
The Controlling Effect of CaCO3 Supersaturation over Zn Carbonate Assemblages: Co-Crystallization in Silica Hydrogel


Abstract

1. Introduction
2. Experimental Methods
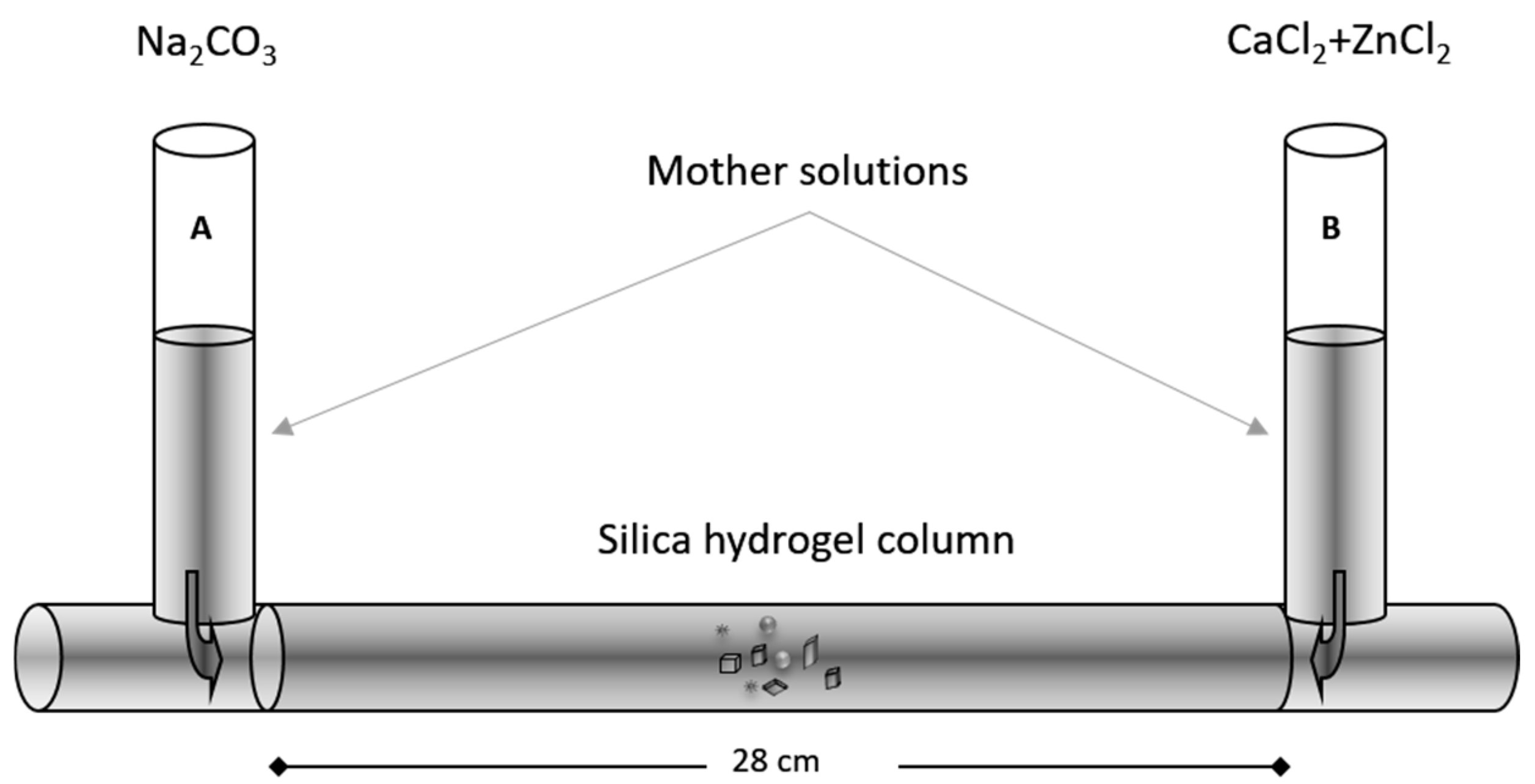
2.1. Crystal Growth Experiments
2.2. Solid Phase Characterization
3. Results
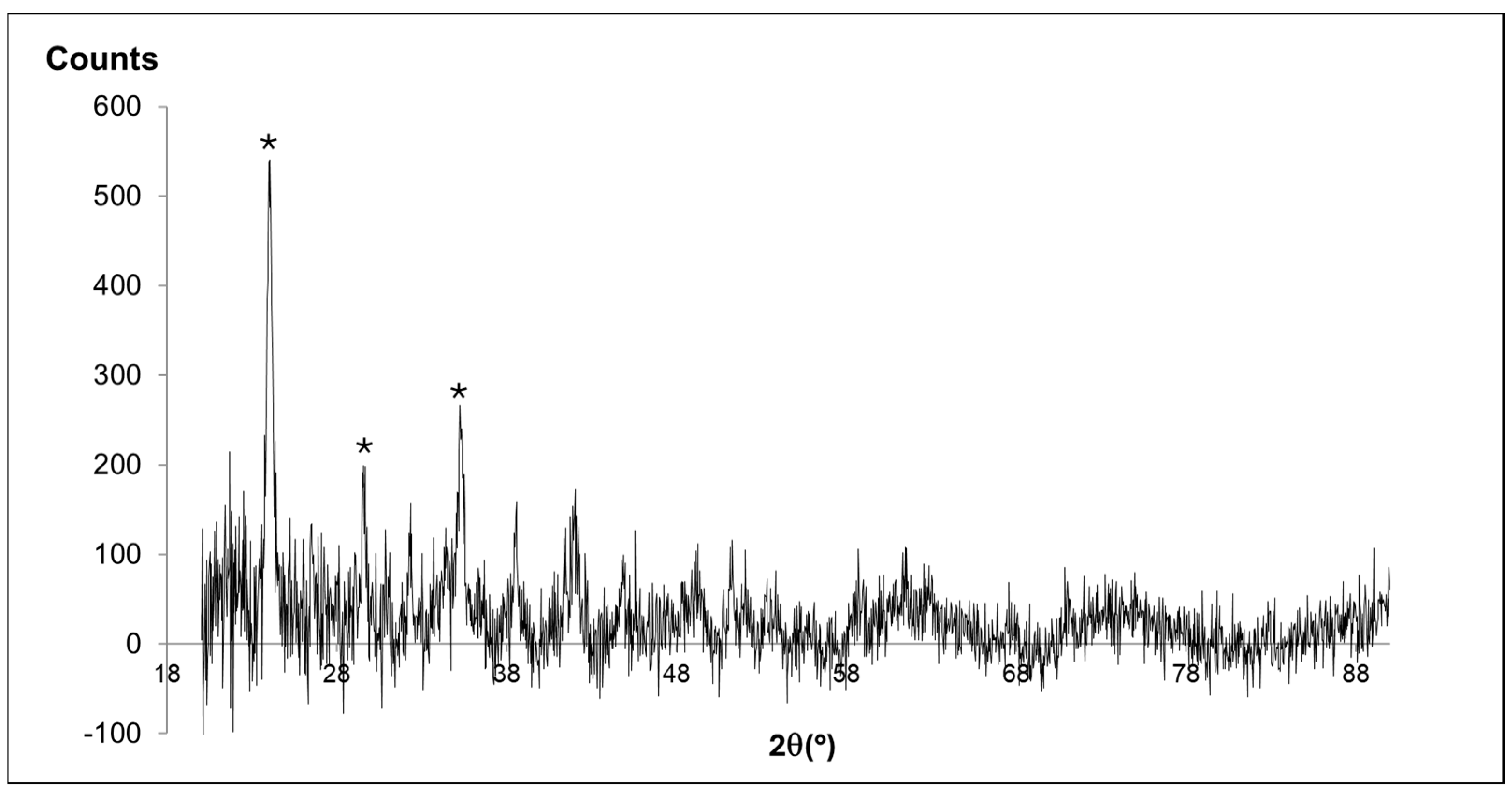
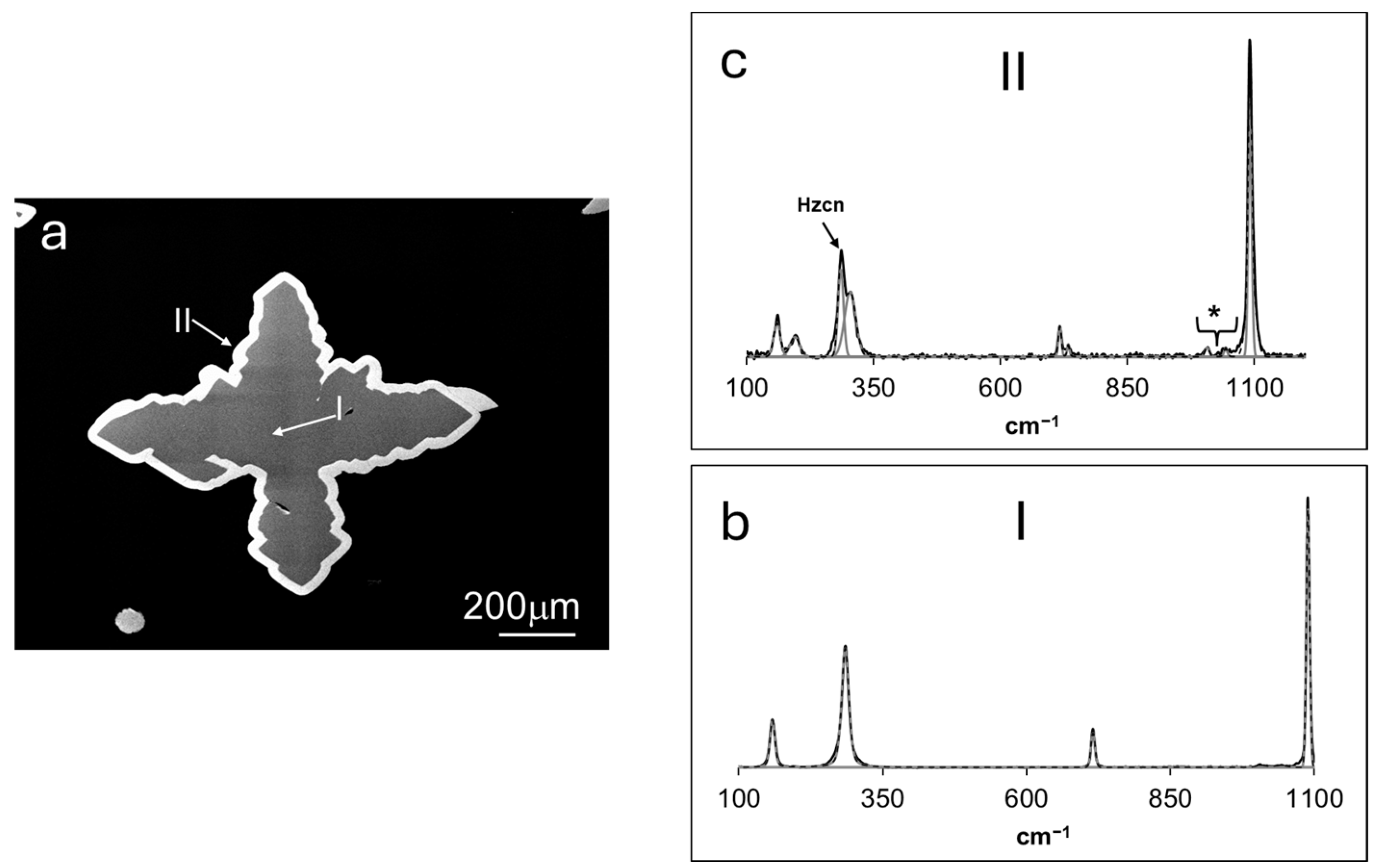
3.1. Crystal–Chemical Features of Grown Solids
3.1.1. Experiments 1–2
3.1.2. Experiment 3
3.1.3. Experiments 4–9
3.1.4. Experiments 10–15
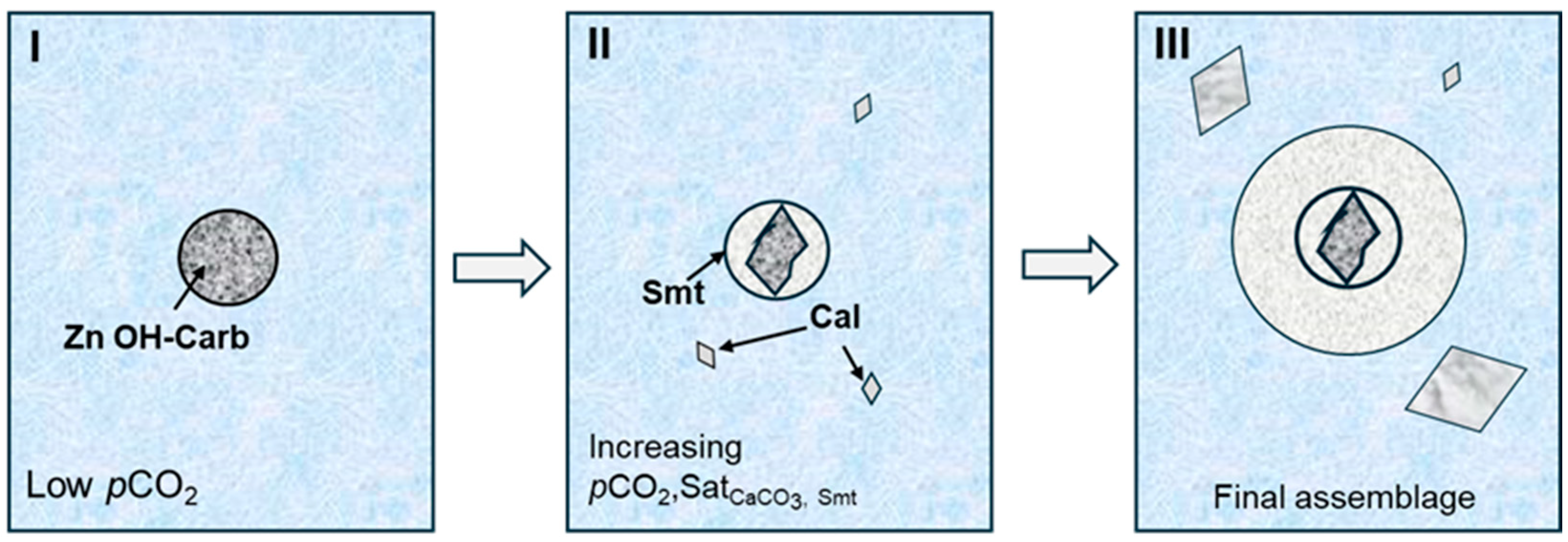
4. Discussion
Zn Carbonate Phases: The Controlling Role of CaCO3 Saturation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, D.; Zheng, L.; Ren, M.; Li, C.; Dong, X.; Wei, X.; Zhou, W.; Cui, J. Zinc in soil reflecting the intensive coal mining activities: Evidence from stable zinc isotopes analysis. Ecotoxicol. Environ. Saf. 2022, 239, 113669. [Google Scholar] [CrossRef]
- Marguí, E.; Queralt, I.; Carvalho, M.; Hidalgo, M. Assessment of metal availability to vegetation (Betula pendula) in Pb-Zn ore concentrate residues with different features. Environ. Pollut. 2007, 145, 179–184. [Google Scholar] [CrossRef]
- Beattie, R.E.; Henke, W.; Campa, M.F.; Hazen, T.C.; McAliley, L.R.; Campbell, J.H. Variation in microbial community structure correlates with heavy-metal contamination in soils decades after mining ceased. Soil Biol. Biochem. 2018, 126, 57–63. [Google Scholar] [CrossRef]
- Schwartz, M. Cadmium in Zinc Deposits: Economic Geology of a Polluting Element. Int. Geol. Rev. 2000, 42, 445–469. [Google Scholar] [CrossRef]
- Giannetta, M.G.; Soler, J.M.; Queralt, I.; Cama, J. Natural attenuation of heavy metals via secondary hydrozincite precipitation in an abandoned Pb-Zn mine. J. Geochem. Explor. 2023, 251, 107236. [Google Scholar] [CrossRef]
- Féher, B.; Szakáll, S.; Zajzon, N.; Miháli, J. Parádsasvárite, a new member of the malachite-rosasite group from Parádsasvárar, Mátra Mountains, Hungary. Mineral. Petrol. 2015, 109, 405–411. [Google Scholar] [CrossRef]
- Livingstone, A.; Champness, P.E. Brianyoungite, a new mineral related to hydrozincite, from the north of England orefield. Mineral. Mag. 1993, 57, 660–670. [Google Scholar] [CrossRef]
- Alwan, A.K.; Williams, P.A. Mineral formation from aqueous solutions. Part, I. The deposition of hydrozincite from natural waters. Transit. Met. Chem. 1979, 4, 128–132. [Google Scholar] [CrossRef]
- Podda, F.; Zuddas, P.; Minacci, A.; Pepi, M.; Baldi, F. Heavy Metal Coprecipitation with Hydrozincite [Zn5(CO3)2(OH)6] from Mine Waters Caused by Photosynthetic Microorganisms. Appl. Environ. Microbiol. 2000, 66, 5092–5098. [Google Scholar] [CrossRef]
- Schindler, P.; Reinert, M.; Gamsjäger, H. Loslichkeitskonstanten und freie Bildungsenthalpien von ZnCO3(s) und Zn5(OH)6(CO3)2 bei 25 °C. Helv. Chim. Acta 1969, 52, 2327–2332. [Google Scholar] [CrossRef]
- Zachara, J.M.; Kittrick, J.A.; Dake, L.S.; Harsh, J.B. Solubility and surface spectroscopy of zinc precipitates on calcite. Geochim. Cosmochim. Acta 1989, 53, 9–19. [Google Scholar] [CrossRef]
- Preis, W.; Gamsjäger, H. Solid + solute phase equilibria in aqueous solution. XIII. Thermodynamic properties of hydrozincite and predominance diagrams for (Zn2+ + H2O + CO2). J. Chem. Thermodyn. 2001, 33, 803–819. [Google Scholar] [CrossRef]
- Medas, D.; De Giudici, G.; Podda, F.; Meneghini, C.; Lattanzi, P. Apparent energy of hydrated biomineral surface and apparent solubility constant: An investigation of hydrozincite. Geochem. Cosmochim. Acta 2014, 140, 349–364. [Google Scholar] [CrossRef]
- Frost, R.L.; López, A.; Wang, L.; Scholz, R.; Sampaio, N.P. SEM, EDX and Raman and infrared spectroscopic study of brianyoungite Zn3(CO3,SO4)(OH)4 from Esperanza Mine, Laurion District, Greece. Spectrochim. Acta Part A 2015, 149, 279–284. [Google Scholar] [CrossRef]
- Prieto, M.; Cubillas, P.; Fernández-González, A. Uptake of dissolved Cd by biogenic and abiogenic aragonite: A comparison with sorption onto calcite. Geochim. Cosmochim. Acta 2003, 67, 3859–3869. [Google Scholar] [CrossRef]
- Hua, B.; Deng, B.L.; Thorton, E.C.; Yang, J.; Amonette, J.E. Incorporation of chromate into calcium carbonate structure coprecipitation. Water Air Soil Pollut. 2007, 179, 381–390. [Google Scholar] [CrossRef]
- Katsikopoulos, D.; Fernández-González, A.; Prieto, M. Crystallization behaviour of the (Mn, Ca)CO3 solid solution in silica gel: Nucleation, growth and zoning phenomena. Mineral. Mag. 2009, 73, 269–284. [Google Scholar] [CrossRef]
- Prieto, M.; Fernández-González, A.; Putnis, A.; Fernández-Díaz, L. Nucleation, growth, and zoning phenomena in crystallizing (Ba,Sr)CO3, Ba(SO4,CrO4), (Ba,Sr)SO4, and (Cd,Ca)CO3 solid solutions from aqueous solutions. Geochim. Cosmochim. Acta 1997, 61, 3383–3397. [Google Scholar] [CrossRef]
- Wada, N.; Yamashita, K.; Umegaki, T. Effects of silver, aluminum, and chromium ions on the polymorphic formation of calcium carbonate under conditions of double diffusion. J. Colloid Interface Sci. 1998, 201, 1–6. [Google Scholar] [CrossRef]
- Henisch, H.K. Crystals in Gels and Liesegang Rings; Cambridge University Press: Cambridge, UK, 1988; p. 197. [Google Scholar]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Perchiazzi, N.; Demitri, N.; Fehér, B.; Vignola, P. On the crystal chemitry of rosasite and parádsasvárite. Can. Mineral. 2017, 55, 1027–1040. [Google Scholar] [CrossRef]
- Whermeister, U.; Soldati, A.L.; Jacob, D.E.; Häger, T.; Hofmeister, W. Raman spectroscopy of synthetic, geological and biological vaterite: A Raman spectroscopic study. J. Raman Spectrosc. 2010, 41, 193–201. [Google Scholar] [CrossRef]
- Hales, M.C.; Frost, R.L. Synthesis and vibrational spectroscopic characterization of synthetic hydrozincite and smithsonite. Polyhedron 2007, 26, 4955–4962. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, L.; Kim, E.J.; Hahn, S.H. Electronic structure and optical properties of Zn(OH)2: LDA+U calculations and intense yellow luminescence. RSC Adv. 2015, 5, 97496. [Google Scholar] [CrossRef]
- Frost, R.L. A Raman spectroscopic study of selected minerals of the rosasite group. J. Raman Spectrosc. 2006, 37, 910–921. [Google Scholar] [CrossRef]
- Ghose, S. The crystal structure of hydrozincite (Zn5(CO3)2(OH)6). Acta Crystallogr. 1964, 17, 1051. [Google Scholar] [CrossRef]
- Prieto, M.; García-Rui, J.M.; Amorós, J.L. Growth of calcite crystals with non-singular faces. J. Cryst. Growth 1981, 52, 864–867. [Google Scholar] [CrossRef]
- Sun, J.; Wu, Z.; Cheng, H.; Zhang, Z.; Frost, R.L. A Raman spectroscopic comparison of calcite and dolomite. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.F.; Edwards, H.G.M.; Korsakov, A.; De Oliveira, L.F.C. Revisiting the Raman spectra of calcium carbonates. Minerals 2023, 13, 1358. [Google Scholar] [CrossRef]
- Williams, P.A. Oxide Zone Geochemistry; Ellis Horwood Ltd.: London, UK, 1999; p. 556. [Google Scholar]
- Sanna, R.; De Guidici, G.; Scorciapiano, A.M.; Floris, C.; Casu, M. Investigation of the hydrozincite structure by infrared and solid-state NMR spectroscopy. Am. Mineral. 2013, 98, 1219–1226. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Parent Solutions | ||
|---|---|---|---|
| Reservoir A | Reservoir B | ||
| Na2CO3 (M) | ZnCl2 (M) | CaCl2 (M) | |
| E1 | 0.5 | 0.5 | 0.1 |
| E2 | 0.5 | 0.5 | 0.2 |
| E3 | 0.5 | 0.5 | 0.3 |
| E4 | 0.5 | 0.5 | 0.4 |
| E5 | 0.5 | 0.5 | 0.5 |
| E6 | 0.5 | 0.4 | 0.5 |
| E7 | 0.5 | 0.3 | 0.5 |
| E8 | 0.5 | 0.2 | 0.5 |
| E9 | 0.5 | 0.1 | 0.5 |
| E10 | 0.3 | 0.5 | 0.5 |
| E11 | 0.1 | 0.5 | 0.5 |
| E12 | 0.3 | 0.1 | 0.5 |
| E13 | 0.1 | 0.1 | 0.5 |
| E14 | 0.3 | 0.05 | 0.5 |
| E15 | 0.1 | 0.05 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorge Pinto, A.; Sánchez-Pastor, N.; Fernández-González, A. The Controlling Effect of CaCO3 Supersaturation over Zn Carbonate Assemblages: Co-Crystallization in Silica Hydrogel. Minerals 2024, 14, 1274. https://doi.org/10.3390/min14121274
Jorge Pinto A, Sánchez-Pastor N, Fernández-González A. The Controlling Effect of CaCO3 Supersaturation over Zn Carbonate Assemblages: Co-Crystallization in Silica Hydrogel. Minerals. 2024; 14(12):1274. https://doi.org/10.3390/min14121274
Chicago/Turabian StyleJorge Pinto, André, Nuria Sánchez-Pastor, and Angeles Fernández-González. 2024. "The Controlling Effect of CaCO3 Supersaturation over Zn Carbonate Assemblages: Co-Crystallization in Silica Hydrogel" Minerals 14, no. 12: 1274. https://doi.org/10.3390/min14121274
APA StyleJorge Pinto, A., Sánchez-Pastor, N., & Fernández-González, A. (2024). The Controlling Effect of CaCO3 Supersaturation over Zn Carbonate Assemblages: Co-Crystallization in Silica Hydrogel. Minerals, 14(12), 1274. https://doi.org/10.3390/min14121274