produced bond-valence parameters for a wide range of ion pairs that have been compiled by Brown [37,38,39]. His latest list of published bond-valence parameters [40] contains 1749 sets of bond-valence parameters for the equation of [40], for 1350 unique ion pairs, and counts 340 sets of bond-valence parameters for 194 cations bonded to oxygen. Several sets of bond-valence parameters are often available for unique ion pairs, and there has been little comparison between different sets of parameters available to determine which is the most suitable for a given ion pair. With regard to the bond-valence parameters, older datasets had the following issues: (1) There was no consistency between parameters from different sources. (2) Different fitting methods have been used by different authors to derive the bond-valence parameters. (3) Very few alternative forms of the bond-valence–bond-length relations have been tested.
dij = R0 − b ln νij
The list of [41] allowed to (1) evaluate published bond-valence parameters for 128 cations bonded to oxygen, using a very large set of bond lengths that have undergone rigorous filtering; (2) investigate many alternative algebraic forms of the bond-valence–bond-length relation; (3) evaluate different fitting methods used in the derivation of bond-valence parameters; and (4) determine new bond-valence parameters for 135 cations bonded to oxygen. Consequently, in order to use the most consistent data, the R0 and b values from [41] for each cation were used.
Empirical structural and chemical data for 64 beryl samples are collected from [1,2,3,4,5,6,7,8].
4. Results
4.1. Bond-Length Calculations
Ideal bond lengths were calculated for each cation usually occurring in the beryl structure including selected trace elements (Table 1). These were calculated from ideal bond valences νij for each cation in the specific coordination body assuming that the bond-valence sum is equal to the nominal charge of the cation. Consequently, no bond-length and angular distortion effects were considered.

Table 1.
Calculated bond lengths (dij) for selected cation–oxygen bonds in specific coordination environments occurring in the beryl structure.
For the interpretation of data and comparison to actual beryl structural data, the average bond-length values for each site were calculated from the set of 64 published beryl structures (Table 2) [1,2,3,4,5,6,7,8]. Only in the case of channel sites, the bond length for 2b was determined as the average from the half the channel diameter—distance of two ring oxygens—and the 2a bond length was derived from Cs-rich beryl and pezzottaite which has a similar structure to beryl. Moreover, for better orientation and distinction of stable and instable bonds, the limits for each site were derived from their average bond lengths as ±15% deviation. However, for the 2a site, the upper limit is the maximal bond length constrained by the size of the cavity between two tetrahedral rings.
Table 2.
Empirical average bond lengths (dij) for selected cation-oxygen bonds in the beryl structural sites (empirical data from [1,2,3,4,5,6,7,8]).
Two tetrahedral sites differ only slightly in the average bond length. The Si tetrahedron is slightly smaller than the Be tetrahedron, but both calculated Si4+–O and Be2+–O bond lengths have values perfectly consistent with their empirical averages (Figure 2). The tetrahedral B3+–O bond is significantly shorter, while bonds of Al3+, Cr3+, V3+, Fe3+, and Mn3+ are longer, but their free substitution is still possible. In contrast, Li+, Mg2+, Fe2+, and Mn2+ form bonds which are less likely to be stable.
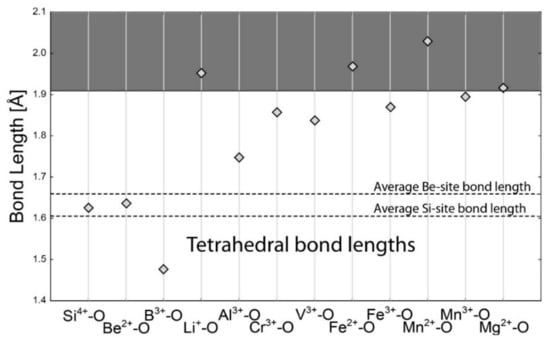
Figure 2.
Calculated bond lengths for each bond in the tetrahedral coordination of cation (Si and Be site). Horizontal dashed lines are for average empirical bond lengths at each site. The horizontal full line is the 15% limit for free substitution. The area of free substitution is white. The substitution is significantly limited for cations in the grey area.
The octahedral site in beryl is dominantly occupied by Al3+. Consequently, the average Al-site bond length is only slightly larger than the calculated ideal d (Al3+–O) (Figure 3). The deviation results from the substitution of other tetravalent (Ti4+), trivalent (Fe3+, Mn3+, Cr3+, V3+, Sc3+), and divalent (Mg2+ and Fe2+) cations, which are well within the area of free octahedral substitution. Divalent Mn is very near the limit of the free substitution resulting in limited substitution, while Li+ is slightly above the boundary.
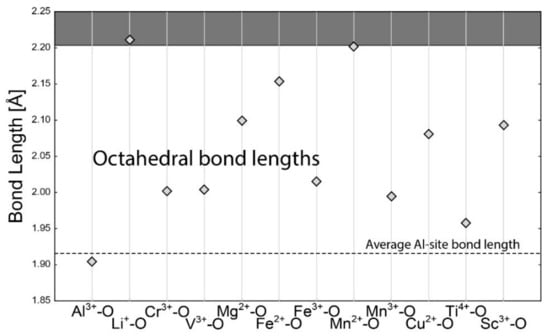
Figure 3.
Calculated bond lengths for each bond in the octahedral coordination of cation (Al site). The horizontal dashed line is for average empirical bond length. The horizontal full line is the 15% limit for free substitution. The area of free substitution is white. The substitution is significantly limited for cations in the grey area.
There are two channel sites in beryl: a smaller one at the center of each tetrahedral ring and a larger one between the pair of rings. Since occupancies of the eight-coordinated 2b-site cations are usually too low for the structural refinement, and as the dataset for the calculation of the average bond length was not sufficient, the average 2b-O distance was calculated as a diameter of effective channel width in the plane of the tetrahedral ring with the value of 2.48 Å. Bond-length calculations revealed that from the cations usually occurring in beryl, only Na+, Ca2+, and Li+ have proper ionic radii to occupy the 2b site. Fe2+ and Fe3+ are too small; K+, Cs+, Rb+, and Ba2+ are too large (Figure 4). Interestingly, if any REE3+ are in the beryl structure, these would be located here.
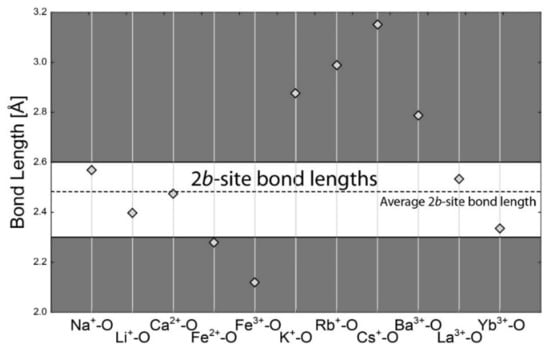
Figure 4.
Calculated bond lengths for each bond in the eightfold coordination of cation (2b site). The horizontal dashed line is for average empirical bond length. The horizontal full line is the 15% limit for free substitution. The free-substitution area is white. The substitution is significantly limited for cations in the grey area.
The 2a site coordination is 12. The maximal bond length is considered to be 3.38 Å, although in special cases, it can be even larger [42]. This is a very large distance, larger than calculated d(Cs+–O) (3.31 Å). This allows the presence of simple molecules, including H2O, CO2, or NH4. These, however, were not included in the calculation because they form no covalent/ionic bonds, only weak hydrogen bonds. Among cations, the large size of the site results in preferring Cs+ at this site, which is in fact the most abundant cation at this site in natural samples. Among others, Rb+, K+, and Ba2+ have calculated ionic radii within the 15% range for free substitution; all others, including Na+, Ca2+, Li+, REE3+, Fe2+ and Fe3+, are too small for the 2a site (Figure 5).
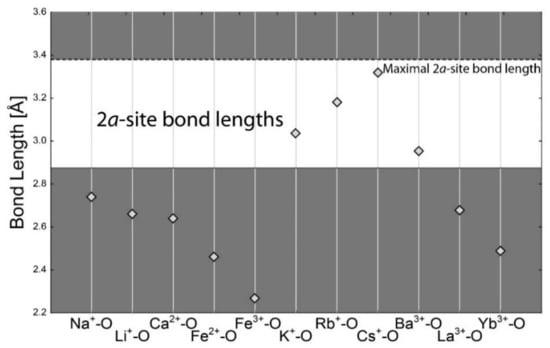
Figure 5.
Calculated bond lengths for each bond in the 12-fold coordination of cation (2a site). The horizontal dashed line is for maximal empirical bond length. The horizontal full line is the 15% limit for free substitution. The free-substitution area is white. The substitution is significantly limited for cations in the grey area.
4.2. Empirical Crystal-Chemical Analysis
The crystal-chemical dataset involving cation proportions, unit-cell dimensions, and bond length at various structural sites was examined to reveal the dependence of structural parameters and chemical composition.
The octahedral Al site has a very good positive correlation of d(Al3+–O) to a (Figure 6a) and negative correlation to c/a (Figure 6c), while there is no obvious dependence on c (Figure 6b), which is consistent with [1]. Compared to major octahedral-site occupants, d(Al3+–O) is negatively correlated to Al (Figure 6d), while positively correlated to divalent cations (Figure 6e) documenting their substitution. The charge balance is achieved by incorporation of Na+ into the channel. It is evidenced by positive correlation of Na and d(Al3+–O) in one group of samples (Figure 6f). However, the second group located upon the correlation trend indicates another substitution mechanism requiring Na+ presence at the 2b site.
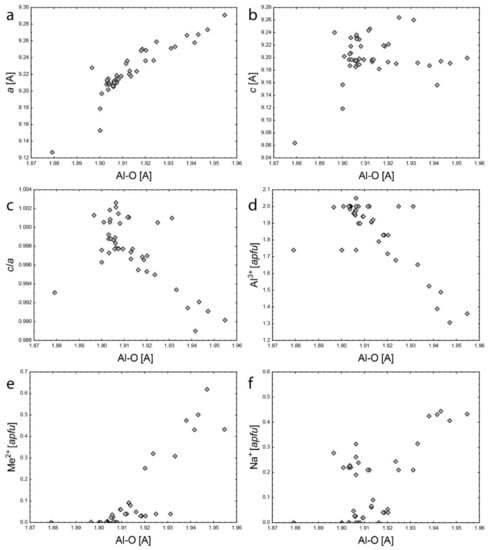
Figure 6.
Comparison of Al3+–O bond length to: (a) a; (b) c; (c) c/a; (d) Al3+; (e) Me2+ (Me = Fe2+ + Mg2+ + Mn2+); (f) Na+.
The tetrahedral Be site has a very good positive correlation of d(Be2+–O) to c (Figure 7b), slightly worse positive correlation to c/a (Figure 7c), and no dependence on a (Figure 7a), which is also in accordance with [1]. This results from Li+ for Be2+ substitution as documented by the positive correlation of Li+ and d(Be2+–O) (Figure 7d), which also requires charge balancing by monovalent cation incorporation into the channel. There is partial correlation of Na+ and d(Be2+–O) (Figure 7e) which suggests that Na+ can partly balance Li–Be substitution. This can also explain deviation of some samples from the d(Al3+–O) vs. Na+ general trend (Figure 6f). However, monovalent cations at the 2b site have a good positive correlation to d(Be2+–O) (Figure 7f), suggesting that these cations charge-balance Li–Be substitution exclusively.
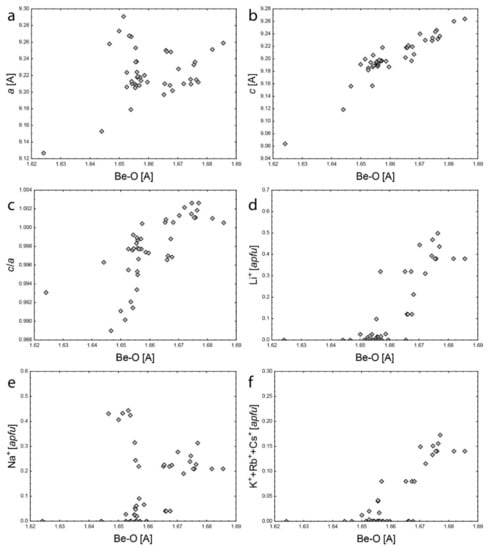
Figure 7.
Comparison of Be2+–O bond length to: (a) a; (b) c; (c) c/a; (d) Li+; (e) Na+; (f) K+ + Rb+ + Cs+.
5. Discussion and Conclusions
The calculation of bond lengths for each site complemented by the empirical crystal-chemical analysis enables determination of main-, minor-, and trace-element distribution at structural sites. The beryl hexagonal structure has a 3D framework of AlO6 octahedra and SiO4 and BeO4 tetrahedra with channels surrounded by six-membered rings of SiO4 and BeO4 tetrahedra [1]. Therefore, beryl structural sites can be divided into: (a) framework sites with strong ionic-covalent bonds; (b) channel sites with weak ionic bonds and hydrogen bonds.
5.1. Framework Sites
Six-membered rings of SiO4 tetrahedra normally have no substitutions which results from very compact Si–O bonds in tetrahedra. The value of d(Si4+–O) was reported as uniformly 1.609 ± 0.002 Å [1]. This is consistent with the d(Si4+–O) calculated from new data, but these show wider variations up to ±0.015 Å, although most of the values are in the interval of ±0.003 Å. However, this is probably not an effect of substitutions because any substitution (except Be) would require significant tetrahedral expansion and distortion. This would result in lowering symmetry similar to cordierite [43,44]. However, the large uniformity in data shows that any substitution of larger cations is below the detection limit. The substitution of Be for Si would not induce any significant distortion, it would be detectable only by lower electron density. However, it is unlikely, due to the large difference in charge, although Be(OH)2Si−1O2O−2 substitution could allow Be to occupy the Si site [45].
However, contraction of d(Si–O) is possible if induced by high pressure. Beryl compressibility is nearly isotropic, with the c axis approximately 20% more compressible than a, and a corresponding small decrease in c/a with increasing pressure. The small difference results from slightly higher Be tetrahedra and Al octahedra compressibility than Si tetrahedra, but the interconnected framework of polyhedra gives beryl high rigidity in compression [3].
In comparison to the SiO4 tetrahedron, the Be sites display a larger variability in the bond lengths of 1.64 to 1.69 Å. This indicates wider substitution than at the Si site, i.e., C+Li+□−1Be2+−1 substitution [1,6,46,47]. The tetrahedral Li–O bond length is 1.952 Å, which is almost 20% larger and is more typical for octahedral sites. This is consistent with the increase in the Be–O bond length and in the c size, which was well described and explained by [1].
However, smaller bond lengths than for Li+ were calculated for Al3+, Cr3+, V3+, Fe3+, Mn3+, and Mg2+, suggesting that these cations could also substitute for Be.
Iron in beryl could be present in both ferrous and ferric states. Ferric iron is usually located in octahedra [1], but some authors suggests its presence in the tetrahedron [32] and channel sites [30,48]. In contrast, Fe2+ is usually located not only in octahedra [1] but also channel sites [14,15,18]. These interpretations locating Fe in other sites than octahedra are based only on the spectroscopic data without thorough structural proof. The Fe3+–O bond length in the tetrahedral coordination is 1.865 Å, which is significantly shorter than the Li+–O bond length. However, Me2+Fe3+Me3+Be2+−1 substitution should be accompanied by substitution of a divalent cation for Al in the octahedron. The pairing of tetrahedral and octahedral Fe3+ and Fe2+ can have a direct impact on the beryl color because it increases the number of possible absorption mechanisms: electron transitions in Fe2+, Fe2+–O, and Fe3+–O ligands to metal charge transfer and Fe2+–Fe3+ intervalence charge transfer. If we omit the structurally unlikely presence of Fe2+ at the channel suggested by several authors [18,26,49], the resultant color could be determined by the proportion of Fe2+ and Fe3+ at octahedral and tetrahedral sites.
Similar assumptions can be made for each of other cations including Al3+, Cr3+, V3+, Mn3+, and Mg2+. In cordierite, Al3+ is major cation at the tetrahedral site similar to the Be site in beryl [44]. Moreover, Mg in high-temperature treated cordierite engendered disorder with Al at octahedral and tetrahedral sites [50], but the specific disorder-inducing environment and the slight difference in cordierite and beryl structure suggests unlikely Mg presence at the untreated beryl tetrahedral site. Moreover, Cu2+ was observed at the Be site of synthetic beryl [51,52]. However, any significant substitution of these cations for Be would result in large structural deformation. Therefore, either none of these cations occupies this site or all of them do. There is no bond-length constraint excluding any of them.
However, based on electron paramagnetic resonance (EPR) data, Li+ and Fe3+ could occupy the site slightly shifted from the Be site (at 0.43, 0.34, 0.17), which is a tetrahedral site neighboring the Be site [31,32,33]. Since there are two neighboring Li sites next to each other and near the Al site, and assuming their possible occupancy by all cations with similar bond lengths to Li+, this could also explain the observed Fe2+–Fe3+ intervalence charge transfers observed in optical absorption spectra (OAS) of beryl [33].
Bond-length calculations at the octahedral site indicate expansion of the octahedron after the substitution of any cation for Al3+, which has the shortest bond lengths of all typical octahedral cations. This is in good agreement with the structural manifestation of the octahedral C+Me2+□−1Al3+−1 substitution which expands the structure in the c direction [1]. Moreover, it is obvious that the homovalent substitution of Fe3+ and Mn3+ also results in the c increase [1,12].
However, the expansion of the Al octahedron may not be homogeneous in all directions. Trivalent Mn at the octahedral site with symmetry 32 induce a local violation of the trigonal symmetry, resulting in a (pseudo-)tetragonal distortion to an elongated octahedron, typical for Jahn–Teller distorted Mn3+, with the elongated O–Mn–O axis corresponding to one of the three O–Al–O axes [12]. It is questionable if this asymmetrical expansion occurs after the substitution of other d elements. In similar structures, Cu2+ and Fe2+ can induce the Jahn–Teller distortion, e.g., in tourmaline-, epidote-, and gadolinite-supergroup minerals [53,54,55,56,57]. However, the identification of such distortion in beryl is limited due to higher symmetry of the octahedral site with three possible equivalent orientations of the elongated O–Me–O axis. Consequently, in the structural refinement, no significant distortion may occur as all orientations have the same probability and, therefore, they may cancel each other out to some average value. However, Jahn–Teller distortion of Cu2+ and Fe2+ occupying octahedron can manifest, similarly to Mn3+, in the optical absorption spectrum of beryl. In the case of the Mn3+ spectrum, the 5E(D) ground state splits into 5B1 and 5A1, and the excited spin-allowed octahedral 5T2(D) state splits into 5B2 and 5E [12].
5.2. Channel Sites
Although the channel sites are not a part of the basic structural framework, their role in the major beryl substitutions requires proper examination.
The smaller 2b site is surrounded by six ring oxygens, but presence of cations at this site requires a H2O molecule at the neighboring to satisfy bond-valence requirements [19]. The bonding of a cation at the 2b site can be described as six electrostatic interactions with ring oxygens and one or two H2O molecules distributed at the opposite cation poles. The bond valences for double-coordinated Na 0.12 vu for Na–Oring and 0.14 vu for Na–Owater are 2.54 and 2.483 Å bond lengths, respectively [19]. This is, in fact, in good agreement with the Na–O bond length of 2.568 Å calculated here. Consequently, the coordination of this site can be assumed as eight.
The actual distance between opposite-ring oxygen centers is approximately 5.2 Å; therefore, the bond length between the ring oxygen and a given cation should be around 2.6 Å. Therefore, we can assume that 2.6 Å is the maximum dij for the occupying cation. This site can therefore be occupied by Na+, Li+, Ca2+, and REE3+.
Ferrous iron is usually located not only in octahedra [1] but also at channel sites based on the spectroscopic data [14,15,18]. Its presence at the channel sites is determined by its ability to form effective bonds with ring oxygens, H2O molecules, or OH groups. The smaller site in the middle of the rings provides the only possibility to accommodate Fe2+ because the larger site is too large even for Na+ [1]. There are a few possible methods of Fe2+ bonding. The most obvious way of Fe2+ bonding is similar to Na bonding; the same scheme enables Fe2+–Oring bonds with the valence of 0.24 vu at a 2.262 Å length and Fe2+–Owater with a 0.28 vu valence of 2.205 Å bond. These lengths are quite realistic for Fe2+ in the eightfold coordination and very similar to the 2.22 Å indicated by spectroscopic data [18] or 2.278 Å calculated here.
However, this scheme has a significant flaw. The actual distance between the ring oxygen and the 2b cation should be around 2.6 Å. Most importantly, the silicate ring is very rigid with almost invariable bond lengths [1], actual variation is between 1.595 and 1.615 Å. Therefore, it is highly unlikely that Fe2+ could induce ring distortion of 0.4 Å because Si4+–O bonds are also significantly stronger. Despite this, we can still calculate bond valences for this arrangement. The 2.6 Å Fe2+–O bond would have 0.096 vu valence; significantly less than the expected 0.24 vu. The remainder of 0.71 vu for double-coordinating H2O is attributable to the 1.860 Å Fe2+–Owater bond. Consequently, the Fe2+ cations would have a flattened ellipsoid shape.
Ferrous iron could also form stable electro-neutral (HO)–Fe2+–(OH) which, however, should be detected in Raman and FTIR spectra as additional O–H bands. In this case, Fe2+ should not participate in any substitution due to the (HO)–Fe2+–(OH) complex charge neutrality.
The possibility of Fe3+ or Fe2+ as (almost) free ions at both channel sites based on EPR spectroscopy [30,48] cannot be tested by the bond-valence approach. However, later interpretation suggests that the observed spectral geometry attributed to Fe3+ at hexagonal symmetrical sites can also be due to the presence of electrons and holes trapped at crystallographically equivalent sites around a threefold axis [32,48]. Moreover, isolated ions with negligible electrostatic interactions with ring oxygens also do not contribute to substitutions in the beryl structural framework.
The 2a site can accommodate bigger cations and even H2O and CO2 molecules [19,20,22,25]. Water is present in two orientations: water type I with the twofold axis oriented perpendicular to the sixfold axis of beryl and water type II associated with alkali and with a twofold axis oriented parallel to the sixfold axis of beryl [14,19,21,22,23,25,26,27,28].
Bond-length considerations for the 2b site excluded cations forming bonds significantly longer than 2.6 Å. These include the largest alkali cations, including K, Rb, Cs, and also Ba. Cesium is typically the most abundant large alkali metal in beryl. This is explained by bond-length considerations; the distance between the cation at the center of the 2a site and ring O is approximately 3.5 Å [42]. Theoretical d(Cs–O) is 3.31 Å. Bonds of all other cations are significantly shorter. Consequently, this site prefers Cs as the largest possible cation.
5.3. Compositional Gap
Division of beryl compositions into “normal”, “tetrahedral”, and “octahedral” series revealed the compositional gap between the “octahedral” and “tetrahedral” beryls. Two beryl series are indicated by the impossibility of finding beryls in which divalent ions substitute for trivalent ions in octahedral sites and monovalent ions simultaneously substitute for divalent ions in tetrahedral sites to an extent near the upper limit [1].
There are a few possible reasons for the compositional gap:
- (1)
- Structural limitations—the rigidity of the 3D structural framework can limit mutual substitutions of larger cations at both Be and Al sites.
- (2)
- Crystal-chemical limitation at the Be and Al sites—substitutions at both sites produce charge deficiency and under-bonding of all O2: −0.25 vu at the Be site with Li+; −0.17 vu at the Al site with Me2+; −0.42 vu in sum. This would increase Si4+–O2 bond valence to 1.42 vu and would induce significant bond shortening to 1.48 Å which would produce a large structural instability.
- (3)
- Channel sites limitations—alkali can charge-balance either octahedral or tetrahedral substitution but not both at the same time. This results from the upper limit for channel sites, which is one apfu (if H2O is not counted) [1], although there are two distinct channel sites. However, the distance between the centers of the 2a and 2b sites is around 2.30 Å [2,4], which disallows the common presence of alkali at both sites. The sum of the most common Na+ and Cs+ ionic radii is 3.06 Å [58]—by far larger than the required 2.30 Å. Only Ca2+ or REE3+ in the channel could be able to charge-balance the combined substitution of Me2+ at the Al site and Li+ at the Be site, but these cations are very rare in beryl. It results from the geochemical properties of the beryl genetic environment. Beryl is the most characteristic and widespread mineral of Be in granitic pegmatites of the LCT (Li–Cs–Ta) suite and is less frequent in the REE-rich NYF (Nb–Y–F) suite [59].
- (4)
- Geochemical limitations—the compositional gap may also result from the chemical composition of the hosting pegmatite environment. The beryl fractionation trend towards a Li-, Cs-rich, and Fe–Mg-poor composition is well documented in rare-element pegmatites worldwide [1,21,60,61,62]. Similarly, the negative correlation between Fe and (Al + Li) and between Fe and Mn in tourmaline is due to fractionation of the pegmatite melt [63]. Other genetic types of beryl are usually Li- and Cs-depleted and, therefore, tend to octahedral or homovalent Me3+Al−1 substitutions.
Author Contributions
Concept of the study, bond-length calculations, compilation and writing of the manuscript, P.B.; statistical crystal-chemical analysis, J.F.
Funding
This work was supported by the Slovak Research and Development Agency under the Contract no. APVV-18-0065.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aurisicchio, C.; Fioravanti, G.; Grubessi, O.; Zanazzi, P.F. Reappraisal of the crystal chemistry of beryl. Am. Miner. 1988, 73, 826–837. [Google Scholar]
- Brown, G.E.; Mills, B.A. High-temperature structure and crystal chemistry of hydrous alkali-rich beryl from the Harding pegmatite, Taos County, New Mexico. Am. Miner. 1986, 71, 547–556. [Google Scholar]
- Hazen, R.M.; Au, A.Y.; Finger, L.W. High-pressure crystal chemistry of beryl (Be3Al2Si6O18) and euclase (BeAlSiO4OH). Am. Miner. 1986, 71, 977–984. [Google Scholar]
- Artioli, G.; Rinaldi, R.; Stahl, K.; Zanazzi, P.F. Structure refinements of beryl by single-crystal neutron and X-ray diffraction. Am. Miner. 1993, 78, 762–768. [Google Scholar]
- Gatta, G.D.; Nestola, F.; Bromiley, G.D.D.; Mattauch, S. The real topological configuration of the extra-framework content in alkali-poor beryl: A multi-methodological study. Am. Miner. 2006, 91, 29–34. [Google Scholar] [CrossRef]
- Hawthorne, F.C.; Cerny, P. The alkali-metal positions in Cs–Li beryl. Can. Miner. 1977, 15, 414–421. [Google Scholar]
- Sherriff, B.L.; Douglas Grundy, H.; Stephen Hartman, J.; Hawthorne, F.C.; Černý, P. The incorporation of alkalis in beryl: Multi-nuclear MAS NMR and crystal-structure study. Can. Miner. 1991, 29, 271–285. [Google Scholar]
- Groat, L.A.; Rossman, G.R.; Dyar, M.D.; Turner, D.; Piccoli, P.M.B.; Schultz, A.J.; Ottolini, L. Crystal chemistry of dark blue aquamarine from the True Blue showing, Yukon Territory, Canada. Can. Miner. 2010, 48, 597–613. [Google Scholar] [CrossRef]
- Taran, M.N.; Rossman, G.R. Optical spectroscopic study of tuhualite and a re-examination of the beryl, cordierite, and osumilite spectra. Am. Miner. 2001, 86, 973–980. [Google Scholar] [CrossRef]
- Taran, M.N.; Vyshnevskyi, O.A. Be, Fe2+-substitution in natural beryl: an optical absorption spectroscopy study. Phys. Chem. Miner. 2019, 46, 795–806. [Google Scholar] [CrossRef]
- Spinolo, G.; Fontana, I.; Galli, A. Optical absorption spectra of Fe2+ and Fe3+ in beryl crystals. Phys. Status Solidi Basic Res. 2007, 244, 4660–4668. [Google Scholar] [CrossRef]
- Fridrichová, J.; Bačík, P.; Ertl, A.; Wildner, M.; Dekan, J.; Miglierini, M. Jahn-Teller distortion of Mn3+-occupied octahedra in red beryl from Utah indicated by optical spectroscopy. J. Mol. Struct. 2018, 1152, 79–86. [Google Scholar] [CrossRef]
- Fridrichová, J.; Bačík, P.; Rusinová, P.; Antal, P.; Škoda, R.; Bizovská, V.; Miglierini, M. Optical and crystal-chemical changes in aquamarines and yellow beryls from Thanh Hoa province, Vietnam induced by heat treatment. Phys. Chem. Miner. 2015, 42, 287–302. [Google Scholar] [CrossRef]
- Wood, D.L.; Nassau, K. The characterization of beryl and emerald by visible and infrared absorption spectroscopy. Am. Miner. 1968, 53, 777–800. [Google Scholar]
- Viana, R.R.; da Costa, G.M.; De Grave, E.; Stern, W.B.; Jordt-Evangelista, H.; Stern, W.B. Characterization of beryl (aquamarine variety) by Mössbauer spectroscopy. Phys. Chem. Miner. 2002, 29, 78–86. [Google Scholar] [CrossRef]
- Přikryl, J.; Novák, M.; Filip, J.; Gadas, P.; Galiová, M.V. Iron+magnesium-bearing beryl from granitic pegmatites: An EMPA, LA-ICP-MS, Mössbauer spectroscopy, and powder XRD study. Can. Miner. 2014, 52, 271–284. [Google Scholar] [CrossRef]
- Taran, M.N.; Dyar, M.D.; Khomenko, V.M. Spectroscopic study of synthetic hydrothermal Fe3+-bearing beryl. Phys. Chem. Miner. 2018, 45, 489–496. [Google Scholar] [CrossRef]
- Don Goldman, S.; Rossman, G.R.; Parkin, K.M. Channel constituents in beryl. Phys. Chem. Miner. 1978, 3, 225–235. [Google Scholar] [CrossRef]
- Fridrichová, J.; Bačík, P.; Bizovská, V.; Libowitzky, E.; Škoda, R.; Uher, P.; Ozdín, D.; Števko, M. Spectroscopic and bond-topological investigation of interstitial volatiles in beryl from Slovakia. Phys. Chem. Miner. 2016, 43, 419–437. [Google Scholar] [CrossRef]
- Fukuda, J.; Shinoda, K.; Nakashima, S.; Miyoshi, N.; Aikawa, N. Polarized infrared spectroscopic study of diffusion of water molecules along structure channels in beryl. Am. Miner. 2009, 94, 981–985. [Google Scholar] [CrossRef]
- Łodziński, M.; Sitarz, M.; Stec, K.; Kozanecki, M.; Fojud, Z.; Jurga, S. ICP, IR, Raman, NMR investigations of beryls from pegmatites of the Sudety Mts. J. Mol. Struct. 2005, 744, 1005–1015. [Google Scholar] [CrossRef]
- Kolesov, B.A.; Geiger, C.A. The orientation and vibrational states of H2O in synthetic alkali-free beryl. Phys. Chem. Miner. 2000, 27, 557–564. [Google Scholar] [CrossRef]
- Charoy, B.; De Donato, P.; Barres, O.; Pinto-Coelho, C. Channel occupancy in an alkali-poor beryl from Serra Branca (Goias, Brazil): Spectroscopic characterization. Am. Miner. 1996, 81, 395–403. [Google Scholar] [CrossRef]
- Hagemann, H.; Lucken, A.; Bill, H.; Gysler-Sanz, J.; Stalder, H.A.A. Polarized Raman spectra of beryl and bazzite. Phys. Chem. Miner. 1990, 17, 395–401. [Google Scholar] [CrossRef]
- Aurisicchio, C.; Grubessi, O.; Zecchini, P. Infrared spectroscopy and crystal chemistry of the beryl group. Can. Miner. 1994, 32, 55–68. [Google Scholar]
- Wood, D.L.; Nassau, K. Infrared spectra of foreign molecules in beryl. J. Chem. Phys. 1967, 47, 2220–2228. [Google Scholar] [CrossRef]
- Fukuda, J.; Shinoda, K. Water molecules in beryl and cordierite: High-temperature vibrational behavior, dehydration, and coordination to cations. Phys. Chem. Miner. 2011, 38, 469–481. [Google Scholar] [CrossRef]
- Fukuda, J.; Shinoda, K. Coordination of water molecules with Na+ cations in a beryl channel as determined by polarized IR spectroscopy. Phys. Chem. Miner. 2008, 35, 347–357. [Google Scholar] [CrossRef]
- Zavarzina, N.I.; Gabuda, S.P.; Bakakin, V.V.; Rylov, G.M. NMR analysis of water in beryls. J. Struct. Chem. 1970, 10, 696–701. [Google Scholar] [CrossRef]
- Blak, A.R.; Isotani, S.; Watanabe, S. Optical absorption and electron spin resonance in blue and green natural beryl. Phys. Chem. Miner. 1982, 8, 161–166. [Google Scholar] [CrossRef]
- Andersson, L.O. The positions of H+, Li+ and Na+ impurities in beryl. Phys. Chem. Miner. 2006, 33, 403–416. [Google Scholar] [CrossRef]
- Andersson, L.O. The yellow color center and trapped electrons in beryl. Can. Miner. 2013, 51, 15–25. [Google Scholar] [CrossRef]
- Andersson, L.O. Comments on Beryl Colors and on Other Observations Regarding Iron-containing Beryls. Can. Miner. 2019, 57, 551–566. [Google Scholar] [CrossRef]
- Lin, J.; Chen, N.; Huang, D.; Pan, Y. Iron pairs in beryl: New insights from electron paramagnetic resonance, synchrotron X-ray absorption spectroscopy, and ab initio calculations. Am. Miner. 2013, 98, 1745–1753. [Google Scholar] [CrossRef]
- Edgar, A.; Hutton, D.R.R. Exchange-coupled pairs of Fe3+ ions in beryl. Solid State Commun. 1982, 41, 195–198. [Google Scholar] [CrossRef]
- Brown, I.D. The Chemical Bond in Inorganic Chemistry; Oxford University Press: Oxford, UK, 2006; ISBN 9780199298815. [Google Scholar]
- Brown, I.D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.D. The Chemical Bond in Inorganic Chemistry: The Bond Valence Model; Oxford University Press: Oxford, UK, 2010; Volume 9780199298, ISBN 9780191708879. [Google Scholar]
- Brown, I.D. (IUCr) Bond Valence Parameters. Available online: https://www.iucr.org/resources/data/datasets/bond-valence-parameters (accessed on 7 October 2019).
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Gagné, O.C.; Hawthorne, F.C. Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 562–578. [Google Scholar] [CrossRef]
- Gatta, G.D.; Adamo, I.; Meven, M.; Lambruschi, E. A single-crystal neutron and X-ray diffraction study of pezzottaite, Cs(Be2Li)Al2Si6O18. Phys. Chem. Miner. 2012, 39, 829–840. [Google Scholar] [CrossRef]
- Gibbs, G.V.V.; Breck, D.W.W.; Meagher, E.P.P. Structural refinement of hydrous and anhydrous synthetic beryl, Al2(Be3Si6)O18 and emerald, Al1.9Cr0.1(Be3Si6)O18. Lithos 1968, 1, 275–285. [Google Scholar] [CrossRef]
- Meagher, E.P.; Gibbs, G.V. The polymorphism of cordierite. II. The crystal structure of indialite. Can. Miner. 1977, 51, 43–49. [Google Scholar]
- Ferraris, G.; Prencipe, M.; Rossi, P. Stoppaniite, a new member of the beryl group: crystal structure and crystal-chemical implications. Eur. J. Miner. 1998, 10, 491–496. [Google Scholar] [CrossRef]
- Bakakin, V.V.; Rilov, G.M.; Belov, N.V. Crystal structure of a lithium-bearing beryl. Dokl. Akad. Nauk SSSR 1969, 188, 659–662. [Google Scholar]
- De Almeida Sampaio Filho, H.; Sighinolfi, G.; Galli, E. Contribution to crystal chemistry of beryl. Contrib. Miner. Petrol. 1973, 38, 279–290. [Google Scholar] [CrossRef]
- Lehmann, G. Optical absorption and electron spin resonance in blue and green natural beryl: A comment. Phys. Chem. Miner. 1983, 9, 278. [Google Scholar] [CrossRef]
- Viana, R.R.; Jordt-Evangelista, H.; Magela da Costa, G.; Stern, W.B. Characterization of beryl (aquamarine variety) from pegmatites of Minas Gerais, Brazil. Phys. Chem. Miner. 2002, 29, 668–679. [Google Scholar] [CrossRef]
- Meagher, E.P.; Gibbs, G.V. Tetrahedral magnesium in cordierite. Geol. Soc. Am. Spec. Pap. 1968, 115, 146. [Google Scholar]
- Schmetzer, K.; Schwarz, D.; Bernhardt, H.-J.; Häger, T. A new type of Tairus hydrothermally-grown synthetic emerald, coloured by vanadium and copper. J. Gemmol. 2006, 30, 59–74. [Google Scholar] [CrossRef]
- Adamo, I.; Gatta, G.D.; Rotiroti, N.; Diella, V.; Pavese, A. Gemmological investigation of a synthetic blue beryl: a multi-methodological study. Miner. Mag. 2008, 72, 799–808. [Google Scholar] [CrossRef]
- Burns, R.G.; Strens, R.G.J. Structural interpretation of polarized absorption spectra of the Al–Fe–Mn–Cr epidotes. Miner. Mag. J. Miner. Soc. 1967, 36, 204–226. [Google Scholar] [CrossRef]
- Ito, J.; Hafner, S.S. Synthesis and study of gadolinites. Am. Miner. 1974, 59, 700–708. [Google Scholar]
- Bačík, P.; Fridrichová, J.; Uher, P.; Pršek, J.; Ondrejka, M. The crystal chemistry of gadolinite-datolite group silicates. Can. Miner. 2014, 52, 625–642. [Google Scholar] [CrossRef]
- Ertl, A.; Vereshchagin, O.S.; Giester, G.; Tillmanns, E.; Meyer, H.-P.P.; Ludwig, T.; Rozhdestvenskaya, I.V.; Frank-Kamenetskaya, O.V. Structural and chemical investigation of a zoned synthetic Cu-rich tourmaline. Can. Miner. 2015, 53, 209–220. [Google Scholar] [CrossRef]
- Bačík, P. The crystal-chemical autopsy of octahedral sites in Na-dominant tourmalines: Octahedral metrics model unconstrained by the Y, Z-site disorder assignment. J. Geosci. 2018, 63, 137–154. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Černý, P.; Ercit, T.S. The classification of granitic pegmatites revisited. Can. Miner. 2005, 43, 2005–2026. [Google Scholar] [CrossRef]
- Uher, P.; Chudík, P.; Bačík, P.; Vaculovič, T.; Galiová, M. Beryl composition and evolution trends: An example from granitic pegmatites of the beryl-columbite subtype, Western Carpathians, Slovakia. J. Geosci. 2010, 55, 69–80. [Google Scholar] [CrossRef][Green Version]
- London, D.; Evensen, J.M. Beryllium in silicic magmas and the origin of Beryl-bearing pegmatites. In Beryllium: Mineralogy, Petrology, and Geochemistry; De Gruyter: Berlin, Germany, 2019; Volume 50, ISBN 9781501508844. [Google Scholar]
- Bačík, P.; Fridrichová, J.; Uher, P.; Rybár, S.; Bizovská, V.; Luptáková, J.; Vrábliková, D.; Pukančík, L.; Vaculovič, T. Octahedral substitution in beryl from weakly fractionated intragranitic pegmatite Predné Solisko, Tatry Mountains (Slovakia): The indicator of genetic conditions. J. Geosci. 2019, 64, 59–72. [Google Scholar] [CrossRef]
- Selway, J.B.; Smeds, S.A.; Černý, P.; Hawthorne, F.C. Compositional evolution of tourmaline in the petalite-subtype Nyköpingsgruvan pegmatites, Utö, Stockholm Archipelago, Sweden. GFF 2002, 124, 93–102. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).