
Time-Dependent Retention of a Mixture of Cs(I), Sm(III), Eu(III) and U(VI) as Waste Cocktail by Calcium Silicate Hydrate (C-S-H) Phases

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standards
2.2. Experiments and ICP-MS Samples
2.3. The ICP-MS System
3. Results
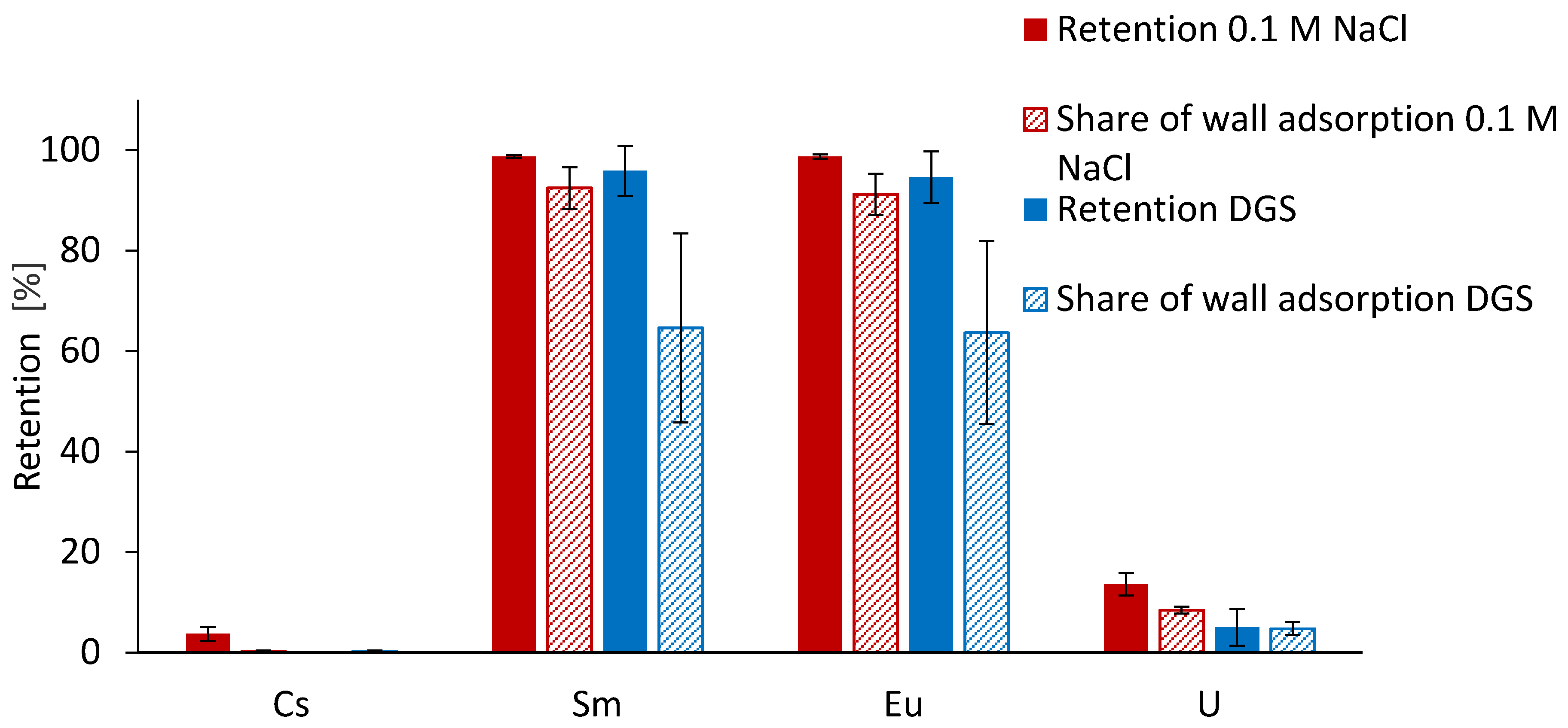
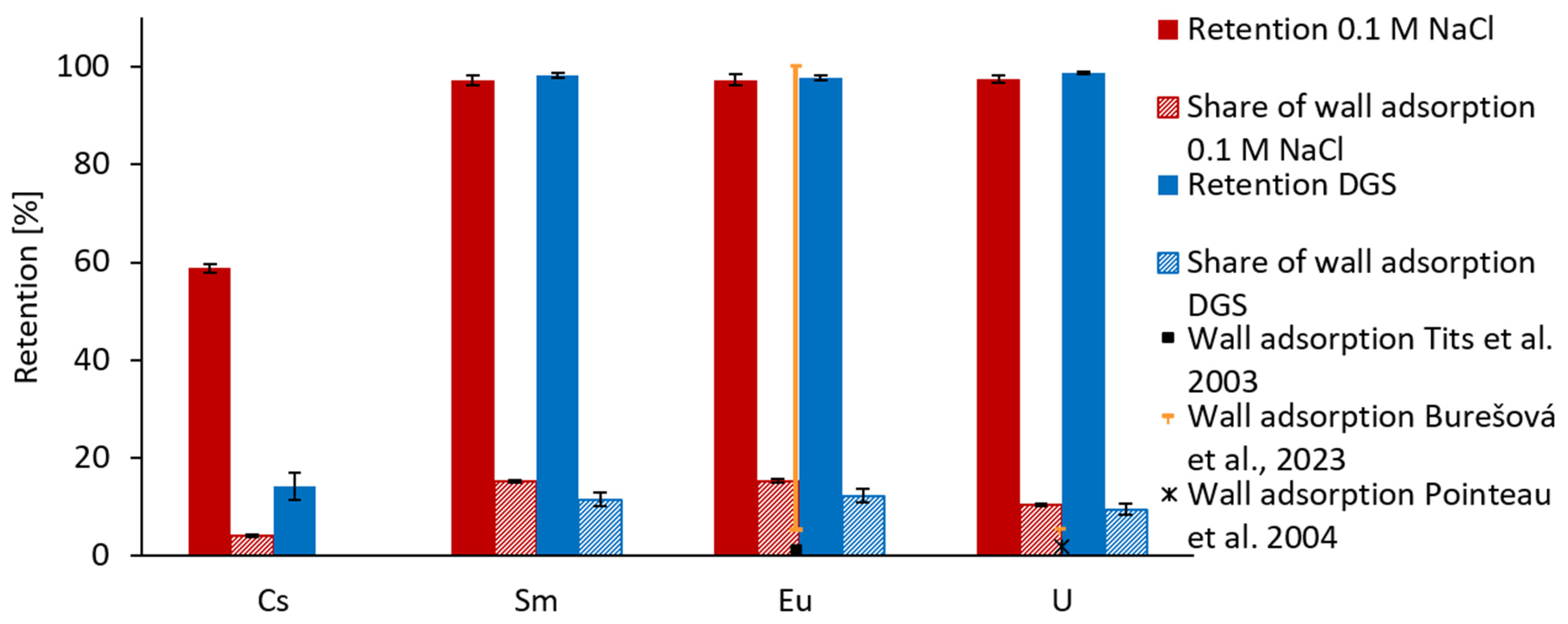
3.1. Precipitation and Wall Adsorption
3.2. Time-Dependent Retention of Cs(I)
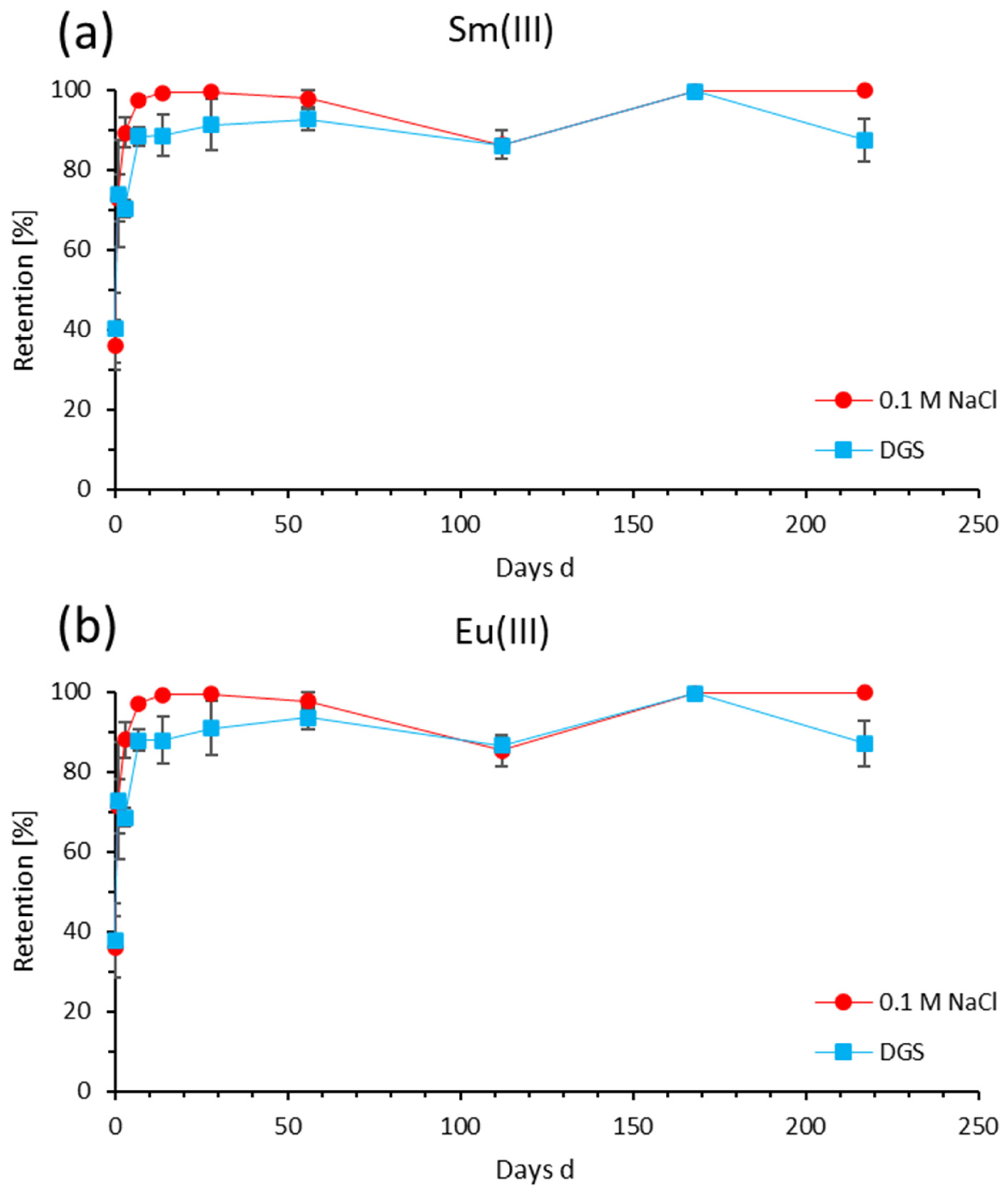
3.3. Time-Dependent Retention of Sm(III) and Eu(III)
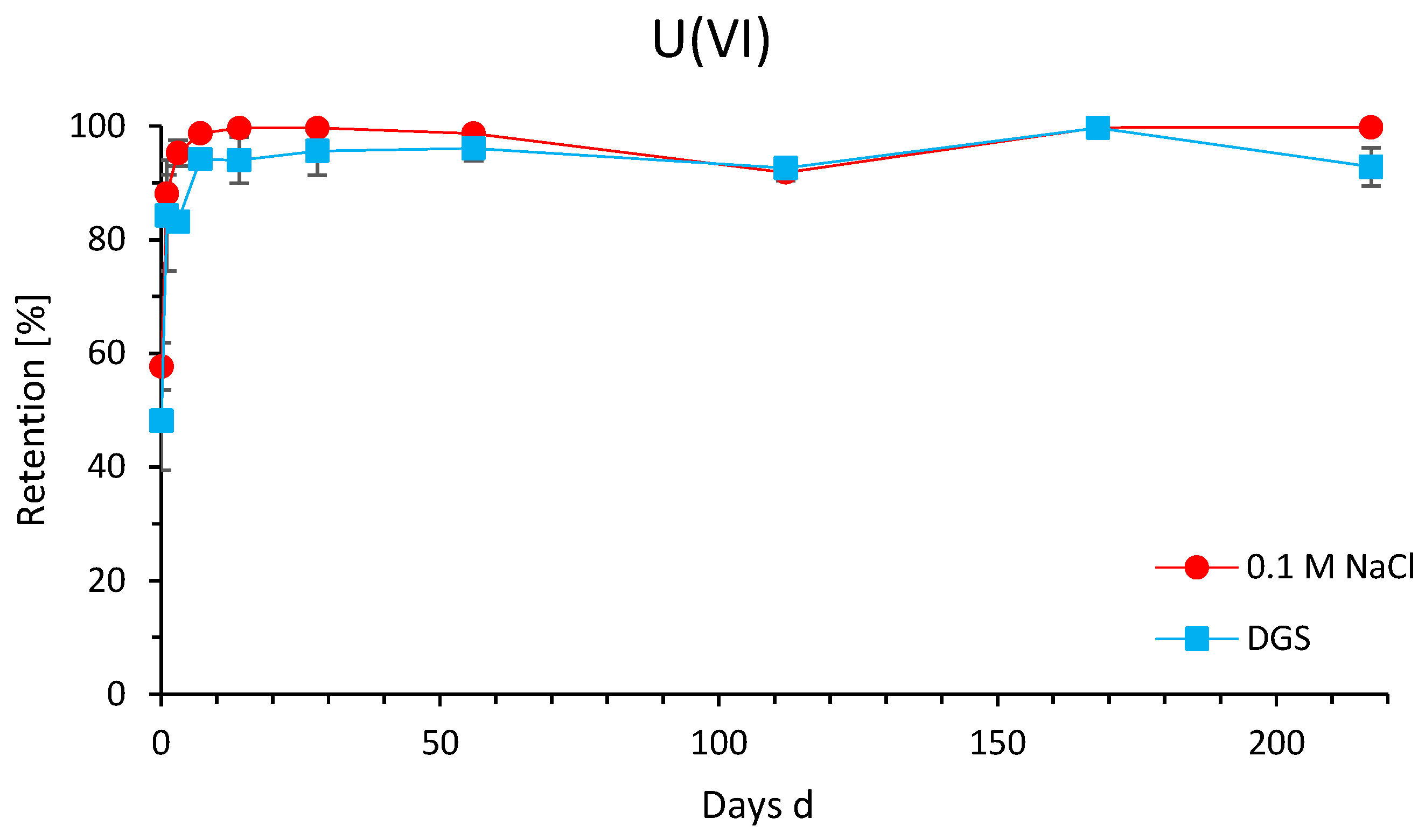
3.4. Time-Dependent Retention of U(VI)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Atkins, M.; Glasser, F.P.; Kindness, A. Cement hydrate phase: Solubility at 25 °C. Cem. Concr. Res. 1992, 22, 241–246. [Google Scholar] [CrossRef]
- Evans, N.D.M. Binding mechanisms of radionuclides to cement. Cem. Concr. Res. 2008, 38, 543–553. [Google Scholar] [CrossRef]
- Ochs, M.; Mallants, D.; Wang, L. Radionuclide and Metal Sorption on Cement and Concrete, 1st ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Jacques, D. Time Dependence of the Geochemical Boundary Conditions for the Cementitious Engineered Barriers of the Belgian Surface Disposal Facility; NIROND-TR 2008-24E; ONDRAF: Brussels, Belgium, 2008.
- Lothenbach, B.; Nonat, A. Calcium silicate hydrates: Solid and liquid phase composition. Cem. Concr. Res. 2015, 78, 57–70. [Google Scholar] [CrossRef]
- Gaona, X.; Dähn, R.; Tits, J.; Scheinost, A.C.; Wieland, E. Uptake of Np(IV) by C–S–H Phases and Cement Paste: An EXAFS Study. Environ. Sci. Technol. 2011, 45, 8765–8771. [Google Scholar] [CrossRef]
- Tits, J.; Wieland, E.; Müller, C.J.; Landesman, C.; Bradbury, M.H. Strontium binding by calcium silicate hydrates. J. Colloid Interface Sci. 2006, 300, 78–87. [Google Scholar] [CrossRef]
- Chen, J.J.; Thomas, J.J.; Taylor, H.F.W.; Jennings, H.M. Solubility and structure of calcium silicate hydrate. Cem. Concr. Res. 2004, 34, 1499–1519. [Google Scholar] [CrossRef]
- Tits, J.; Fujita, T.; Tsukamoto, M.; Wieland, E. Uranium(VI) Uptake by Synthetic Calcium Silicate Hydrates. Mater. Res. Soc. Symp. Proc. 2008, 1107, 467. [Google Scholar] [CrossRef]
- Kaja, A.M.; Delsing, A.; van der Laan, S.R.; Brouwers, H.J.H.; Yu, Q. Effects of carbonation on the retention of heavy metals in chemically activated BOF slag pastes. Cem. Concr. Res. 2021, 148, 106534. [Google Scholar] [CrossRef]
- Ke, X.; Bernal, S.A.; Provis, J.L.; Lothenbach, B. Thermodynamic modelling of phase evolution in alkali-activated slag cements exposed to carbon dioxide. Cem. Concr. Res. 2020, 136, 106158. [Google Scholar] [CrossRef]
- Steiner, S.; Lothenbach, B.; Proske, T.; Borgschulte, A.; Winnefeld, F. Effect of relative humidity on the carbonation rate of portlandite, calcium silicate hydrates and ettringite. Cem. Concr. Res. 2020, 135, 106116. [Google Scholar] [CrossRef]
- Cirkel GmbH & Co. KG. Product Information: Circosil®; Cirkel GmbH & Co. KG: Emsdetten, Germany, 2019. [Google Scholar]
- Baur, S.; Brix, K.; Feuerstein, A.; Janka, O.; Kautenburger, R. Retention of waste cocktail elements onto characterised calcium silicate hydrate (C–S–H) phases: A kinetic study under highly saline and hyperalkaline conditions. Appl. Geochem. 2022, 143, 105319. [Google Scholar] [CrossRef]
- Schade, T. Analyse und Modellierung der Einflussfaktoren Schnell Erstarrender Hochfester Anorganischer Verbundmörtel; Kassel University Press: Kassel, Germany, 2021. [Google Scholar]
- Richardson, I. Model structures for C-(A)-S-H(I). Acta Crystallogr. B 2014, 70, 903–923. [Google Scholar] [CrossRef] [PubMed]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Jackson, M.D.; Chae, S.R.; Mulcahy, S.R.; Meral, C.; Taylor, R.; Li, P.; Emwas, A.-H.; Moon, J.; Yoon, S.; Vola, G.; et al. Unlocking the secrets of Al-tobermorite in Roman seawater concrete. Am. Miner. 2013, 98, 1669–1687. [Google Scholar] [CrossRef]
- Jackson, M.D.; Mulcahy, S.R.; Chen, H.; Li, Y.; Li, Q.; Cappelletti, P.; Wenk, H.-R. Phillipsite and Al-tobermorite mineral cements produced through low-temperature water-rock reactions in Roman marine concrete. Am. Miner. 2017, 102, 1435–1450. [Google Scholar] [CrossRef]
- Collins, E.D.; DelCul, G.D.; Spencer, B.B.; Brunson, R.R.; Johnson, J.A.; Terekhov, D.S.; Emmanuel, N.V. Process Development Studies for Zirconium Recovery/Recycle from used Nuclear Fuel Cladding. Procedia Chem. 2012, 7, 72–76. [Google Scholar] [CrossRef]
- Motta, A.T.; Couet, A.; Comstock, R.J. Corrosion of Zirconium Alloys Used for Nuclear Fuel Cladding. Annu. Revi. Mater. Res. 2015, 45, 311–343. [Google Scholar] [CrossRef]
- Anderson, W.K. Broad Aspects of Absorber Materials Selection for Reactor Control. Nucl. Sci. Eng. 1958, 4, 357–372. [Google Scholar] [CrossRef]
- Johnston, H.F.; Russell, J.L.; Silvernail, W.L. Relative Control Rod Worths of Some Rare Earth Oxides. Nucl. Sci. Eng. 1959, 6, 93–96. [Google Scholar] [CrossRef]
- Eisenbud, M.; Krauskopf, K.; Franca, E.P.; Lei, W.; Ballad, R.; Linsalata, P.; Fujimori, K. Natural analogues for the transuranic actinide elements: An investigation in Minas Gerais, Brazil. Environ. Geol. Water Sci. 1984, 6, 1–9. [Google Scholar] [CrossRef]
- Krauskopf, K.B. Thorium and rare-earth metals as analogs for actinide elements. Chem. Geol. 1986, 55, 323–335. [Google Scholar] [CrossRef]
- Tits, J.; Stumpf, T.; Rabung, T.; Wieland, E.; Fanghänel, T. Uptake of Cm(III) and Eu(III) by Calcium Silicate Hydrates: A Solution Chemistry and Time-Resolved Laser Fluorescence Spectroscopy Study. Environ. Sci. Technol. 2003, 37, 3568–3573. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Kunimaru, T.; Metcalfe, R.; Martin, A.J. The hydrogeochemistry of argillaceous rock formations at the Horonobe URL site, Japan. Phys. Chem. Earth Parts A/B/C 2007, 32, 170–180. [Google Scholar] [CrossRef]
- Nguyen, X.P.; Cui, Y.J.; Tang, A.M.; Deng, Y.F.; Li, X.L.; Wouters, L. Effects of pore water chemical composition on the hydro-mechanical behavior of natural stiff clays. Eng. Geol. 2013, 166, 52–64. [Google Scholar] [CrossRef]
- Van Loon, L.R.; Baeyens, B.; Bradbury, M.H. Diffusion and retention of sodium and strontium in Opalinus clay: Comparison of sorption data from diffusion and batch sorption measurements, and geochemical calculations. Appl. Geochem. 2005, 20, 2351–2363. [Google Scholar] [CrossRef]
- Brix, K.; Baur, S.; Haben, A.; Kautenburger, R. Building the bridge between U(VI) and Ca-bentonite—Influence of concentration, ionic strength, pH, clay composition and competing ions. Chemosphere 2021, 285, 131445. [Google Scholar] [CrossRef]
- Brix, K.; Hein, C.; Haben, A.; Kautenburger, R. Adsorption of caesium on raw Ca-bentonite in high saline solutions: Influence of concentration, mineral composition, other radionuclides and modelling. Appl. Clay Sci. 2019, 182, 105275. [Google Scholar] [CrossRef]
- Kautenburger, R.; Brix, K.; Hein, C. Insights into the retention behaviour of europium(III) and uranium(VI) onto Opalinus Clay influenced by pore water composition, temperature, pH and organic compounds. Appl. Geochem. 2019, 109, 104404. [Google Scholar] [CrossRef]
- Liu, C.; Zachara, J.M.; Smith, S.C. A cation exchange model to describe Cs+ sorption at high ionic strength in subsurface sediments at Hanford site, USA. J. Contam. Hydrol. 2004, 68, 217–238. [Google Scholar] [CrossRef]
- Yan, Y.; Bernard, E.; Miron, G.D.; Rentsch, D.; Ma, B.; Scrivener, K.; Lothenbach, B. Kinetics of Al uptake in synthetic calcium silicate hydrate (C-S-H). Cem. Concr. Res. 2023, 172, 107250. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, B.; Miron, G.D.; Kulik, D.A.; Scrivener, K.; Lothenbach, B. Al uptake in calcium silicate hydrate and the effect of alkali hydroxide. Cem. Concr. Res. 2022, 162, 106957. [Google Scholar] [CrossRef]
- Iwaida, T.; Nagasaki, S.; Tanaka, S.; Yaita, T.; Tachimori, S. Structure alteration of C-S-H (calcium silicate hydrated phases) caused by sorption of caesium. Radiochim. Acta 2002, 90, 677–681. [Google Scholar] [CrossRef]
- Missana, T.; García-Gutiérrez, M.; Mingarro, M.; Alonso, U. Comparison between cesium and sodium retention on calcium silicate hydrate (CSH) phases. Appl. Geochem. 2018, 98, 36–44. [Google Scholar] [CrossRef]
- Atkinson, A.; Nickerson, A.K. Diffusion and Sorption of Cesium, Strontium, and Iodine in Water-Saturated Cement. Nucl. Technol. 1988, 81, 100–113. [Google Scholar] [CrossRef]
- Duque-Redondo, E.; Kazuo, Y.; López-Arbeloa, I.; Manzano, H. Cs-137 immobilization in C-S-H gel nanopores. Phys. Chem. Chem. Phys. 2018, 20, 9289–9297. [Google Scholar] [CrossRef]
- Heath, T.G.; Ilett, D.J.; Tweed, C.J. Thermodynamic Modelling of the Sorption of Radioelements onto Cementitious Materials. Mater. Res. Soc. Symp. Proc. OPL 1995, 412, 443. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, P.; Hou, D. The mechanism of cesium ions immobilization in the nanometer channel of calcium silicate hydrate: A molecular dynamics study. Phys. Chem. Chem. Phys. 2017, 19, 27974–27986. [Google Scholar] [CrossRef]
- Missana, T.; García-Gutiérrez, M.; Mingarro, M.; Alonso, U. Analysis of barium retention mechanisms on calcium silicate hydrate phases. Cem. Concr. Res. 2017, 93, 8–16. [Google Scholar] [CrossRef]
- Viallis, H.; Faucon, P.; Petit, J.C.; Nonat, A. Interaction between Salts (NaCl, CsCl) and Calcium Silicate Hydrates (C–S–H). J. Phys. Chem. B 1999, 103, 5212–5219. [Google Scholar] [CrossRef]
- Burešová, M.; Kittnerová, J.; Drtinová, B. Comparative study of Eu and U sorption on cementitious materials in the presence of organic substances. J. Radioanal. Nucl. Chem. 2023, 332, 1499–1504. [Google Scholar] [CrossRef]
- Dettmann, S.; Huittinen, N.M.; Jahn, N.; Kretzschmar, J.; Kumke, M.U.; Kutyma, T.; Lohmann, J.; Reich, T.; Schmeide, K.; Shams Aldin Azzam, S.; et al. Influence of gluconate on the retention of Eu(III), Am(III), Th(IV), Pu(IV), and U(VI) by C-S-H (C/S = 0.8). Front. Nucl. Eng. 2023, 2, 1124856. [Google Scholar] [CrossRef]
- Mandaliev, P.; Stumpf, T.; Tits, J.; Dähn, R.; Walther, C.; Wieland, E. Uptake of Eu(III) by 11Å tobermorite and xonotlite: A TRLFS and EXAFS study. Geochim. Cosmochim. Acta 2011, 75, 2017–2029. [Google Scholar] [CrossRef]
- Pointeau, I.; Piriou, B.; Fedoroff, M.; Barthes, M.G.; Marmier, N.; Fromage, F. Sorption Mechanisms of Eu(3+) on CSH Phases of Hydrated Cements. J. Colloid Interface Sci. 2001, 236, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.L.; Pointeau, I.; Coreau, N.; Reiller, P. Mechanism of Europium Retention by Calcium Silicate Hydrates: An EXAFS Study. Environ. Sci. Technol. 2004, 38, 4423–4431. [Google Scholar] [CrossRef]
- Macé, N.; Page, J.; Reiller, P.E. Uranium(VI) Sorption onto Hardened Cement Paste under High Saline and Alkaline Conditions. Minerals 2023, 13, 325. [Google Scholar] [CrossRef]
- Philipp, T.; Shams Aldin Azzam, S.; Rossberg, A.; Huittinen, N.; Schmeide, K.; Stumpf, T. U(VI) sorption on Ca-bentonite at (hyper)alkaline conditions—Spectroscopic investigations of retention mechanisms. Sci. Total Environ. 2019, 676, 469–481. [Google Scholar] [CrossRef]
- Pointeau, I.; Landesman, C.; Giffaut, E.; Reiller, P. Reproducibility of the uptake of U(VI) onto degraded cement pastes and calcium silicate hydrate phases. Radiochim. Acta 2004, 92, 645–650. [Google Scholar] [CrossRef]
- Harfouche, M.; Wieland, E.; Dähn, R.; Fujita, T.; Tits, J.; Kunz, D.; Tsukamoto, M. EXAFS study of U(VI) uptake by calcium silicate hydrates. J. Colloid Interface Sci. 2006, 303, 195–204. [Google Scholar] [CrossRef]
- Androniuk, I.; Landesman, C.; Henocq, P.; Kalinichev, A.G. Adsorption of gluconate and uranyl on C-S-H phases: Combination of wet chemistry experiments and molecular dynamics simulations for the binary systems. Phys. Chem. Earth Parts A/B/C 2017, 99, 194–203. [Google Scholar] [CrossRef]
- Baqer, Y.; Thornton, S.; Stewart, D.I.; Norris, S.; Chen, X. Analysis of Uranium Sorption in a Laboratory Column Experiment Using a Reactive Transport and Surface Complexation Model. Transp. Porous Media 2023, 149, 423–452. [Google Scholar] [CrossRef]
- Androniuk, I.; Kalinichev, A.G. Molecular dynamics simulation of the interaction of uranium (VI) with the C–S–H phase of cement in the presence of gluconate. Appl. Geochem. 2020, 113, 104496. [Google Scholar] [CrossRef]
- Philipp, T.; Huittinen, N.; Shams Aldin Azzam, S.; Stohr, R.; Stietz, J.; Reich, T.; Schmeide, K. Effect of Ca(II) on U(VI) and Np(VI) retention on Ca-bentonite and clay minerals at hyperalkaline conditions—New insights from batch sorption experiments and luminescence spectroscopy. Sci. Total Environ. 2022, 842, 156837. [Google Scholar] [CrossRef] [PubMed]
- Tits, J.; Walther, C.; Stumpf, T.; Macé, N.; Wieland, E. A luminescence line-narrowing spectroscopic study of the uranium(vi) interaction with cementitious materials and titanium dioxide. Dalton Trans. 2015, 44, 966–976. [Google Scholar] [CrossRef]
- Altmaier, M.; Metz, V.; Neck, V.; Müller, R.; Fanghänel, T. Solid-liquid equilibria of Mg(OH)2(cr) and Mg2(OH)3Cl·4H2O(cr) in the system Mg-Na-H-OH-Cl-H2O at 25 °C. Geochim. Cosmochim. Acta 2003, 67, 3595–3601. [Google Scholar] [CrossRef]
- Hein, C.; Sander, J.M.; Kautenburger, R. New approach of a transient ICP-MS measurement method for samples with high salinity. Talanta 2017, 164, 477–482. [Google Scholar] [CrossRef]
- Ball, J.W.; Nordstrom, D.K. User’s Manual for WATEQ4F, with Revised Thermodynamic Data Base and Text Cases for Calculating Speciation of Major, Trace, and Redox Elements in Natural Waters; US Geological Survey: Reston, VA, USA, 1991; pp. 91–183.
- Diakonov, I.I.; Ragnarsdottir, K.V.; Tagirov, B.R. Standard thermodynamic properties and heat capacity equations of rare earth hydroxides:: II. Ce(III)-, Pr-, Sm-, Eu(III)-, Gd-, Tb-, Dy-, Ho-, Er-, Tm-, Yb-, and Y-hydroxides. Comparison of thermochemical and solubility data. Chem. Geol. 1998, 151, 327–347. [Google Scholar] [CrossRef]
- Spahiu, K.; Bruno, J. A Selected Thermodynamic Database for REE to be Used in HLNW Performance Assessment Exercises; 0284-3757; Fuel and Waste Management Co.: Stockholm, Sweden, 1995; p. 90. [Google Scholar]
- Altmaier, M.; Yalçıntaş, E.; Gaona, X.; Neck, V.; Müller, R.; Schlieker, M.; Fanghänel, T. Solubility of U(VI) in chloride solutions. I. The stable oxides/hydroxides in NaCl systems, solubility products, hydrolysis constants and SIT coefficients. J. Chem. Thermodyn. 2017, 114, 2–13. [Google Scholar] [CrossRef]
- Plancque, G.; Moulin, V.; Toulhoat, P.; Moulin, C. Europium speciation by time-resolved laser-induced fluorescence. Anal. Chim. Acta 2003, 478, 11–22. [Google Scholar] [CrossRef]
- Pointeau, I.; Marmier, N.; Fromage, F.; Fedoroff, M.; Giffaut, E. Cesium and Lead Uptake by CSH Phases of Hydrated Cement. Mater. Res. Soc. Symp. Proc. 2000, 663, 97. [Google Scholar] [CrossRef]
- Arayro, J.; Dufresne, A.; Zhou, T.; Ioannidou, K.; Ulm, J.-F.; Pellenq, R.; Béland, L.K. Thermodynamics, kinetics, and mechanics of cesium sorption in cement paste: A multiscale assessment. Phys. Rev. Mater. 2018, 2, 053608. [Google Scholar] [CrossRef]
- Duque-Redondo, E.; Yamada, K.; Manzano, H. Cs retention and diffusion in C-S-H at different Ca/Si ratio. Cem. Concr. Res. 2021, 140, 106294. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, J.G. Carbonation-induced weathering effect on cesium retention of cement paste. J. Nucl. Mater. 2018, 505, 159–164. [Google Scholar] [CrossRef]
- Komarneri, S.; Roy, D.M.; Roy, R. Al-substituted tobermorite: Shows cation exchange. Cem. Concr. Res. 1982, 12, 773–780. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nishimoto, S.; Kameshima, Y.; Miyake, M. Hydrothermal preparation of tobermorite from blast furnace slag for Cs+ and Sr2+ sorption. J. Hazard. Mater. 2014, 266, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Bach, T.T.H.; Chabas, E.; Pochard, I.; Cau Dit Coumes, C.; Haas, J.; Frizon, F.; Nonat, A. Retention of alkali ions by hydrated low-pH cements: Mechanism and Na+/K+ selectivity. Cem. Concr. Res. 2013, 51, 14–21. [Google Scholar] [CrossRef]
- Cornell, R.M. Adsorption of cesium on minerals: A review. J. Radioanal. Nucl. Chem. 1993, 171, 483–500. [Google Scholar] [CrossRef]
- Vejsada, J.; Hradil, D.; Řanda, Z.; Jelínek, E.; Štulík, K. Adsorption of cesium on Czech smectite-rich clays—A comparative study. Appl. Clay Sci. 2005, 30, 53–66. [Google Scholar] [CrossRef]
- Bu, J.; Gonzalez Teresa, R.; Brown, K.G.; Sanchez, F. Adsorption mechanisms of cesium at calcium-silicate-hydrate surfaces using molecular dynamics simulations. J. Nucl. Mater. 2019, 515, 35–51. [Google Scholar] [CrossRef]
- Li, K.; Pang, X. Sorption of radionuclides by cement-based barrier materials. Cem. Concr. Res. 2014, 65, 52–57. [Google Scholar] [CrossRef]
- Ali, O.I.M.; Osman, H.H.; Sayed, S.A.; Shalabi, M.E.H. The removal of some rare earth elements from their aqueous solutions on by-pass cement dust (BCD). J. Hazard. Mater. 2011, 195, 62–67. [Google Scholar] [CrossRef]
- Häußler, V.; Amayri, S.; Beck, A.; Platte, T.; Stern, T.A.; Vitova, T.; Reich, T. Uptake of actinides by calcium silicate hydrate (C-S-H) phases. Appl. Geochem. 2018, 98, 426–434. [Google Scholar] [CrossRef]
- Pointeau, I.; Coreau, N.; Reiller, P.E. Uptake of anionic radionuclides onto degraded cement pastes and competing effect of organic ligands. Radiochim. Acta 2008, 96, 367–374. [Google Scholar] [CrossRef]
- Zhang, N.; Carrez, P.; Shahsavari, R. Screw-Dislocation-Induced Strengthening–Toughening Mechanisms in Complex Layered Materials: The Case Study of Tobermorite. ACS Appl. Mater. Interfaces 2017, 9, 1496–1506. [Google Scholar] [CrossRef]
- Boulard, L.; Kautenburger, R. Short-term elemental release from Portland cement concrete in hypersaline leaching conditions. Adv. Cem. Res. 2020, 32, 148–157. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equilibration Time [d] | Rd(Cs(I)) in 0.1 M NaCl [103 L·kg−1] | Rd(Cs(I)) in DGS [103 L·kg−1] |
|---|---|---|
| 0.04 | 0.123 ± 0.004 | 0.023 ± 0.004 |
| 1 | 0.46 ± 0.02 | 0.038 ± 0.002 |
| 3 | 0.65 ± 0.05 | 0.041 ± 0.004 |
| 7 | 0.77 ± 0.04 | 0.046 ± 0.007 |
| 14 | 0.82 ± 0.02 | 0.043 ± 0.009 |
| 28 | 0.89 ± 0.03 | 0.041 ± 0.006 |
| 56 | 0.86 ± 0.05 | 0.038 ± 0.006 |
| 112 | 0.74 ± 0.03 | 0.046 ± 0.006 |
| 168 | 0.88 ± 0.03 | 0.034 ± 0.004 |
| 217 | 0.82 ± 0.04 | 0.024 ± 0.006 |
| Equilibration Time [d] | Rd(Sm(III)) in 0.1 M NaCl [103 L·kg−1] | Rd(Eu(III)) in 0.1 M NaCl [103 L·kg−1] | Rd(Sm(III)) in DGS [103 L·kg−1] | Rd(Eu(III)) in DGS [103 L·kg−1] |
|---|---|---|---|---|
| 0.04 | 0.14 ± 0.04 | 0.14 ± 0.05 | 0.18 ± 0.07 | 0.16 ± 0.06 |
| 1 | 0.7 ± 0.2 | 0.6 ± 0.2 | 0.9 ± 0.6 | 0.9 ± 0.6 |
| 3 | 2 ± 0.7 | 1.8 ± 0.7 | 0.60 ± 0.06 | 0.55 ± 0.06 |
| 7 | 9 ± 4 | 8.5 ± 3.4 | 1.9 ± 0.4 | 1.8 ± 0.4 |
| 14 | 37 ± 9 | 35 ± 8 | 2.3 ± 1.2 | 2.2 ± 1.1 |
| 28 | 41 ± 16 | 38 ± 13 | 4 ± 3 | 4 ± 3 |
| 56 | 11 ± 12 | 11 ± 11 | 3.4 ± 1.3 | 4 ± 2 |
| 112 | 1.6 ± 0.4 | 1.4 ± 0.4 | 1.6 ± 0.2 | 1.6 ± 0.3 |
| 168 | 79 ± 60 | 82 ± 66 | >100 * | >50 * |
| 217 | 290 ± 380 | 770 ± 1200 | 2 ± 1 | 2 ± 1 |
| Equilibration Time [d] | Rd(U(VI)) in 0.1 M NaCl [103 L·kg−1] | Rd(U(VI)) in DGS [103 L·kg−1] |
|---|---|---|
| 0.04 | 0.34 ± 0.06 | 0.24 ± 0.09 |
| 1 | 1.8 ± 0.7 | 1.9 ± 1.4 |
| 3 | 5 ± 2.4 | 1.2 ± 0.1 |
| 7 | 19 ± 10 | 4.2 ± 1.1 |
| 14 | 71 ± 17 | 5.3 ± 3.2 |
| 28 | 71 ± 20 | 12 ± 12 |
| 56 | 19 ± 20 | 7 ± 4 |
| 112 | 2.8 ± 0.5 | 3.2 ± 0.7 |
| 168 | 110 ± 76 | >100 * |
| 217 | 210 ± 200 | 4 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brix, K.; Haben, A.; Kautenburger, R. Time-Dependent Retention of a Mixture of Cs(I), Sm(III), Eu(III) and U(VI) as Waste Cocktail by Calcium Silicate Hydrate (C-S-H) Phases. Minerals 2023, 13, 1469. https://doi.org/10.3390/min13121469
Brix K, Haben A, Kautenburger R. Time-Dependent Retention of a Mixture of Cs(I), Sm(III), Eu(III) and U(VI) as Waste Cocktail by Calcium Silicate Hydrate (C-S-H) Phases. Minerals. 2023; 13(12):1469. https://doi.org/10.3390/min13121469
Chicago/Turabian StyleBrix, Kristina, Aaron Haben, and Ralf Kautenburger. 2023. "Time-Dependent Retention of a Mixture of Cs(I), Sm(III), Eu(III) and U(VI) as Waste Cocktail by Calcium Silicate Hydrate (C-S-H) Phases" Minerals 13, no. 12: 1469. https://doi.org/10.3390/min13121469
APA StyleBrix, K., Haben, A., & Kautenburger, R. (2023). Time-Dependent Retention of a Mixture of Cs(I), Sm(III), Eu(III) and U(VI) as Waste Cocktail by Calcium Silicate Hydrate (C-S-H) Phases. Minerals, 13(12), 1469. https://doi.org/10.3390/min13121469