

Structural and Thermodynamic Properties of Magnesium-Rich Liquids at Ultrahigh Pressure



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Simulation Methods
3. Results
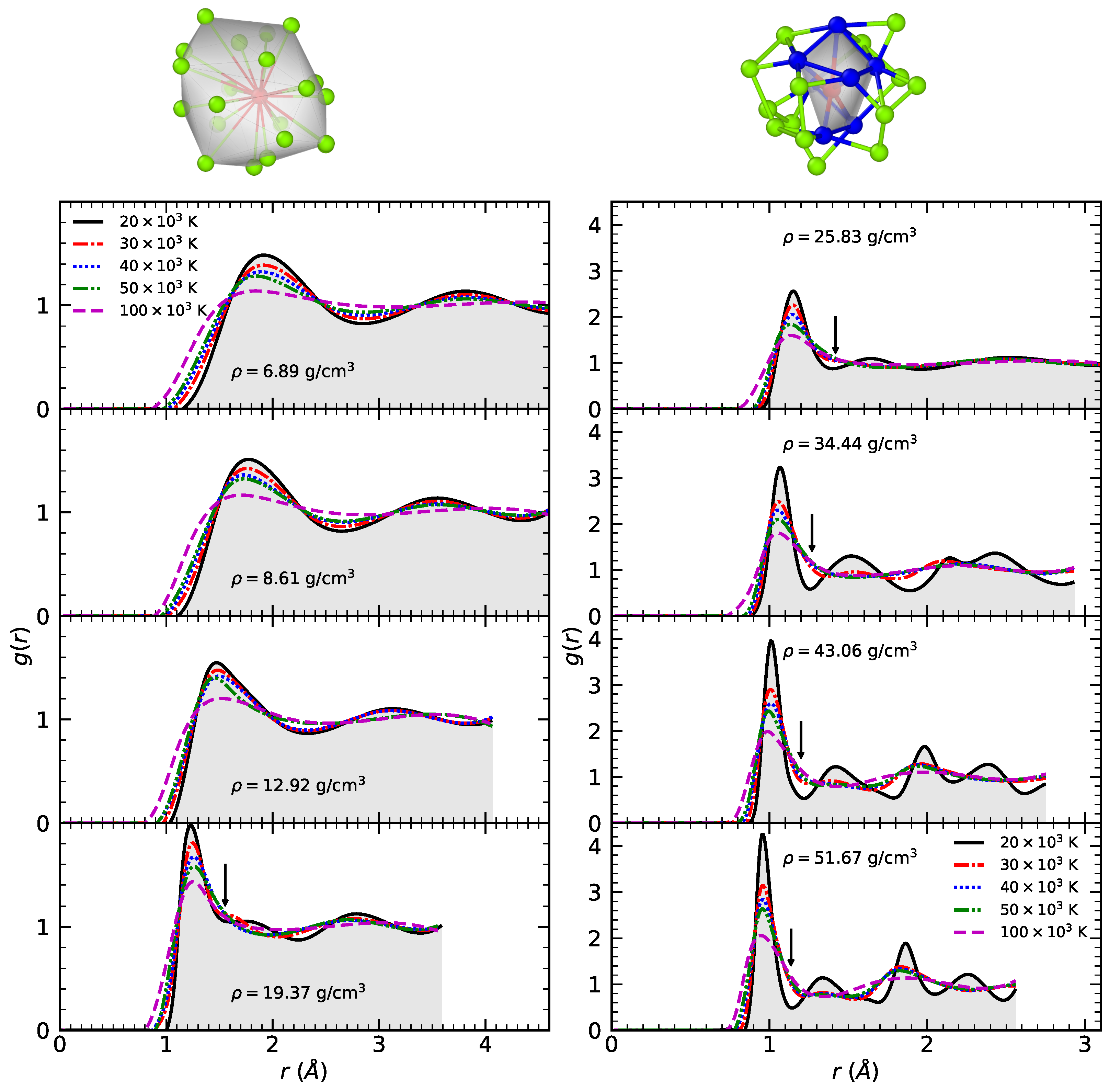
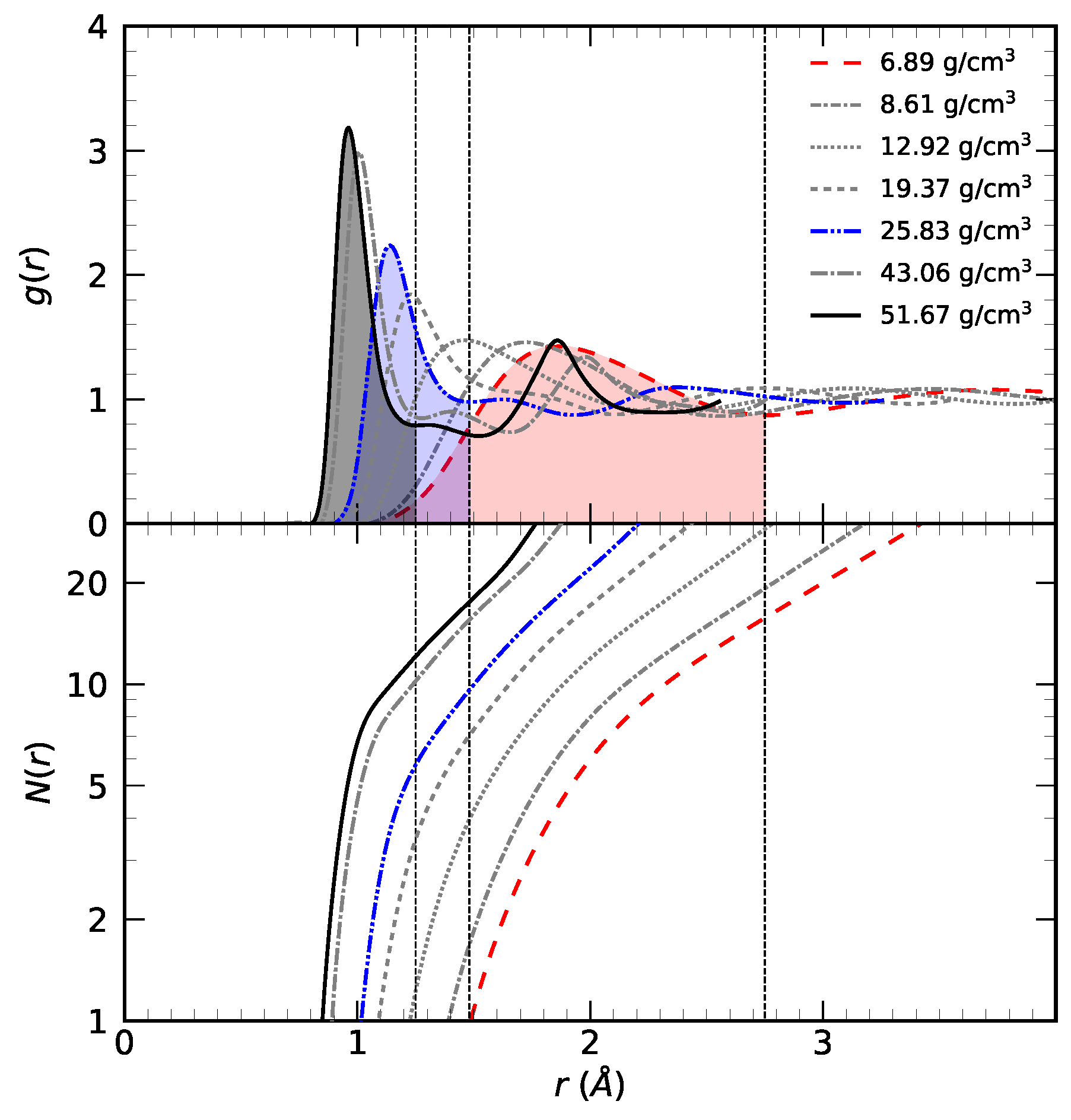
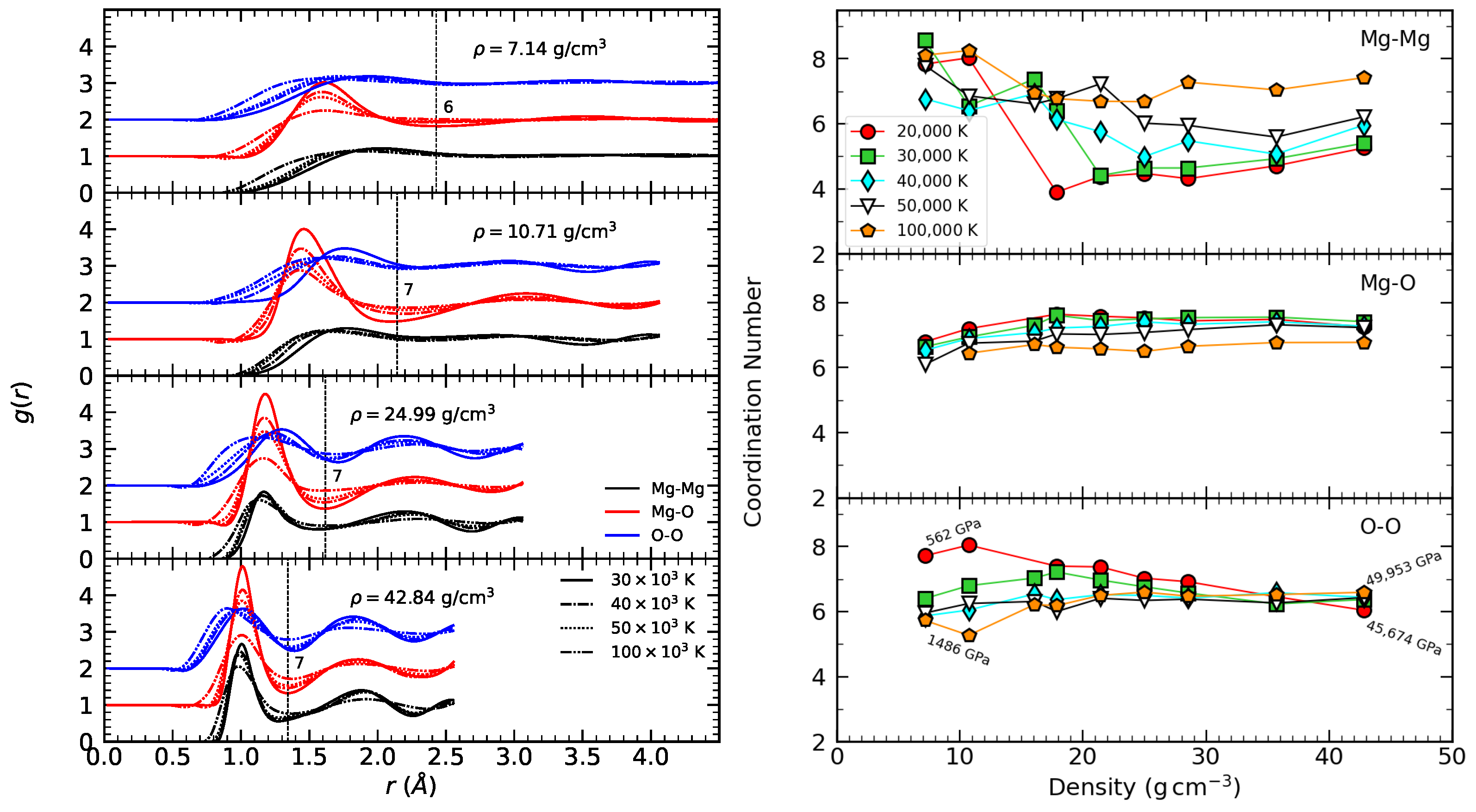
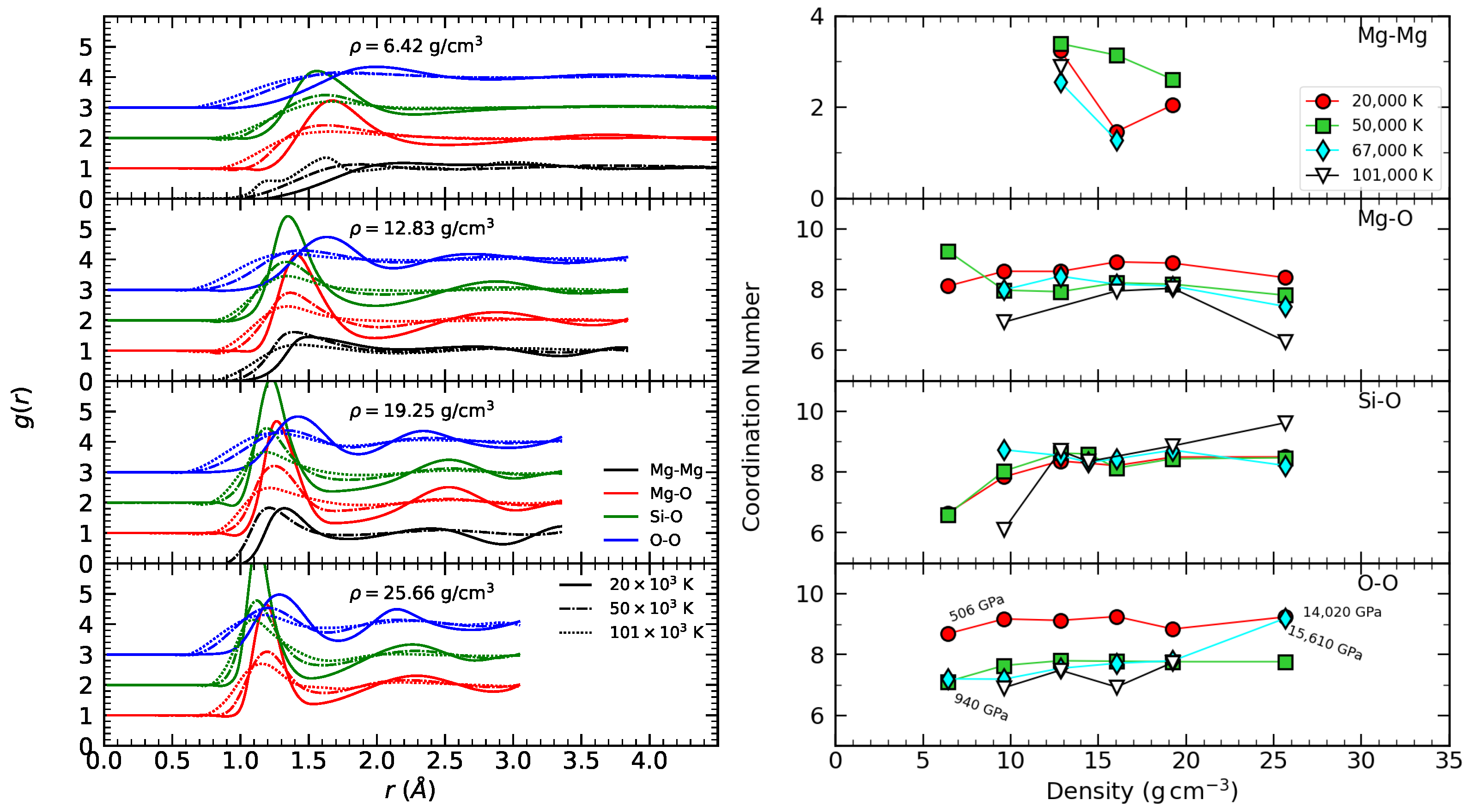
3.1. Radial Distribution Function
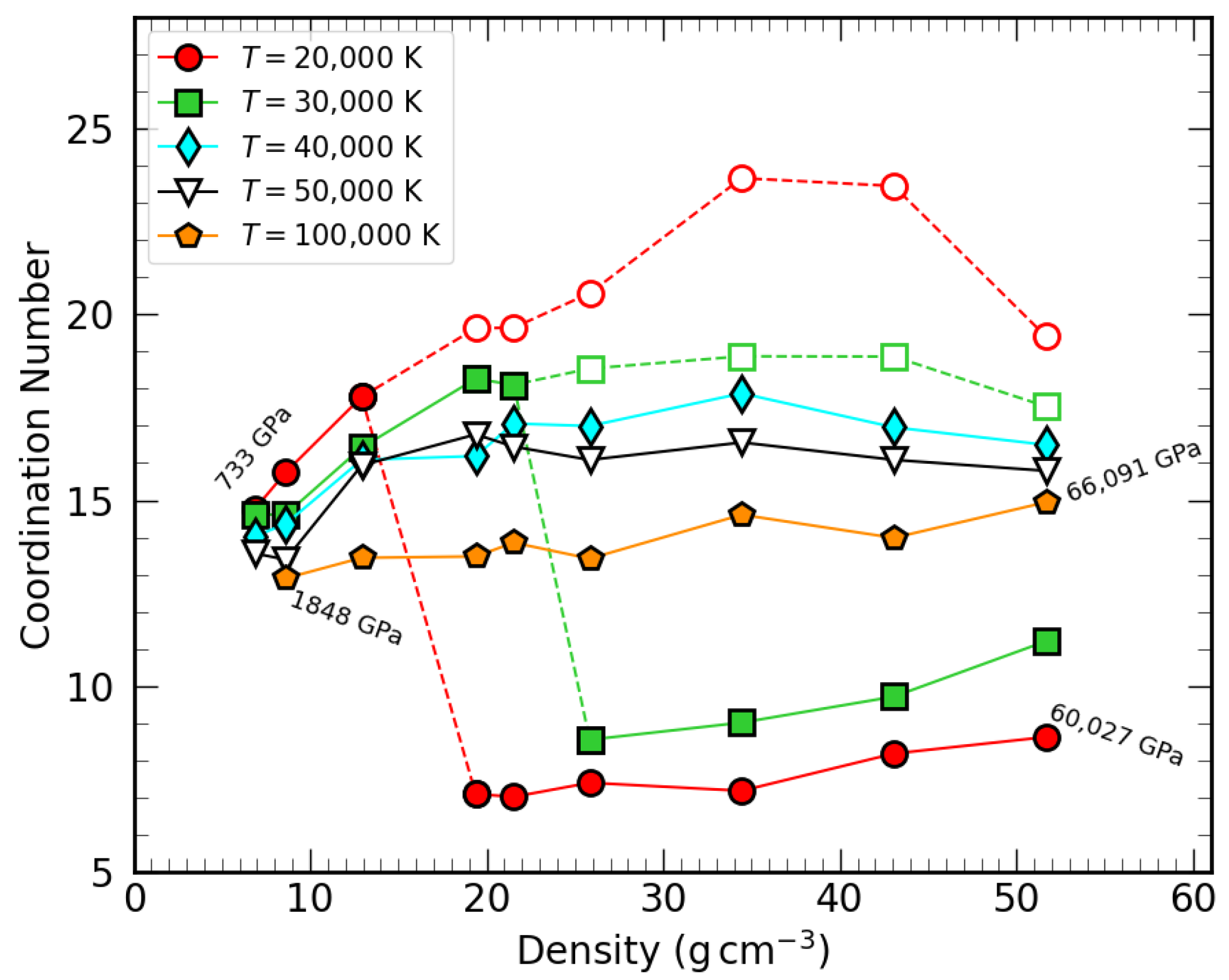
3.2. Coordination Number
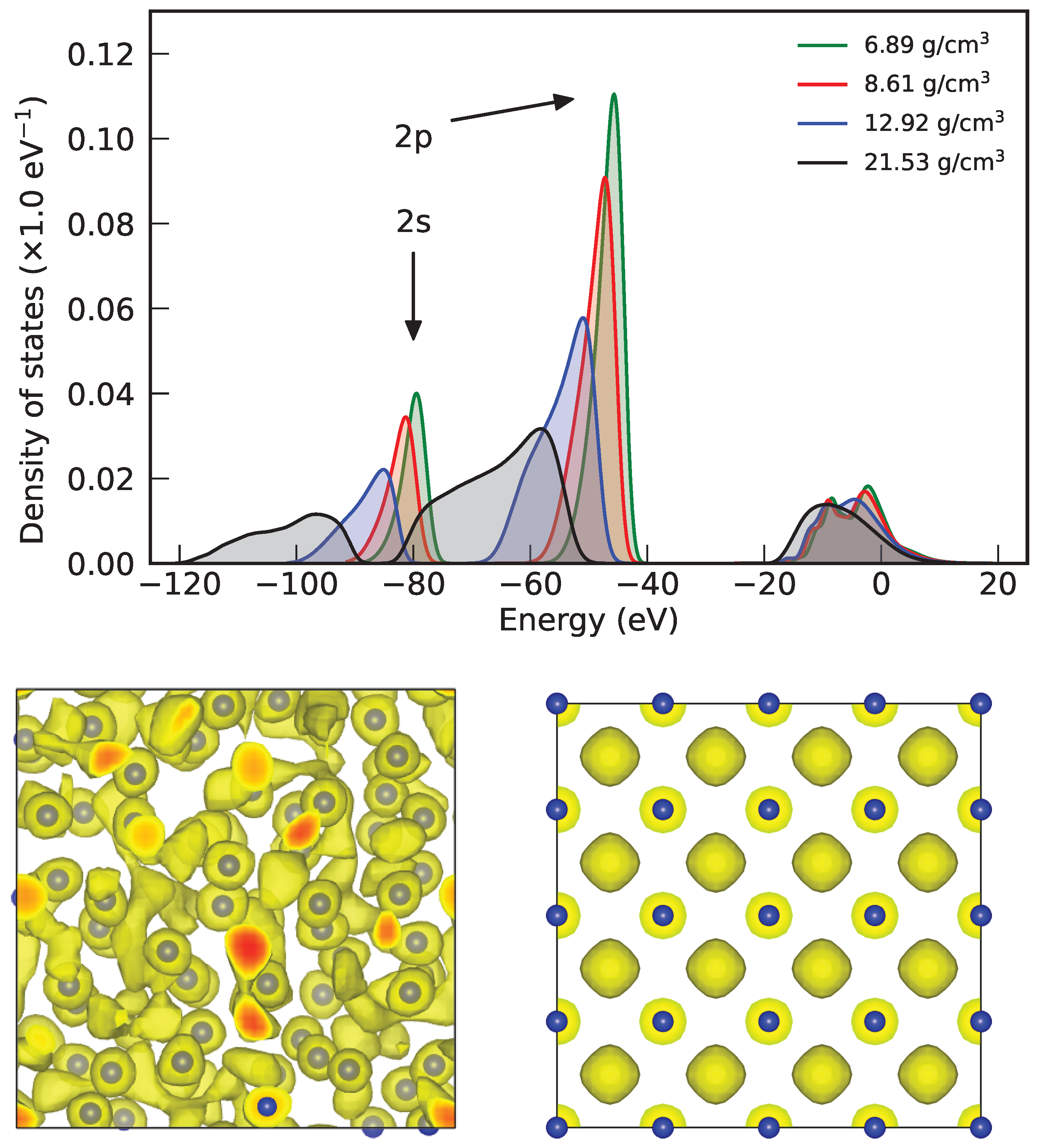
3.3. Electronic Density of States
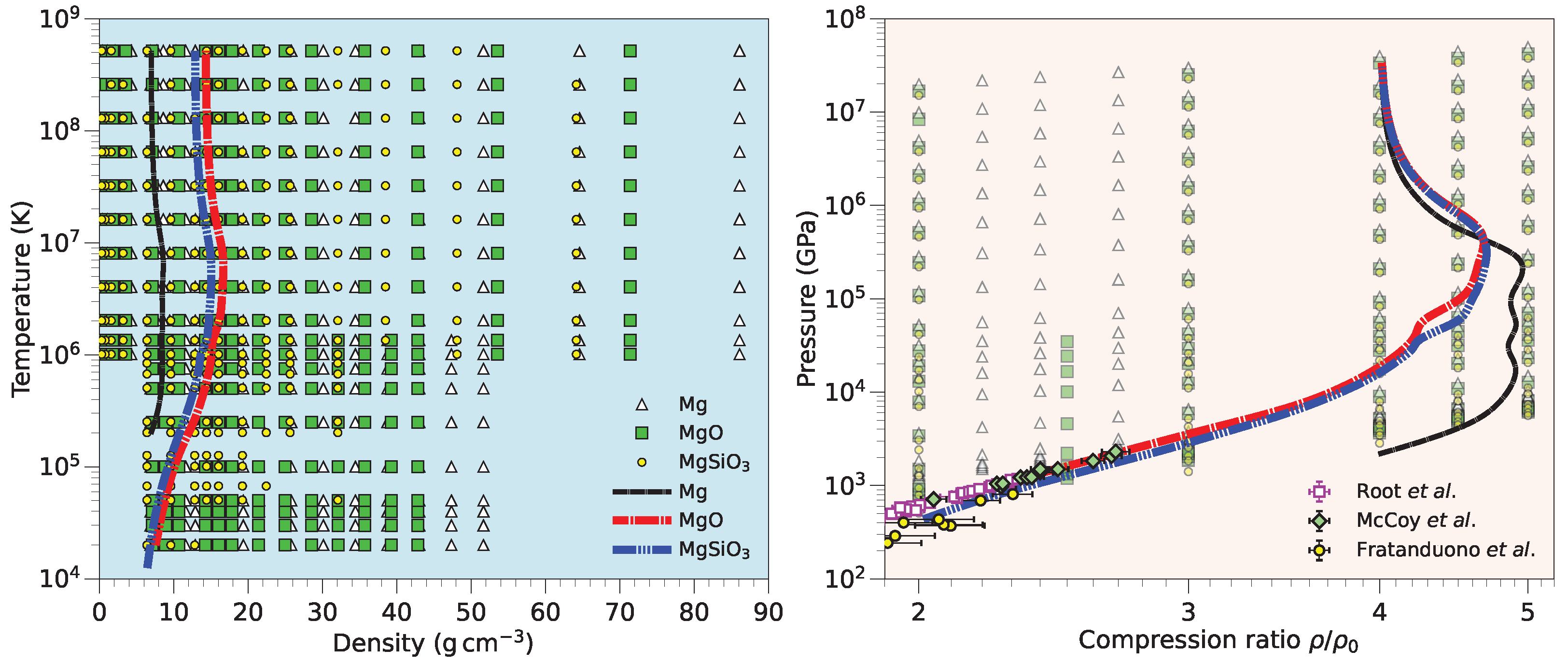
3.4. MgO and MgSiO3
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT-MD | Density Functional Theory Molecular Dynamics |
| PIMC | Path-Integral Monte Carlo |
| VASP | Vienna Ab initio Simulation Package |
References
- Labrosse, S.; Hernlund, J.; Coltice, N. A crystallizing dense magma ocean at the base of the Earth’s mantle. Nature 2007, 450, 866–869. [Google Scholar] [CrossRef]
- Rogers, L.A. Most 1.6 earth-radius planets are not rocky. Astrophys. J. 2015, 801, 41. [Google Scholar] [CrossRef]
- Kovačević, T.; González-Cataldo, F.; Stewart, S.T.; Militzer, B. Miscibility of rock and ice in the interiors of water worlds. Sci. Rep. 2022, 12, 13055. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, T.; González-Cataldo, F.; Militzer, B. The homogeneous mixing of MgO and H2O at extreme conditions. Contrib. Plasma Phys. 2023, e202300017. [Google Scholar] [CrossRef]
- Soubiran, F.; Militzer, B. Electrical conductivity and magnetic dynamos in magma oceans of Super-Earths. Nat. Commun. 2018, 9, 3883. [Google Scholar] [CrossRef] [PubMed]
- Boujibar, A.; Driscoll, P.; Fei, Y. Super-Earth Internal Structures and Initial Thermal States. J. Geophys. Res. Planets 2020, 125, e2019JE006124. [Google Scholar] [CrossRef]
- Smith, R.F.; Eggert, J.H.; Jeanloz, R.; Duffy, T.S.; Braun, D.G.; Patterson, J.R.; Rudd, R.E.; Biener, J.; Lazicki, A.E.; Hamza, A.V.; et al. Ramp compression of diamond to five terapascals. Nature 2014, 511, 330–333. [Google Scholar] [CrossRef]
- Smith, R.F.; Fratanduono, D.E.; Braun, D.G.; Duffy, T.S.; Wicks, J.K.; Celliers, P.M.; Ali, S.J.; Fernandez-Pañella, A.; Kraus, R.G.; Swift, D.C.; et al. Equation of state of iron under core conditions of large rocky exoplanets. Nat. Astron. 2018, 2, 452–458. [Google Scholar] [CrossRef]
- González-Cataldo, F.; Godwal, B.K.; Driver, K.; Jeanloz, R.; Militzer, B. Model of ramp compression of diamond from ab initio simulations. Phys. Rev. B 2021, 104, 134104. [Google Scholar] [CrossRef]
- Saumon, D.; Blouin, S.; Tremblay, P.E. Current challenges in the physics of white dwarf stars. Phys. Rep. 2022, 988, 1–63. [Google Scholar] [CrossRef]
- Coppari, F.; Smith, R.F.; Wang, J.; Millot, M.; Kim, D.; Rygg, J.R.; Hamel, S.; Eggert, J.H.; Duffy, T.S. Implications of the iron oxide phase transition on the interiors of rocky exoplanets. Nat. Geosci. 2021, 14, 121–126. [Google Scholar] [CrossRef]
- Zurkowski, C.; Fei, Y. Mineralogy of Planetary Cores. In Celebrating the International Year of Mineralogy: Progress and Landmark Discoveries of the Last Decades; Springer: Cham, Switzerland, 2023; pp. 207–247. [Google Scholar]
- Militzer, B.; Hubbard, W.B.; Wahl, S.; Lunine, J.I.; Galanti, E.; Kaspi, Y.; Miguel, Y.; Guillot, T.; Moore, K.M.; Parisi, M.; et al. Juno Spacecraft Measurements of Jupiter’s Gravity Imply a Dilute Core. Planet. Sci. J. 2022, 3, 185. [Google Scholar] [CrossRef]
- González-Cataldo, F.; Wilson, H.F.; Militzer, B. Ab Initio Free Energy Calculations of the Solubility of Silica in Metallic Hydrogen and Application to Giant Planet Cores. Astrophys. J. 2014, 787, 79. [Google Scholar] [CrossRef]
- Abu-Shawareb, H.; Acree, R.; Adams, P.; Adams, J.; Addis, B.; Aden, R.; Adrian, P.; Afeyan, B.; Aggleton, M.; Aghaian, L.; et al. Lawson criterion for ignition exceeded in an inertial fusion experiment. Phys. Rev. Lett. 2022, 129, 075001. [Google Scholar] [CrossRef]
- de Koker, N.; Stixrude, L. Self-consistent thermodynamic description of silicate liquids, with application to shock melting of MgO periclase and MgSiO3 perovskite. Geophys. J. Int. 2009, 178, 162–179. [Google Scholar] [CrossRef]
- González-Cataldo, F.; Soubiran, F.; Peterson, H.; Militzer, B. Path integral Monte Carlo and density functional molecular dynamics simulations of warm dense MgSiO3. Phys. Rev. B 2020, 101, 024107. [Google Scholar] [CrossRef]
- Stixrude, L.; Scipioni, R.; Desjarlais, M.P. A silicate dynamo in the early Earth. Nat. Commun. 2020, 11, 935. [Google Scholar] [CrossRef] [PubMed]
- Karki, B.B.; Bhattarai, D.; Stixrude, L. First-principles calculations of the structural, dynamical, and electronic properties of liquid MgO. Phys. Rev. B 2006, 73, 174208. [Google Scholar] [CrossRef]
- Karki, B.B.; Bhattarai, D.; Stixrude, L. First-principles simulations of liquid silica: Structural and dynamical behavior at high pressure. Phys. Rev. B 2007, 76, 104205. [Google Scholar] [CrossRef]
- González-Cataldo, F.; Davis, S.; Gutiérrez, G. Z method calculations to determine the melting curve of silica at high pressures. In Proceedings of the Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2016; Volume 720, p. 012032. [Google Scholar]
- Mookherjee, M.; Stixrude, L.; Karki, B. Hydrous silicate melt at high pressure. Nature 2008, 452, 983–986. [Google Scholar] [CrossRef] [PubMed]
- González-Cataldo, F.; Militzer, B. Thermal and pressure ionization in warm, dense MgSiO3 studied with first-principles computer simulations. AIP Conf. Proc. 2020, 2272, 090001. [Google Scholar] [CrossRef]
- Liu, C.; Shi, J.; Gao, H.; Wang, J.; Han, Y.; Lu, X.; Wang, H.T.; Xing, D.; Sun, J. Mixed Coordination Silica at Megabar Pressure. Phys. Rev. Lett. 2021, 126, 035701. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M. Fermion nodes. J. Stat. Phys. 1991, 63, 1237–1267. [Google Scholar] [CrossRef]
- Ceperley, D.M. Path integrals in the theory of condensed helium. Rev. Mod. Phys. 1995, 67, 279–355. [Google Scholar] [CrossRef]
- Ceperley, D. Path integral Monte Carlo methods for fermions. In Monte Carlo and Molecular Dynamics of Condensed Matter Systems; Binder, K., Ciccotti, G., Eds.; Editrice Compositori: Bologna, Italy, 1996; Volume 49, p. 443. [Google Scholar]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Driver, K.P.; Soubiran, F.; Militzer, B. Path integral Monte Carlo simulations of warm dense aluminum. Phys. Rev. E 2018, 97, 063207. [Google Scholar] [CrossRef]
- Soubiran, F.; González-Cataldo, F.; Driver, K.P.; Zhang, S.; Militzer, B. Magnesium oxide at extreme temperatures and pressures studied with first-principles simulations. J. Chem. Phys. 2019, 151, 214104. [Google Scholar] [CrossRef]
- González-Cataldo, F.; Soubiran, F.; Militzer, B. Equation of state of hot, dense magnesium derived with first-principles computer simulations. Phys. Plasmas 2020, 27, 092706. [Google Scholar] [CrossRef]
- Driver, K.P.; Militzer, B. First-principles simulations of warm dense lithium fluoride. Phys. Rev. E 2017, 95, 043205. [Google Scholar] [CrossRef]
- Zhang, S.; Militzer, B.; Gregor, M.C.; Caspersen, K.; Yang, L.H.; Gaffney, J.; Ogitsu, T.; Swift, D.; Lazicki, A.; Erskine, D.; et al. Theoretical and experimental investigation of the equation of state of boron plasmas. Phys. Rev. E 2018, 98, 023205. [Google Scholar] [CrossRef]
- Driver, K.P.; Soubiran, F.; Zhang, S.; Militzer, B. First-principles equation of state and electronic properties of warm dense oxygen. J. Chem. Phys. 2015, 143, 164507. [Google Scholar] [CrossRef] [PubMed]
- Militzer, B.; Driver, K.P. Development of Path Integral Monte Carlo Simulations with Localized Nodal Surfaces for Second-Row Elements. Phys. Rev. Lett. 2015, 115, 176403. [Google Scholar] [CrossRef]
- Driver, K.P.; Soubiran, F.; Zhang, S.; Militzer, B. Comparison of path integral Monte Carlo simulations of helium, carbon, nitrogen, oxygen, water, neon, and silicon plasmas. High Energy Density Phys. 2017, 23, 81–89. [Google Scholar] [CrossRef]
- Zhang, S.; Militzer, B.; Benedict, L.X.; Soubiran, F.; Sterne, P.A.; Driver, K.P. Path integral Monte Carlo simulations of dense carbon-hydrogen plasmas. J. Chem. Phys. 2018, 148, 102318. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.F.; Wong, M.L.; Militzer, B. Superionic to Superionic Phase Change in Water: Consequences for the Interiors of Uranus and Neptune. Phys. Rev. Lett. 2013, 110, 151102. [Google Scholar] [CrossRef]
- Millot, M.; Hamel, S.; Rygg, J.R.; Celliers, P.M.; Collins, G.W.; Coppari, F.; Fratanduono, D.E.; Jeanloz, R.; Swift, D.C.; Eggert, J.H. Experimental evidence for superionic water ice using shock compression. Nat. Phys. 2018, 14, 297–302. [Google Scholar] [CrossRef]
- Militzer, B. Path Integral Monte Carlo Simulations of Hot Dense Hydrogen. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2000. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Price, G.; Alfè, D. Ab initio thermodynamics and phase diagram of solid magnesium: A comparison of the LDA and GGA. J. Chem. Phys. 2006, 125, 194507. [Google Scholar] [CrossRef]
- Driver, K.P.; Militzer, B. First-principles simulations and shock Hugoniot calculations of warm dense neon. Phys. Rev. B 2015, 91, 045103. [Google Scholar] [CrossRef]
- Militzer, B.; González-Cataldo, F.; Zhang, S.; Driver, K.P.; Soubiran, F. First-principles equation of state database for warm dense matter computation. Phys. Rev. E 2021, 103, 013203. [Google Scholar] [CrossRef]
- McCoy, C.A.; Marshall, M.C.; Polsin, D.N.; Fratanduono, D.E.; Celliers, P.M.; Meyerhofer, D.D.; Boehly, T.R. Hugoniot, sound velocity, and shock temperature of MgO to 2300 GPa. Phys. Rev. B 2019, 100, 014106. [Google Scholar] [CrossRef]
- Root, S.; Shulenburger, L.; Lemke, R.W.; Dolan, D.H.; Mattsson, T.R.; Desjarlais, M.P. Shock Response and Phase Transitions of MgO at Planetary Impact Conditions. Phys. Rev. Lett. 2015, 115, 198501. [Google Scholar] [CrossRef] [PubMed]
- Fratanduono, D.E.; Millot, M.; Kraus, R.G.; Spaulding, D.K.; Collins, G.W.; Celliers, P.M.; Eggert, J.H. Thermodynamic properties of MgSiO3 at super-Earth mantle conditions. Phys. Rev. B 2018, 97, 214105. [Google Scholar] [CrossRef]
- Militzer, B.; González-Cataldo, F.; Zhang, S.; Whitley, H.D.; Swift, D.C.; Millot, M. Nonideal mixing effects in warm dense matter studied with first-principles computer simulations. J. Chem. Phys. 2020, 153, 184101. [Google Scholar] [CrossRef]
- Karki, B.B.; Ghosh, D.B.; Bajgain, S.K. Simulation of Silicate Melts Under Pressure. In Magmas Under Pressure; Elsevier: Amsterdam, The Netherlands, 2018; pp. 419–453. [Google Scholar] [CrossRef]
- Allen, M.; Tildesley, D. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Gorman, M.G.; Elatresh, S.; Lazicki, A.; Cormier, M.M.E.; Bonev, S.A.; McGonegle, D.; Briggs, R.; Coleman, A.L.; Rothman, S.D.; Peacock, L.; et al. Experimental observation of open structures in elemental magnesium at terapascal pressures. Nat. Phys. 2022, 18, 1307–1311. [Google Scholar] [CrossRef]
- Li, P.; Gao, G.; Wang, Y.; Ma, Y. Crystal structures and exotic behavior of magnesium under pressure. J. Phys. Chem. C 2010, 114, 21745–21749. [Google Scholar] [CrossRef]
- Zong, H.; Robinson, V.N.; Hermann, A.; Zhao, L.; Scandolo, S.; Ding, X.; Ackland, G.J. Free electron to electride transition in dense liquid potassium. Nat. Phys. 2021, 17, 955–960. [Google Scholar] [CrossRef]
- Miao, M.S.; Hoffmann, R. High pressure electrides: A predictive chemical and physical theory. Accounts Chem. Res. 2014, 47, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Kang, D.; Zhao, Z.; Wu, Y.; Yuan, J. Dynamic ionic clusters with flowing electron bubbles from warm to hot dense iron along the Hugoniot curve. Phys. Rev. Lett. 2012, 109, 175701. [Google Scholar] [CrossRef] [PubMed]
- Woolman, G.; Robinson, V.N.; Marqués, M.; Loa, I.; Ackland, G.J.; Hermann, A. Structural and electronic properties of the alkali metal incommensurate phases. Phys. Rev. Mater. 2018, 2, 053604. [Google Scholar] [CrossRef]
- Ayrinhac, S.; Robinson, V.N.; Decremps, F.; Gauthier, M.; Antonangeli, D.; Scandolo, S.; Morand, M. High-pressure transformations in liquid rubidium. Phys. Rev. Mater. 2020, 4, 113611. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Rogan, J. Structure of liquid GeO2 from a computer simulation model. Phys. Rev. E 2004, 69, 031201. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Menéndez-Proupin, E.; Loyola, C.; Peralta, J.; Davis, S. Computer simulation study of amorphous compounds: Structural and vibrational properties. J. Mater. Sci. 2010, 45, 5124–5134. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Cataldo, F.; Militzer, B. Structural and Thermodynamic Properties of Magnesium-Rich Liquids at Ultrahigh Pressure. Minerals 2023, 13, 885. https://doi.org/10.3390/min13070885
González-Cataldo F, Militzer B. Structural and Thermodynamic Properties of Magnesium-Rich Liquids at Ultrahigh Pressure. Minerals. 2023; 13(7):885. https://doi.org/10.3390/min13070885
Chicago/Turabian StyleGonzález-Cataldo, Felipe, and Burkhard Militzer. 2023. "Structural and Thermodynamic Properties of Magnesium-Rich Liquids at Ultrahigh Pressure" Minerals 13, no. 7: 885. https://doi.org/10.3390/min13070885
APA StyleGonzález-Cataldo, F., & Militzer, B. (2023). Structural and Thermodynamic Properties of Magnesium-Rich Liquids at Ultrahigh Pressure. Minerals, 13(7), 885. https://doi.org/10.3390/min13070885