
Theoretical Study on Impact of Chemical Composition and Water Content on Mechanical Properties of Stratlingite Mineral


Abstract

1. Introduction
2. Computational and Structural Details
2.1. Computational Details
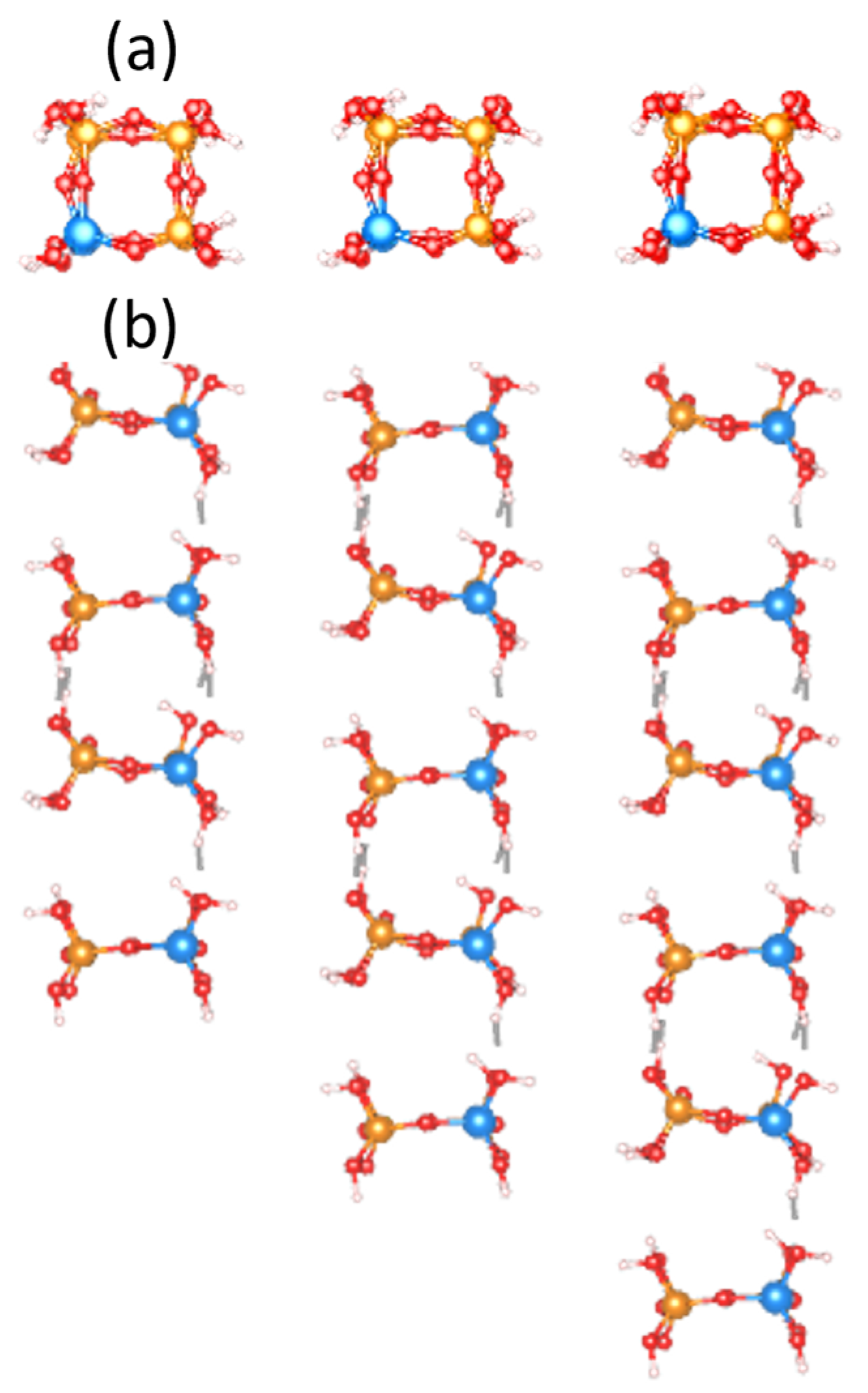
2.2. Structural Models
3. Results and Discussion
3.1. Structural and Bonding Properties
3.2. Mechanical Properties: Silicon and Water Content Effects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford: London, UK, 1997. [Google Scholar]
- L’Hôpital, E.; Lothenbach, B.; Le Saout, G.; Kulik, D.; Scrivener, K. Incorporation of aluminium in calcium-silicate-hydrates. Cem. Concr. Res. 2015, 75, 91–103. [Google Scholar] [CrossRef]
- Matschei, T.; Lothenbach, B.; Glasser, F.P. Thermodynamic properties of Portland cement hydrates in the system CaO–Al2O3–SiO2–CaSO4–CaCO3–H2O. Cem. Concr. Res. 2007, 37, 1379–1410. [Google Scholar] [CrossRef]
- Hentschel, G.; Kuzel, H. Stratlingite, 2CaO.AI2O3-SiO2-8H2O, A new mineral. Neues Jahrb. Mineral. Monat. 1976, 326–331. [Google Scholar]
- Jambor, J. New mineral names. Am. Min. 1992, 77, 670–675. [Google Scholar]
- Passaglia, E.; Turconi, B. Silicati e altri minerali di Montalto di Castro (Viterbo). Riv. Miner. Ital. 1982, 4, 97–110. [Google Scholar]
- Rinaldi, R.; Sacerdoti, M.; Passaglia, E. Stratlingite: Crystal structure, chemistry, and a reexamination of its polytype vertumnite. Eur. J. Min. 1990, 2, 841–849. [Google Scholar] [CrossRef]
- Midgley, H.G.; Rao, P.B. Formation of Strätlingite, 2CaO·SiO2 Al2O3 8H2O, in relation to the hydration of high alumina cement. Cem. Concr. Res. 1978, 8, 169–172. [Google Scholar] [CrossRef]
- Kwan, S.; LaRosa, J.; Grutzeck, M.W. 29Si and 27Al MASNMR study of strätlingite. J. Am. Ceram. Soc. 1995, 78, 1921–1926. [Google Scholar] [CrossRef]
- Aranda, M.A.G.; De la Torre, Á.G.; León-Reina, L. Rietveld quantitative phase analysis of OPC clinkers, cement and hydration products. Rev. Miner. Geochem. 2012, 74, 169–209. [Google Scholar] [CrossRef]
- Jackson, M.D.; Landis, E.N.; Brune, P.F.; Vitti, M.; Chena, H.; Lia, Q.; Kunz, M.; Wenk, H.-R.; Monteiro, P.J.M.; Ingraffea, A.R. Mechanical resilience and cementitious processes in Imperial Roman architectural mortar. Proc. Natl. Acad. Sci. USA 2014, 11, 18484–18489. [Google Scholar] [CrossRef]
- Oh, J.E.; Clark, S.M.; Monteiro, P.J.M. Does the Al substitution in C-S-H(I) change its mechanical property? Cem. Concr. Res. 2011, 41, 102–106. [Google Scholar] [CrossRef]
- Matschei, T.; Lothenbach, B.; Glasser, F.P. The AFm phase in Portland cement. Cem. Concr. Res. 2007, 37, 118–130. [Google Scholar] [CrossRef]
- Balonis, M.; Glasser, F.P. The density of cement phases. Cem. Concr. Res. 2009, 39, 733–739. [Google Scholar] [CrossRef]
- Santacruz, I.; De la Torre, Á.G.; Sanz, J.; Alvarez-Pinazo, G.; Aranda, M.A.G.; Cabeza, A. Structure of stratlingite and effect of hydration methodology on microstructure. Adv. Cem. Res. 2016, 28, 13–22. [Google Scholar] [CrossRef]
- Moon, J.; Oh, J.E.; Balonis, M.; Glasser, F.P.; Clark, S.M.; Monteiro, P.J.M. Pressure induced reactions amongst calcium aluminate phases. Cem. Concr. Res. 2012, 41, 571–578. [Google Scholar] [CrossRef]
- Cuesta, A.; Rejmak, P.; Ayuela, A.; De la Torre, A.G.; Santacruz, I.; Carrasco, L.F.; Popescu, C.; Aranda, M.A.G. Experimental and theoretical high pressure study of calcium hydroxyaluminate phases. Cem. Concr. Res. 2017, 97, 1–10. [Google Scholar] [CrossRef]
- Baquerizo, L.G.; Matschei, T.; Scrivener, K.L.; Saeidpour, M.; Wadsö, L. Hydration states of AFm cement phases. Cem. Concr. Res. 2015, 73, 143–157. [Google Scholar] [CrossRef]
- Scholtzová, E.; Tunega, D.; Speziale, S. Mechanical properties of ettringite and thaumasite—DFT and experimental study. Cem. Concr. Res. 2015, 77, 9–15. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Tunega, D.; Bučko, T.; Zaoui, A. Assessment of ten DFT methods in predicting structures of sheet silicates: Importance of dispersion corrections. J. Chem. Phys. 2012, 137, 114105. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic coefficients from ab initio total energy calculations. Phys. Rev. B 2001, 63, 174103. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Marmier, A.; Lethbridge, Z.A.D.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding. A Theoretical Perspective; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Voigt, W. Lehrbuch der Kristallphysik; Teubner: Berlin, Germany, 1928. [Google Scholar]
- Reuss, A. Berechnung der Fliessgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2009, 45, 823–843. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 2013, 8, 345–349. [Google Scholar] [CrossRef]
- Kleinman, L. Deformation potentials in silicon. I. Uniaxial strain. Phys. Rev. 1962, 128, 2614. [Google Scholar] [CrossRef]
- Thompson, R.P.; Clegg, W.J. Predicting whether a material is ductile or brittle. Curr. Opin. Solid State Mater. Sci. 2018, 22, 100–108. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | CA Layer Formula | AS Layer Formula | Sum Chem. Formula/UC | Ca:Al:Si Ratio |
|---|---|---|---|---|
| Str1 | [Ca2Al(OH)6·2H2O] | [(Si3AlO8)·H2O] | Ca6Al6Si9O51H36 | 2:2:3 |
| Str2 | [Ca2Al(OH)7·H2O] | [(Si4O8)·H2O] | Ca6Al3Si12O51H33 | 2:1:4 |
| Str1-w | [Ca2Al(OH)6·2H2O] | [(Si3AlO8)] | Ca6Al6Si9O48H30 | 2:2:3 |
| Str1-a | [Ca2Al(OH)6] | [(Si3AlO8)] | Ca6Al6Si9O42H18 | 2:2:3 |
| Str1_d | [Ca8Al4(OH)24·8H2O] | [(Si6Al2O10(OH)12)·4H2O] | Ca24Al18Si18O174H174 | 2:1.5:1.5 |
| Str2_d | [Ca8Al4(OH)24·8H2O] | [(Si8O12(OH)10)·4H2O] | Ca24Al12Si24O174H168 | 2:1:2 |
| a/Å | b/Å | c/Å | α/° | β/° | γ/° | V/Å3 | dev/% b | |
|---|---|---|---|---|---|---|---|---|
| Exp. | 5.745 | 5.745 | 37.770 | 90.00 | 90.00 | 120.00 | 1079.587 | 0.0 |
| Str1 | 5.509 | 5.503 | 38.064 | 89.67 | 90.09 | 119.94 | 1000.031 | −7.4 |
| Str1-w | 5.509 | 5.487 | 38.171 | 89.70 | 98.99 | 119.81 | 1000.973 | −7.3 |
| Str1-a | 5.464 | 5.463 | 36.072 | 90.23 | 89.93 | 120.01 | 932.431 | −13.6 |
| Str2 | 5.377 | 5.436 | 40.096 | 92.28 | 85.60 | 120.08 | 1011.098 | −6.3 |
| Str1_d a | 5.686 | 5.672 | 37.052 | 89.72 | 90.28 | 120.29 | 1031.622 | −4.4 |
| Str2_d a | 5.727 | 5.674 | 37.194 | 92.01 | 89.10 | 120.61 | 1039.553 | −3.7 |
| Str1 | Interval | Mean ± Std |
|---|---|---|
| AlIV–O | 1.718–1.763 | 1.743 ± 0.014 |
| AlVI–O | 1.885–1.903 | 1.894 ± 0.005 |
| Si–O | 1.576–1.723 | 1.653 ± 0.046 |
| Ca–O | 2.313–2.419 | 2.369 ± 0.037 |
| O–H | 0.969–0.985 | 0.975 ± 0.005 |
| H···O | 1.771–2.610 | 2.237 ± 0.230 |
| Elastic Constants | Str1 | Str1-w | Str1-a | Str2 | Str1_d | Str2_d |
|---|---|---|---|---|---|---|
| C11 | 154.1 | 153.8 | 146.3 | 160.1 | 60.4 | 54.6 |
| C22 ≈ C11 | 148.4 | 165.1 | 145.8 | 176.2 | 70.9 | 64.3 |
| C33 | 32.4 | 44.3 | 135.3 | 20.3 | 60.6 | 48.9 |
| C44 | 20.2 | 22.8 | 15.2 | 5.0 | 12.0 | 8.8 |
| C55 ≈ C44 | 21.5 | 24.1 | 14.3 | 13.0 | 8.6 | 8.1 |
| C66 ≈ 1/2 (C11–C12) | 47.8 | 47.2 | 47.3 | 49.1 | 18.4 | 15.4 |
| C12 | 66.0 | 71.4 | 48.9 | 75.2 | 19.3 | 15.3 |
| C13 | 9.6 | 14.6 | 4.9 | 0.7 | 13.4 | 12.6 |
| C14 | 0.5 | 3.1 | 0.7 | 2.1 | −0.0 | 0.0 |
| C15 ≈ 0 | 3.1 | −0.5 | 0.9 | 4.6 | 1.9 | 3.2 |
| C16 | 8.6 | 4.5 | −0.7 | 0.3 | 0.6 | 0.1 |
| C23 ≈ C13 | 13.2 | 19.6 | 7.3 | 5.6 | 16.6 | 9.9 |
| C24 ≈ C14 | 0.6 | 3.1 | 0.0 | −0.6 | 2.0 | 0.6 |
| C25 | −4.2 | −5.7 | −1.3 | −1.4 | −1.3 | 1.4 |
| C26 | 3.5 | −0.4 | 0.4 | −2.8 | −0.8 | −2.0 |
| C34 | −2.4 | 2.1 | 0.6 | 0.4 | 0.6 | 0.2 |
| C35 | 0.8 | −2.8 | 1.2 | −2.5 | 1.3 | 3.6 |
| C36 | 2.4 | −0.9 | −1.5 | 0.8 | 0.4 | 0.3 |
| C45 | 1.8 | 2.7 | 1.1 | 4.6 | −0.5 | −0.8 |
| C46 | −4.1 | −4.2 | −0.6 | 1.2 | −2.0 | −1.3 |
| C56 ≈ C14 | 0.2 | 0.0 | 1.4 | −0.8 | 0.1 | −1.0 |
| Property | Str1 | Str1-w | Str1-a | Str2 | Str1-d | Str2-d |
|---|---|---|---|---|---|---|
| BH/GPa | 42.6 | 50.8 | 60.3 | 37.5 | 32.0 | 26.3 |
| EH/GPa | 72.7 | 80.3 | 83.4 | 52.0 | 40.9 | 34.9 |
| GH/GPa | 29.9 | 32.5 | 32.9 | 20.5 | 15.9 | 13.6 |
| Poisson’s Ratio νH | 0.215 | 0.236 | 0.269 | 0.269 | 0.287 | 0.279 |
| Pugh’s Ratio BH/GH | 1.42 | 1.56 | 1.84 | 1.83 | 2.02 | 1.93 |
| Vickers Hardness/GPa | 6.8 | 6.5 | 5.5 | 3.91 | 2.93 | 2.77 |
| Universal Elast. Anisotropy | 2.73 | 1.92 | 2.69 | 14.6 | 0.99 | 1.26 |
| Cauchy Pressure pC/GPa | −45.7 | −48.6 | −33.6 | −70.2 | −7.4 | −6.5 |
| Kleinman parameter ξ | 0.72 | 0.78 | 0.58 | 0.79 | 0.56 | 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tunega, D.; Zaoui, A. Theoretical Study on Impact of Chemical Composition and Water Content on Mechanical Properties of Stratlingite Mineral. Minerals 2025, 15, 648. https://doi.org/10.3390/min15060648
Tunega D, Zaoui A. Theoretical Study on Impact of Chemical Composition and Water Content on Mechanical Properties of Stratlingite Mineral. Minerals. 2025; 15(6):648. https://doi.org/10.3390/min15060648
Chicago/Turabian StyleTunega, Daniel, and Ali Zaoui. 2025. "Theoretical Study on Impact of Chemical Composition and Water Content on Mechanical Properties of Stratlingite Mineral" Minerals 15, no. 6: 648. https://doi.org/10.3390/min15060648
APA StyleTunega, D., & Zaoui, A. (2025). Theoretical Study on Impact of Chemical Composition and Water Content on Mechanical Properties of Stratlingite Mineral. Minerals, 15(6), 648. https://doi.org/10.3390/min15060648