
Mineral Physicochemistry Underlying Feature-Based Extraction of Mineral Abundance and Composition from Shortwave, Mid and Thermal Infrared Reflectance Spectra

 ,
,  ,
,  ,
,
Abstract
1. Introduction

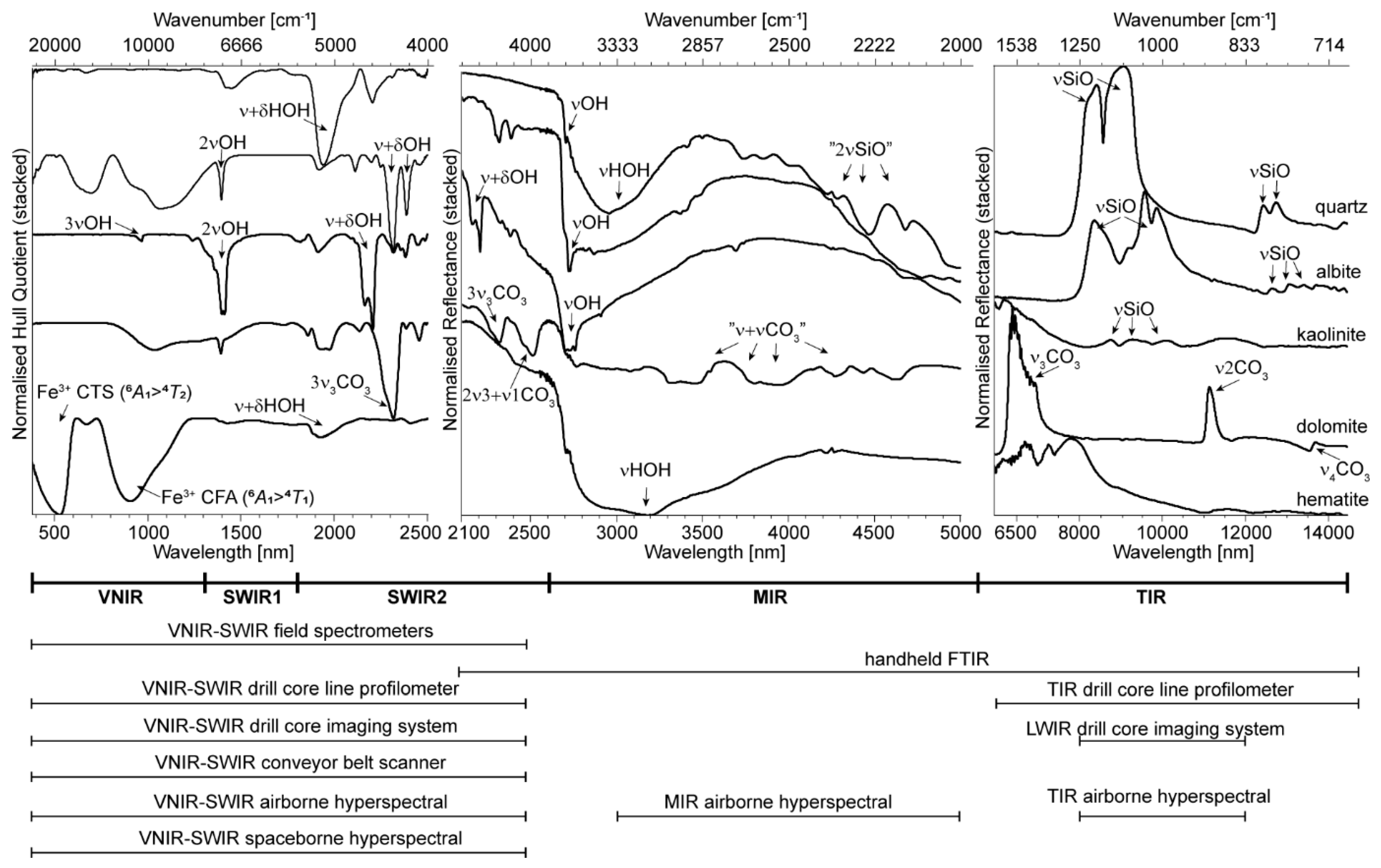
2. Mineral Diagnostic Features in Reflectance Spectra
2.1. SWIR 1 Functional Groups (1300 to 1850 nm)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral Group | Mineral Species (Examples) | Assignment of Absorption | Lower Limit (nm) | Upper Limit (nm) | Literature | Intensity Script | Wavelength Script |
|---|---|---|---|---|---|---|---|
| sheet silicate | kaolinite | 2νMnOH | 1392 to 1415 | [41] | 1400D | 1400W | |
| sheet silicate | muscovite | 1406 to 1415 | [31] | ||||
| sheet silicate | lepidolite | 1387 to 1425 | [31] | ||||
| sheet silicate | talc | 1392 to 1412 | [28] | ||||
| sheet silicate | smectites | 1394 to 1445 | [28,42] | ||||
| sheet silicate | palygorskite | 1386 to 1443 | [43] | ||||
| sheet silicate | chlorite | 1391 to 1415 | [44] | ||||
| double chain silicate | calcic amphibole | 1392 to 1413 | [45] | ||||
| sulphate | jarosite | 2νMnOH, ν + 2δH2O | 1471 | [35] | 1480D | 1480W | |
| sulphate | alunite | 2νMnOH, ν + 2δH2O | 1473 (K-rich) | 1491 (Na-rich) | [35] | ||
| sheet silicate | prehnite | 2νM2OH | 1476 (Fe-rich) | 1478 (Mg-rich) | [32] | ||
| sorosilicate | epidote, clinozoisite, pistasite | 2νM2OH | 1545 (Fe-rich) | 1563 (Al-rich) | [30,32,33] | 1550D | 1550W |
| NH4 | alunite, montmorillonite, buddingtonite, white mica | ν1 + ν3NH4 | 1520 to 1560 | [34] | |||
| sulphate | gypsum | ν + δOH/H2O and rotational fundamentals or δS-O overtones | 1751 | [46] | 1760D | - | |
| sulphate | alunite | ν + 2δMnOH | 1762 (Na-rich) | 1764 (K-rich) | [35,37] | 1760W | |
| sulphate | jarosite | ν + 2δMnOH | 1849 | [35] | 1850D | - | |
2.2. SWIR 2 Functional Groups (1850 to 2600 nm)
- di-octahedral 1:1 phyllosilicate (“7 Å phase”): e.g., kaolinite;
- tri-octahedral 2:1 phyllosilicate (“9 Å phase”): e.g., talc;
- di-octahedral 2:1 phyllosilicate (“10 Å phase”): e.g., muscovite, phengite, celadonite;
- tri-octahedral 2:1 phyllosilicate (“10 Å phase”): e.g., biotite;
- tri-octahedral 2:1:1 phyllosilicate (“14 Å phase”): e.g., chlorite.
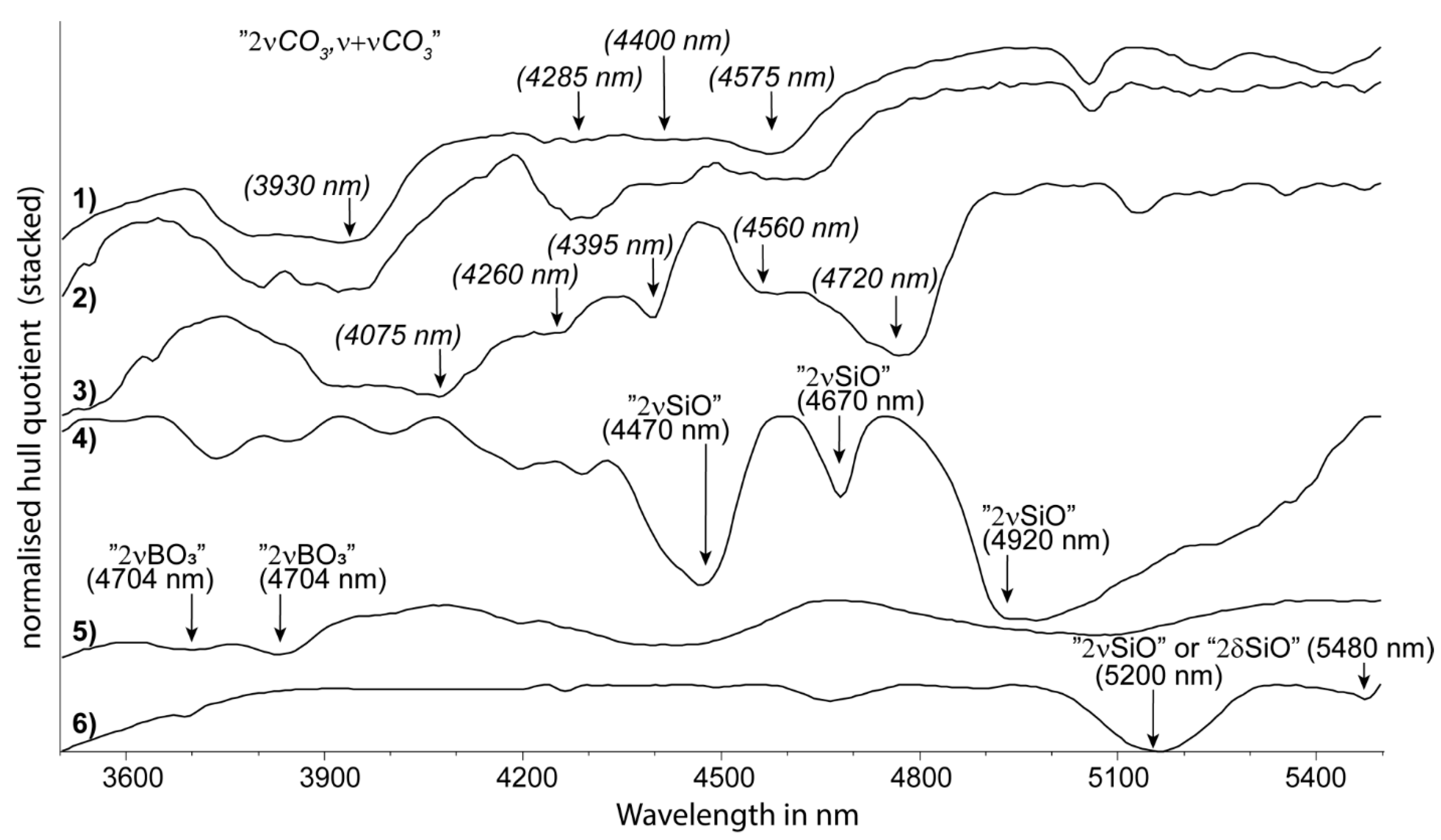
2.3. MIR Functional Groups (2600 to 5500 nm)
2.4. TIR Functional Groups (5500 to 15,000 nm)

3. Feature Extraction
3.1. Single Feature Extraction Scripts—SWIR and MIR
3.2. Binary Discrimination Diagrams—SWIR
3.3. Single Feature Extraction Scripts and Binary Discrimination Diagrams—TIR
3.4. Impact of Grain Size Variations on SWIR, MIR and TIR Scripts
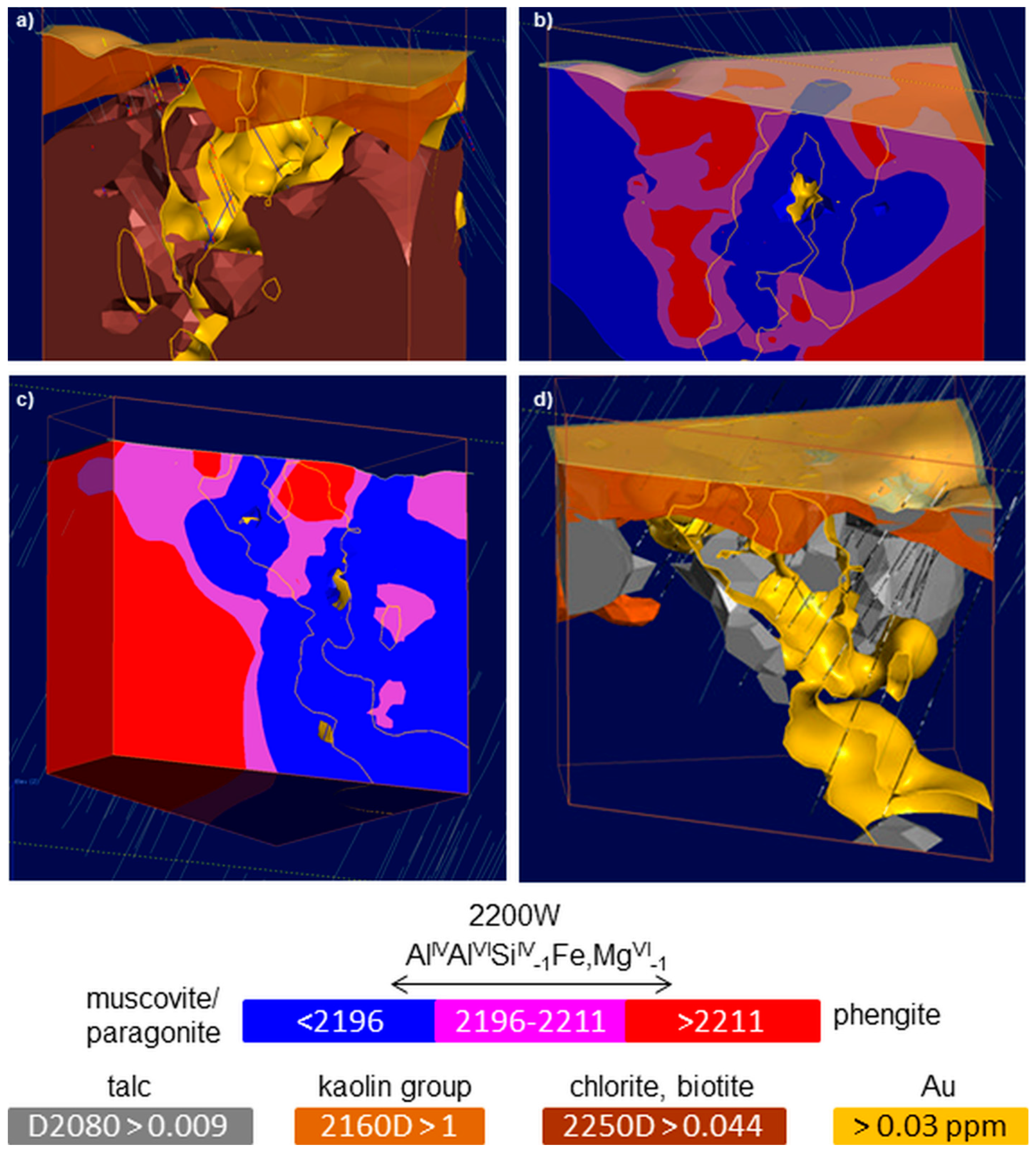
3.5. 3D Mineral Mapping
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clark, R.N.; King, T.V.V.; Klejwa, M.; Swayze, G.A.; Vergo, N. High Spectral Resolution Reflectance Spectroscopy of Minerals. J. Geophys. Res. 1990, 95, 12653. [Google Scholar] [CrossRef]
- Murray, H.H. Correlation of Paper-Coating Quality with Degree of Crystal Perfection of Kaolinite. Clays Clay Miner. 1955, 4, 31–40. [Google Scholar] [CrossRef]
- Vedder, W.; McDonald, R.S. Vibrations of the OH Ions in Muscovite. J. Chem. Phys. 1963, 38, 1583–1590. [Google Scholar] [CrossRef]
- Cudahy, T.; Jones, M.; Thomas, M.; Laukamp, C.; Caccetta, M.; Hewson, R.; Rodger, A.; Verrall, M. Next Generation Mineral Mapping: Queensland Airborne HyMap and Satellite ASTER Surveys 2006–2008; CSIRO Report P2007/364; CSIRO: Canberra, Australia, 2008; p. 161. [Google Scholar]
- Laukamp, C.; Cudahy, T.; Caccetta, M.; Chia, J.; Gessner, K.; Haest, M.; Liu, Y.C.; Rodger, A. The Uses, Abuses and Opportunities for Hyperspectral Technologies and Derived Geoscience Information; AIG Bulletin: Crows Nest, NSW, Ausatralia, 2010; Volume 51, pp. 73–76. [Google Scholar]
- Laukamp, C.; LeGras, M.; Lau, I.C. Hyperspectral Proximal Sensing Instruments and Their Applications for Exploration through Cover. In Portable Spectroscopy and Spectrometry 2: Applications; Crocombe, R., Ed.; Wiley: Hoboken, NJ, USA, 2021. [Google Scholar]
- Lyon, R.J.P. Stanford Research Institute; United States; National Aeronautics and Space Administration. In Evaluation of Infrared Spectrophotometry for Compositional Analysis of Lunar and Planetary Soils; National Aeronautics and Space Administration: Washington, DC, USA, 1963. [Google Scholar]
- The Layer Silicates. The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: London, UK, 1974; pp. 331–363. [Google Scholar] [CrossRef]
- Hunt, G.R. Spectral Signatures in Particulate Minerals in the Visible and near Infrared. GEOPHYSICS 1977, 42, 501–513. [Google Scholar] [CrossRef]
- Cudahy, T.; Caccetta, M.; Thomas, M.; Hewson, R.; Abrams, M.; Kato, M.; Kashimura, O.; Ninomiya, Y.; Yamaguchi, Y.; Collings, S.; et al. Satellite-Derived Mineral Mapping and Monitoring of Weathering, Deposition and Erosion. Sci. Rep. 2016, 6, 23702. [Google Scholar] [CrossRef] [PubMed]
- Laukamp, C.; Cudahy, T.; Thomas, M.; Jones, M.; Cleverley, J.S.; Oliver, N.H.S. Hydrothermal Mineral Alteration Patterns in the Mount Isa Inlier Revealed by Airborne Hyperspectral Data. Aust. J. Earth Sci. 2011, 58, 917–936. [Google Scholar] [CrossRef]
- Van Ruitenbeek, F.J.A.; Debba, P.; van der Meer, F.D.; Cudahy, T.; van der Meijde, M.; Hale, M. Mapping White Micas and Their Absorption Wavelengths Using Hyperspectral Band Ratios. Remote Sens. Environ. 2006, 102, 211–222. [Google Scholar] [CrossRef]
- Van der Meer, F.D.; van der Werff, H.M.A.; van Ruitenbeek, F.J.A.; Hecker, C.A.; Bakker, W.H.; Noomen, M.F.; van der Meijde, M.; Carranza, E.J.M.; de Smeth, J.B.; Woldai, T. Multi- and Hyperspectral Geologic Remote Sensing: A Review. Int. J. Appl. Earth Obs. Geoinf. 2012, 14, 112–128. [Google Scholar] [CrossRef]
- Burns, R.G. Mineralogical Applications of Crystal Field Theory, 2nd ed.; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar] [CrossRef]
- Chukanov, N.V. Infrared Spectra of Mineral Species: Extended Library, 1st ed.; Springer Geochemistry/Mineralogy; Springer Netherlands: Imprint; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Povarennykh, A.S. The Use of Infrared Spectra for the Determination of Minerals. Am. Miner. 1978, 63, 956–959. [Google Scholar]
- McLeod, R.L.; Gabell, A.R.; Green, A.A.; Gardavsky, V. Chlorite Infrared Spectral Data as Proximity Indicators of Volcanogenic Massive Sulphide Mineralisation; Pascal and Francis: Gold Coast, Australia, 1987. [Google Scholar]
- Lypaczewski, P.; Rivard, B. Estimating the Mg# and AlVI Content of Biotite and Chlorite from Shortwave Infrared Reflectance Spectroscopy: Predictive Equations and Recommendations for Their Use. Int. J. Appl. Earth Obs. Geoinf. 2018, 68, 116–126. [Google Scholar] [CrossRef]
- Gaffey, S.J. Spectral Reflectance of Carbonate Minerals in the Visible and near Infrared (0.35–2.55 Microns); Calcite, Aragonite, and Dolomite. Am. Miner. 1986, 71, 151–162. [Google Scholar]
- Schodlok, M.C.; Whitbourn, L.; Huntington, J.; Mason, P.; Green, A.; Berman, M.; Coward, D.; Connor, P.; Wright, W.; Jolivet, M.; et al. HyLogger-3, a Visible to Shortwave and Thermal Infrared Reflectance Spectrometer System for Drill Core Logging: Functional Description. Aust. J. Earth Sci. 2016, 63, 929–940. [Google Scholar] [CrossRef]
- Loizzo, R.; Guarini, R.; Longo, F.; Scopa, T.; Formaro, R.; Facchinetti, C.; Varacalli, G. Prisma: The Italian Hyperspectral Mission. In Proceedings of the IGARSS 2018-2018 IEEE International Geoscience and Remote Sensing Symposium, Valencia, Spain, 22–27 July 2018; pp. 175–178. [Google Scholar]
- Kaufmann, H.; Segl, K.; Guanter, L.; Hofer, S.; Foerster, K.P.; Stuffler, T.; Mueller, A.; Richter, R.; Bach, H.; Hostert, P.; et al. Environmental Mapping and Analysis Program (EnMAP)—Recent Advances and Status. In Proceedings of the IGARSS 2008-2008 IEEE International Geoscience and Remote Sensing Symposium, Boston, MA, USA, 7–11 July 2008; pp. 109–112. [Google Scholar]
- Moenke, H.H.W. Vibrational Spectra and the Crystal-Chemical Classification of Minerals. In The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: Londan, UK, 1974; pp. 111–118. [Google Scholar] [CrossRef]
- Agar, R.A. REMOTE SENSING: Geoscan Airborne Multi-Spectral Scanners as Exploration Tools for Western Australian Diamond and Gold Deposits. ASEG Ext. Abstr. 1994, 1994, 433–448. [Google Scholar] [CrossRef]
- Vincent, R.K.; Hunt, G.R. Infrared Reflectance from Mat Surfaces. Appl. Opt. 1968, 7, 53. [Google Scholar] [CrossRef]
- Salisbury, J.W.; Wald, A. The Role of Volume Scattering in Reducing Spectral Contrast of Reststrahlen Bands in Spectra of Powdered Minerals. Icarus 1992, 96, 121–128. [Google Scholar] [CrossRef]
- Kustov, L. Spectra of Hydroxyl Groups in Zeolites in the Near-Infrared Region. J. Catal. 1981, 72, 149–159. [Google Scholar] [CrossRef]
- Petit, S.; Martin, F.; Wiewiora, A.; De Parseval, P.; Decarreau, A. Crystal-Chemistry of Talc: A near Infrared (NIR) Spectroscopy Study. Am. Miner. 2004, 89, 319–326. [Google Scholar] [CrossRef]
- Strens, R.G.J. The Common Chain, Ribbon, and Ring Silicates. In The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: Londan, UK, 1974; pp. 305–330. [Google Scholar] [CrossRef]
- Langer, K.; Raith, M. Infrared Spectra of Al-Fe(III)-Epidotes and Zoisites, Ca2(Al1-p Fe3+p)Al2O(OH)[Si2O7][SiO4]. Am. Miner. 1974, 59, 1249–1258. [Google Scholar]
- LeGras, M.; Laukamp, C.; Lau, I.; Mason, P. NVCL Spectral Reference Library—Phyllosilicates Part 2: Micas; CSIRO Report EP183095; CSIRO: Canberra, Australia, 2018. [Google Scholar]
- White, A.J.R.; Laukamp, C.; Stokes, M.A.; Legras, M.; Pejcic, B. Vibrational Spectroscopy of Epidote, Pumpellyite and Prehnite Applied to Low-Grade Regional Metabasites. Geochem. Explor. Environ. Anal. 2017, 17, 315–333. [Google Scholar] [CrossRef]
- Roache, T.J.; Walshe, J.L.; Huntington, J.F.; Quigley, M.A.; Yang, K.; Bil, B.W.; Blake, K.L.; Hyvärinen, T. Epidote–Clinozoisite as a Hyperspectral Tool in Exploration for Archean Gold. Aust. J. Earth Sci. 2011, 58, 813–822. [Google Scholar] [CrossRef]
- Krohn, M.D.; Altaner, S.P. Near-infrared Detection of Ammonium Minerals. GEOPHYSICS 1987, 52, 924–930. [Google Scholar] [CrossRef]
- Bishop, J.L.; Murad, E. The Visible and Infrared Spectral Properties of Jarosite and Alunite. Am. Miner. 2005, 90, 1100–1107. [Google Scholar] [CrossRef]
- Stoffregen, R.E.; Cygan, G.L. An Experimental Study of Na-K Exchange between Alunite and Aqueous Sulfate Solutions. Am. Miner. 1990, 75, 209–220. [Google Scholar]
- Chang, Z.; Hedenquist, J.W.; White, N.C.; Cooke, D.R.; Roach, M.; Deyell, C.L.; Garcia, J.; Gemmell, J.B.; McKnight, S.; Cuison, A.L. Exploration Tools for Linked Porphyry and Epithermal Deposits: Example from the Mankayan Intrusion-Centered Cu-Au District, Luzon, Philippines*. Econ. Geol. 2011, 106, 1365–1398. [Google Scholar] [CrossRef]
- Turner, D.J.; Rivard, B.; Groat, L.A. Visible and Short-Wave Infrared Reflectance Spectroscopy of REE Fluorocarbonates. Am. Miner. 2014, 99, 1335–1346. [Google Scholar] [CrossRef]
- Turner, D.J.; Rivard, B.; Groat, L.A. Visible and Short-Wave Infrared Reflectance Spectroscopy of REE Phosphate Minerals. Am. Miner. 2016, 101, 2264–2278. [Google Scholar] [CrossRef]
- Turner, D.J.; Rivard, B.; Groat, L.A. Visible and Short-Wave Infrared Reflectance Spectroscopy of Selected REE-Bearing Silicate Minerals. Am. Miner. 2018, 103, 927–943. [Google Scholar] [CrossRef]
- Frost, R.L.; Johansson, U. Combination Bands in the Infrared Spectroscopy of Kaolins—A Drift Spectroscopic Study. Clays Clay Miner. 1998, 46, 466–477. [Google Scholar] [CrossRef]
- Bishop, J.; Madejová, J.; Komadel, P.; Fröschl, H. The Influence of Structural Fe, Al and Mg on the Infrared OH Bands in Spectra of Dioctahedral Smectites. Clay Miner. 2002, 37, 607–616. [Google Scholar] [CrossRef]
- Gionis, V. On the Structure of Palygorskite by Mid- and near-Infrared Spectroscopy. Am. Miner. 2006, 91, 1125–1133. [Google Scholar] [CrossRef]
- Mathian, M.; Hebert, B.; Baron, F.; Petit, S.; Lescuyer, J.-L.; Furic, R.; Beaufort, D. Identifying the Phyllosilicate Minerals of Hypogene Ore Deposits in Lateritic Saprolites Using the Near-IR Spectroscopy Second Derivative Methodology. J. Geochem. Explor. 2018, 186, 298–314. [Google Scholar] [CrossRef]
- Laukamp, C.; Termin, K.A.; Pejcic, B.; Haest, M.; Cudahy, T. Vibrational Spectroscopy of Calcic Amphiboles—Applications for Exploration and Mining. Eur. J. Miner. 2012, 24, 863–878. [Google Scholar] [CrossRef]
- Cloutis, E.; Hawthorne, F.; Mertzman, S.; Krenn, K.; Craig, M.; Marcino, D.; Methot, M.; Strong, J.; Mustard, J.; Blaney, D. Detection and Discrimination of Sulfate Minerals Using Reflectance Spectroscopy. Icarus 2006, 184, 121–157. [Google Scholar] [CrossRef]
- Felzer, B.; Hauff, P.; Goetz, A.F.H. Quantitative Reflectance Spectroscopy of Buddingtonite from the Cuprite Mining District, Nevada. J. Geophys. Res. Solid Earth 1994, 99, 2887–2895. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Hirai, S.; Redfern, S.A.T.; Salje, E.K.H. Dehydroxylation and CO2 Incorporation in Annealed Mica (Sericite): An Infrared Spectroscopic Study. Am. Miner. 2005, 90, 173–180. [Google Scholar] [CrossRef]
- Reddy, B.J.; Frost, R.L.; Martens, W.N.; Wain, D.L.; Kloprogge, J.T. Spectroscopic Characterization of Mn-Rich Tourmalines. Vib. Spectrosc. 2007, 44, 42–49. [Google Scholar] [CrossRef]
- Bishop, J.L.; Lane, M.D.; Dyar, M.D.; Brown, A.J. Reflectance and Emission Spectroscopy Study of Four Groups of Phyllosilicates: Smectites, Kaolinite-Serpentines, Chlorites and Micas. Clay Miner. 2008, 43, 35–54. [Google Scholar] [CrossRef]
- Ruan, H.D.; Frost, R.L.; Kloprogge, J.T. Application of Near-Infrared Spectroscopy to the Study of Alumina Phases. Appl. Spectrosc. 2001, 55, 190–196. [Google Scholar] [CrossRef]
- Meunier, A. Clays; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2010. [Google Scholar]
- Crowley, J.K. Near-Infrared Reflectance Spectra of Mixtures of Kaolin-Group Minerals: Use in Clay Mineral Studies. Clays Clay Miner. 1988, 36, 310–316. [Google Scholar] [CrossRef]
- Petit, S. Characterization of Octahedral Substitutions in Kaolinites Using Near Infrared Spectroscopy. Clays Clay Miner. 1999, 47, 103–108. [Google Scholar] [CrossRef]
- Post, J.L.; Noble, P.N. The Near-Infrared Combination Band Frequencies of Dioctahedral Smectites, Micas, and Illites. Clays Clay Miner. 1993, 41, 639–644. [Google Scholar] [CrossRef]
- Huntington, J.F.; Cudahy, T.J.; Yang, K.; Scott, K.M.; Mason, P.; Gray, D.J.; Berman, M.; Bischoff, L.; Reston, M.S.; Mauger, A.J. Mineral Mapping with Field Spectroscopy for Exploration; CSIRO Exploration and Mining Report 419R AMIRA Project, P435; CSIRO: Canberra, Australia, 1999; p. 45. [Google Scholar]
- Scott, K.M.; Yang, K. Spectral Reflectance Studies of White Micas; CSIRO Report 439R AMIRA Project P435; CSIRO: Canberra, Australia, 1997; p. 35. [Google Scholar]
- Martínez-Alonso, S.; Rustad, J.R.; Goetz, A.F.H. Ab Initio Quantum Mechanical Modeling of Infrared Vibrational Frequencies of the OH Group in Dioctahedral Phyllosilicates. Part I: Methods, Results and Comparison to Experimental Data. Am. Miner. 2002, 87, 1215–1223. [Google Scholar] [CrossRef]
- Tuddenham, W.M.; Lyon, R.J.P. Relation of Infrared Spectra and Chemical Analysis for Some Chlorites and Related Minerals. Anal. Chem. 1959, 31, 377–380. [Google Scholar] [CrossRef]
- Stubican, V.; Roy, R. Infrared Spectra of Layer-Structure Silicates. J. Am. Ceram. Soc. 1961, 44, 625–627. [Google Scholar] [CrossRef]
- Doublier, M.P.; Roache, T.; Potel, S.; Laukamp, C. Short-Wavelength Infrared Spectroscopy of Chlorite Can Be Used to Determine Very Low Metamorphic Grades. Eur. J. Miner. 2012, 24, 891–902. [Google Scholar] [CrossRef]
- Bierwirth, P.N. Laboratory and Imaging Spectroscopy of Tourmaline—A Tool for Mineral Exploration. In Proceedings of the 14th Australasian Remote Sensing & Photogrammetry Conference, Darwin, Australia, 27 September 2008. [Google Scholar]
- Spear, F.S. Metamorphic Phase Equilibria and Pressure-Temperature-Time Paths, 2nd ed.; Monograph/Mineralogical Society of America; Mineralogical Society of America: Washington, DC, USA, 1995. [Google Scholar]
- Gaffey, S.J. Spectral Reflectance of Carbonate Minerals and Rocks in the Visible and Near Infrared (0.35 to 2.55 MUM) and Its Applications in Carbonate Petrology; University of Hawaii: Honolulu, HI, USA, 1984. [Google Scholar]
- Moenke, H.H.W. Silica, the Three-Dimensional Silicates, Borosilicates and Beryllium Silicates. In The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society of Great Britain and Ireland: Londan, UK, 1974; pp. 365–382. [Google Scholar] [CrossRef]
- Ruan, H.D.; Frost, R.L.; Kloprogge, J.T. The Behavior of Hydroxyl Units of Synthetic Goethite and Its Dehydroxylated Product Hematite. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2001, 57, 2575–2586. [Google Scholar] [CrossRef]
- Burns, R.G.; Strens, R.G.J. Infrared Study of the Hydroxyl Bands in Clinoamphiboles. Science 1966, 153, 890–892. [Google Scholar] [CrossRef]
- Robert, J.L.; Kodama, H. Generalization of the Correlations between Hydroxyl-Stretching Wavenumbers and Composition of Micas in the System K2O-Mg2O-Al2O3-SiO2-H2O: A Single Model for Trioctahedral and Dioctahedral Micas. Am. J. Sci. 1988, 288, 196–212. [Google Scholar]
- Besson, G. Refined Relationships between Chemical Composition of Dioctahedral Fine-Grained Mica Minerals and Their Infrared Spectra within the OH Stretching Region. Part I: Identification of the OH Stretching Bands. Clays Clay Miner. 1997, 45, 158–169. [Google Scholar] [CrossRef]
- Besson, G. Refined Relationships between Chemical Composition of Dioctahedral Fine-Grained Micaceous Minerals and Their Infrared Spectra within the OH Stretching Region. Part II: The Main Factors Affecting OH Vibrations and Quantitative Analysis. Clays Clay Miner. 1997, 45, 170–183. [Google Scholar] [CrossRef]
- Makreski, P.; Jovanovski, G.; Gajović, A. Minerals from Macedonia. Vib. Spectrosc. 2006, 40, 98–109. [Google Scholar] [CrossRef]
- Gonzalez-Carreño, T.; Fernandez, M.; Sanz, J. Infrared and Electron Microprobe Analysis of Tourmalines. Phys. Chem. Miner. 1988, 15, 452–460. [Google Scholar] [CrossRef]
- Madejová, J. FTIR Techniques in Clay Mineral Studies. Vib. Spectrosc. 2003, 31, 1–10. [Google Scholar] [CrossRef]
- Aines, R.D.; Rossman, G.R. Water in Minerals? A Peak in the Infrared. J. Geophys. Res. Solid Earth 1984, 89, 4059–4071. [Google Scholar] [CrossRef]
- Igisu, M.; Ueno, Y.; Shimojima, M.; Nakashima, S.; Awramik, S.M.; Ohta, H.; Maruyama, S. Micro-FTIR Spectroscopic Signatures of Bacterial Lipids in Proterozoic Microfossils. Precambrian Res. 2009, 173, 19–26. [Google Scholar] [CrossRef]
- Green, D.; Schodlok, M. Characterisation of Carbonate Minerals from Hyperspectral TIR Scanning Using Features at 14,000 and 11,300 Nm. Aust. J. Earth Sci. 2016, 63, 951–957. [Google Scholar] [CrossRef]
- Chester, R.; Elderfield, H. The Application of Infra-Red Absorption Spectroscopy to Carbonate Mineralogy. Sedimentology 1967, 9, 5–21. [Google Scholar] [CrossRef]
- Yitagesu, F.A.; van der Meer, F.; van der Werff, H.; Hecker, C. Spectral Characteristics of Clay Minerals in the 2.5–14 μm Wavelength Region. Appl. Clay Sci. 2011, 53, 581–591. [Google Scholar] [CrossRef]
- Rubens, H.; Nichols, E.F. Versuche mit Wärmestrahlen von grosser Wellenlänge. Ann. Phys. Chem. 1897, 296, 418–462. [Google Scholar] [CrossRef]
- Spitzer, W.G.; Kleinman, D.A. Infrared Lattice Bands of Quartz. Phys. Rev. 1961, 121, 1324–1335. [Google Scholar] [CrossRef]
- Thompson, C.S.; Wadsworth, M.E. Determination of the Composition of Plagio-Clase Feldspars by Means of Infrared Spectroscopy. Am. Miner. 1957, 42, 334–341. [Google Scholar]
- Vincent, R.K.; Rowan, L.C.; Gillespie, R.E.; Knapp, C. Thermal-Infrared Spectra and Chemical Analyses of Twenty-Six Igneous Rock Samples. Remote Sens. Environ. 1975, 4, 199–209. [Google Scholar] [CrossRef]
- Walter, L.S.; Salisbury, J.W. Spectral Characterization of Igneous Rocks in the 8- to 12-Μm Region. J. Geophys. Res. 1989, 94, 9203. [Google Scholar] [CrossRef]
- Salisbury, J.W.; Walter, L.S. Thermal Infrared (2.5–13.5 Μm) Spectroscopic Remote Sensing of Igneous Rock Types on Particulate Planetary Surfaces. J. Geophys. Res. 1989, 94, 9192. [Google Scholar] [CrossRef]
- Cudahy, T.; Hewson, R.; Caccetta, M.; Roache, A.; Whitbourn, L.; Connor, P.; Coward, D.; Mason, P.; Yang, K.; Huntington, J.; et al. Drill Core Logging of Plagioclase Feldspar Composition and Other Minerals Associated with Archean Gold Mineralization at Kambalda, Western Australia, Using a Bidirectional Thermal Infrared Reflectance System. Remote Sens. Spectr. Geol. 2009, 223–235. [Google Scholar] [CrossRef]
- Harlov, D.E.; Andrut, M.; Pöter, B. Characterisation of Buddingtonite (NH4)[AlSi3O8] and ND4 -Buddingtonite (ND4)[AlSi3O8] Using IR Spectroscopy and Rietveld Refinement of XRD Spectra. Phys. Chem. Miner. 2001, 28, 188–198. [Google Scholar] [CrossRef]
- Makreski, P.; Jovanovski, G. Minerals from Macedonia. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 73, 460–467. [Google Scholar] [CrossRef]
- Lane, M.D. Mid-Infrared Emission Spectroscopy of Sulfate and Sulfate-Bearing Minerals. Am. Miner. 2007, 92, 1–18. [Google Scholar] [CrossRef]
- Adler, H.H.; Kerr, P.F. Variations in Infrared Spectra, Molecular Symmetry and Site Symmetry of Sulfate Minerals. Am. Miner. 1965, 50, 132–147. [Google Scholar]
- Mauger, A.J.; Ehrig, K.; Kontonikas-Charos, A.; Ciobanu, C.L.; Cook, N.J.; Kamenetsky, V.S. Alteration at the Olympic Dam IOCG–U Deposit: Insights into Distal to Proximal Feldspar and Phyllosilicate Chemistry from Infrared Reflectance Spectroscopy. Aust. J. Earth Sci. 2016, 63, 959–972. [Google Scholar] [CrossRef]
- Laukamp, C.; LeGras, M.; Montenegro, V.; Windle, S.; McFarlane, A.J. Grandite-Based Resource Characterisation of the Skarn-Hosted Cu-Zn-Mo Deposit of Antamina, Peru. Miner. Depos. 2021. [Google Scholar] [CrossRef]
- LeGras, M.; Laukamp, C. NVCL Spectral Reference Library—Pyroxenes and Pyroxenoids; CSIRO Report EP184276; CSIRO: Canberra, Australia, 2019. [Google Scholar]
- Schodlok, M.C.; Green, A.; Huntington, J. A Reference Library of Thermal Infrared Mineral Reflectance Spectra for the HyLogger-3 Drill Core Logging System. Aust. J. Earth Sci. 2016, 63, 941–949. [Google Scholar] [CrossRef]
- Adler, H.H. Infrared Spectra of Phosphate Minerals: Symmetry and Substitutional Effects in the Pyromorphite Series. Am. Miner. 1964, 49, 1002–1015. [Google Scholar]
- Clavier, N.; Mesbah, A.; Szenknect, S.; Dacheux, N. Monazite, Rhabdophane, Xenotime & Churchite: Vibrational Spectroscopy of Gadolinium Phosphate Polymorphs. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 205, 85–94. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Thermal Decomposition of Ferrian Chamosite: An Infrared Emission Spectroscopic Study. Contrib. Miner. Pet. 2000, 138, 59–67. [Google Scholar] [CrossRef]
- Hamilton, V.E. Thermal Infrared (Vibrational) Spectroscopy of Mg–Fe Olivines: A Review and Applications to Determining the Composition of Planetary Surfaces. Geochemistry 2010, 70, 7–33. [Google Scholar] [CrossRef]
- Madejová, J.; Komadel, P. Baseline Studies of the Clay Minerals Society Source Clays: Infrared Methods. Clays Clay Miner. 2001, 49, 410–432. [Google Scholar] [CrossRef]
- Panikorovskii, T.; Chukanov, N.; Rusakov, V.; Shilovskikh, V.; Mazur, A.; Balassone, G.; Ivanyuk, G.; Krivovichev, S. Vesuvianite from the Somma-Vesuvius Complex: New Data and Revised Formula. Minerals 2017, 7, 248. [Google Scholar] [CrossRef]
- McAloon, B.P.; Hofmeister, A.M. Single-Crystal IR Spectroscopy of Grossular-Andradite Garnets. Am. Miner. 1995, 80, 1145–1156. [Google Scholar] [CrossRef]
- Lane, M.D.; Christensen, P.R. Thermal Infrared Emission Spectroscopy of Anhydrous Carbonates. J. Geophys. Res. Planets 1997, 102, 25581–25592. [Google Scholar] [CrossRef]
- Liebscher, A. Spectroscopy of Epidote Minerals. Rev. Miner. Geochem. 2004, 56, 125–170. [Google Scholar] [CrossRef]
- Conel, J.E. Infrared Emissivities of Silicates: Experimental Results and a Cloudy Atmosphere Model of Spectral Emission from Condensed Particulate Mediums. J. Geophys. Res. 1969, 74, 1614–1634. [Google Scholar] [CrossRef]
- Hook, S.J.; Dmochowski, J.E.; Howard, K.A.; Rowan, L.C.; Karlstrom, K.E.; Stock, J.M. Mapp. ing Variations in Weight Percent Silica Measured from Multispectral Thermal Infrared Imagery—Examples from the Hiller Mountains, Nevada, USA and Tres Virgenes-La Reforma, Baja California Sur, Mexico. Remote Sens. Environ. 2005, 95, 273–289. [Google Scholar] [CrossRef]
- Sonntag, I.; Laukamp, C.; Hagemann, S.G. Low Potassium Hydrothermal Alteration in Low Sulfidation Epithermal Systems as Detected by IRS and XRD: An Example from the Co–O Mine, Eastern Mindanao, Philippines. Ore Geol. Rev. 2012, 45, 47–60. [Google Scholar] [CrossRef]
- Van Ruitenbeek, F.J.A.; Bakker, W.H.; van der Werff, H.M.A.; Zegers, T.E.; Oosthoek, J.H.P.; Omer, Z.A.; Marsh, S.H.; van der Meer, F.D. Mapping the Wavelength Position of Deepest Absorption Features to Explore Mineral Diversity in Hyperspectral Images. Planet. Space Sci. 2014, 101, 108–117. [Google Scholar] [CrossRef]
- Haest, M.; Cudahy, T.; Laukamp, C.; Gregory, S. Quantitative Mineralogy from Infrared Spectroscopic Data. I. Validation of Mineral Abundance and Composition Scripts at the Rocklea Channel Iron Deposit in Western Australia. Econ. Geol. 2012, 107, 209–228. [Google Scholar] [CrossRef]
- Haest, M.; Cudahy, T.; Laukamp, C.; Gregory, S. Quantitative Mineralogy from Infrared Spectroscopic Data. II. Three-Dimensional Mineralogical Characterization of the Rocklea Channel Iron Deposit, Western Australia. Econ. Geol. 2012, 107, 229–249. [Google Scholar] [CrossRef]
- Lampinen, H.M.; Laukamp, C.; Occhipinti, S.A.; Hardy, L. Mineral Footprints of the Paleoproterozoic Sediment-Hosted Abra Pb-Zn-Cu-Au Deposit Capricorn Orogen, Western Australia. Ore Geol. Rev. 2019, 104, 436–461. [Google Scholar] [CrossRef]
- Yang, K.; Browne, P.R.L.; Huntington, J.F.; Walshe, J.L. Characterising the Hydrothermal Alteration of the Broadlands–Ohaaki Geothermal System, New Zealand, Using Short-Wave Infrared Spectroscopy. J. Volcanol. Geotherm. Res. 2001, 106, 53–65. [Google Scholar] [CrossRef]
- Sunshine, J.M.; Pieters, C.M.; Pratt, S.F. Deconvolution of Mineral Absorption Bands: An Improved Approach. J. Geophys. Res. 1990, 95, 6955. [Google Scholar] [CrossRef]
- Rodger, A.; Fabris, A.; Laukamp, C. Feature Extraction and Clustering of Hyperspectral Drill Core Measurements to Assess Potential Lithological and Alteration Boundaries. Minerals 2021, 11, 136. [Google Scholar] [CrossRef]
- Rodger, A.; Laukamp, C.; Haest, M.; Cudahy, T. A Simple Quadratic Method of Absorption Feature Wavelength Estimation in Continuum Removed Spectra. Remote Sens. Environ. 2012, 118, 273–283. [Google Scholar] [CrossRef]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef]
- Clark, R.N.; Roush, T.L. Reflectance Spectroscopy: Quantitative Analysis Techniques for Remote Sensing Applications. J. Geophys. Res. Solid Earth 1984, 89, 6329–6340. [Google Scholar] [CrossRef]
- Doublier, M.P.; Roache, A.; Potel, S. Geological Survey of Western Australia. App. lication of SWIR Spectroscopy in Very Low-Grade Metamorphic Environments: A Comparison with XRD Methods; Geological Survey of Western Australia: East Perth, Australia, 2010. [Google Scholar]
- Wells, M.; Laukamp, C.; Hancock, E. Reflectance Spectroscopic Characterisation of Mineral Alteration Footprints Associated with Sediment-Hosted Gold Mineralisation at Mt Olympus (Ashburton Basin, Western Australia). Aust. J. Earth Sci. 2016, 63, 987–1002. [Google Scholar] [CrossRef]
- Simpson, M.P.; Rae, A.J. Short-Wave Infrared (SWIR) Reflectance Spectrometric Characterisation of Clays from Geothermal Systems of the Taupō Volcanic Zone, New Zealand. Geothermics 2018, 73, 74–90. [Google Scholar] [CrossRef]
- LeGras, M.; Laukamp, C.; Pejcic, B.; Shelton, T.; Lau, I.; Stromberg, J. MIR Spectral Library, 2021. CSIRO Data Access Portal. Available online: https://data.csiro.au/collections/collection/CIcsiro:49193 (accessed on 18 February 2021).
- Burley, L.L.; Barnes, S.J.; Laukamp, C.; Mole, D.R.; Le Vaillant, M.; Fiorentini, M.L. Rapid Mineralogical and Geochemical Characterisation of the Fisher East Nickel Sulphide Prospects, Western Australia, Using Hyperspectral and PXRF Data. Ore Geol. Rev. 2017, 90, 371–387. [Google Scholar] [CrossRef]
- Moltzen, J. HyLoggerTM Results from Chips; A RoXplorer® Pilot Study: Hobart, Australia, 2021; p. 1. [Google Scholar]
- Geiger, C.A.; Winkler, B.; Langer, K. Infrared Spectra of Synthetic Almandine–Grossular and Almandine–Pyrope Garnet Solid Solutions: Evidence for Equivalent Site Behaviour. Miner. Mag. 1989, 53, 231–237. [Google Scholar] [CrossRef][Green Version]
- Sutherland, D.N.; Gottlieb, P. Application of Automated Quantitative Mineralogy in Mineral Processing. Miner. Eng. 1991, 4, 753–762. [Google Scholar] [CrossRef]
- Laukamp, C.; Lau, I.C.; Stolf, M.; Green, A.A. Impact of Grain Size Variations of Quartz on Estimation of Kaolinite Content from Infrared Spectral Signatures; IMA 2018, 13.-17.08.2018, Melbourne; CSIRO: Clayton, Australia, 2018. [Google Scholar]
- Laukamp, C.; White, A.J.R.; Rodger, A.; Gum, J.; Metelka, V.; Lau, I.C.; Gordon, G.; Fonteneau, L. Mapping Mineral Footprints Associated with Gold in the Bulong Area, Western Australai, Using Airborne and Drill Core Hyper-Spectral and Geochemcial Data; IGARSS 2018, 23-27.07.2018; CSIRO: Clayton, Australia, 2018; p. 4. [Google Scholar]
- Elias, M. Lateritic Nickel Mineralization of the Yilgarn Craton. Soc. Econ. Geol. Spec. Publ. 2006, 13, 195–210. [Google Scholar]
- Elias, M.; Donaldson, M.J.; Giorgetta, N.E. Geology, Mineralogy, and Chemistry of Lateritic Nickel-Cobalt Deposits near Kalgoorlie, Western Australia. Econ. Geol. 1981, 76, 1775–1783. [Google Scholar] [CrossRef]
- Duke, E.F. Near Infrared Spectra of Muscovite, TschermaCSIRk Substitution, and Metamorphic Reaction Progress: Implications for Remote Sensing. Geology 1994, 22, 621–624. [Google Scholar] [CrossRef]
- Laukamp, C.; Salama, W.; González-Álvarez, I. Proximal and Remote Spectroscopic Characterisation of Regolith in the Albany–Fraser Orogen (Western Australia). Ore Geol. Rev. 2016, 73, 540–554. [Google Scholar] [CrossRef]
- Tappert, M.C.; Rivard, B.; Tappert, R.; Feng, J. Using reflectance spectroscopy to estimate the orientation of quartz crystals in rocks. Can. Miner. 2013, 51, 405–413. [Google Scholar] [CrossRef]









| Mineral Group | Mineral Species (Examples) | Assignment of Absorption | Lower Limit (nm) | Upper Limit (nm) | Literature | Intensity Script | Wavelength Script |
|---|---|---|---|---|---|---|---|
| talc | talc | ν + δM3OH | 2080 | [45] | 2080D | - | |
| NH4 | alunite, montmorillonite, buddingtonite, white mica | ν1 + ν4NH4 | 2020 to 2120 | [34] | - | - | |
| di-oct. sheet silicate | kaolin group | ν + δM2OHo | 2159 | [41] | 2160D | - | |
| di-oct. sheet silicate | pyrophyllite | ν + δMnOH | 2166 | [48] | |||
| sulphate | alunite | ν + δMnOH | 2172 | [35] | - | ||
| cyclosilicate | tourmaline | ν + δMnOH | 2174 | [49] | |||
| di-oct. sheet silicate | kaolin group | ν + δM2OHi | 2209 | [41] | 2200D | 2200W | |
| di-oct. sheet silicate | muscovite, phengite, paragonite | ν + δ(M)2OH | 2185 ([VI]Al-rich) | 2215 ([VI]Al-poor) | [3] | ||
| cyclosilicate | tourmaline | ν + δMnOH | 2204 | [49] | |||
| sulphate | jarosite | ν + δMnOH | 2212 | [35] | |||
| di-oct. sheet silicate | prehnite | ν + δMnOH | 22353), 2280, 2333, 2358, 2384 | [32] | |||
| tri-oct. sheet silicate | clinochlore, chamosite, ripidolite | ν + δM2OH | 2248 (Mg-rich) | 2261 (Fe2+-rich) | [17,50] | 2250D | 2250W |
| tri-oct. sheet silicate | biotite, annite, phlogopite (only when AlVI is present) | ν + δM2OH | 2248 (Mg-rich) | 2259 (Fe2+-rich) | [18,31,50] | ||
| sorosilicate | epidote group | ν + δMnOH | 2250 | [32] | |||
| cyclosilicate | tourmaline | ν + δMnOH | 2256 | [49] | |||
| sulphate | jarosite | ν + δMnOH | 2263 | [35] | |||
| hydroxide | gibbsite | ν + δMnOH | 2268 | [51] | |||
| talc | talc | ν + δM3OH | 2279 | [45] | 2290D | 2290W | |
| tri-oct. sheet silicate | Fe-smectite | ν + δM2OH | 2288 | 2317 | [50] | ||
| talc | talc | ν + δM3OH | 2300 | [45] | |||
| cyclosilicate | tourmaline | ν + δMnOH | 2302 | [49] | |||
| Ca-amphibole | tremolite, actinolite | ν + δMg3OH | 2296 (Mg-rich) | 2318 (Fe2+-rich) | [45] | ||
| Ca-amphibole | tremolite, actinolite | ν + δMg3OH | 2318 (Mg-rich) | 2338 (Fe2+-rich) | [45] | ||
| carbonate | ankerite, calcite, dolomite, magnesite, siderite | 3n3CO3 | 2300 (Mg-rich) | 2340 (Ca only) | [19] | 2320D | 2320W |
| chlorite | clinochlore, chamosite, ripidolite | ν + δMg3OH | 2331 (Mg-rich) | 2358 (Fe2+-rich) | [50] | 2340D | 2340W |
| di-oct. sheet silicate | muscovite, phengite, paragonite | ν + δMg3OH | 2348 ([VI]Al-rich) | 2366 ([VI]Al-poor) | [50] | ||
| tri-oct. sheet silicate | biotite, annite, phlogopite | ν + δM2OH | 2320 (Mg-rich) | 2350 (Fe2+-rich) | [18,31] | ||
| sorosilicate | epidote group | ν + δM2OH | 2340 | [32] | |||
| cyclosilicate | tourmaline | ν + δMnOH | 2359 (ca. 5% to 11% MgO) | 2375 (0% MgO) | [49] | - | - |
| tri-oct. sheet silicate | biotite, annite, phlogopite | ν + δM2OH | 2377 (Mg-rich) | 2390 (Fe2+-rich) | [18,31] | 2390D | 2390W |
| talc | talc | ν + δMg3OH | 2380 | [45] | |||
| Ca-amphibole | tremolite, actinolite | ν + δMg3OH | 2382 (Mg-rich) | 2406 (Fe2+-rich) | [45] | ||
| carbonate | ankerite, calcite, dolomite, magnesite, siderite | 2ν3 + ν1 | 2505 (Mg-rich) | 2541 (Ca only) | [19] | - | - |
| Mineral Group | Mineral Species (Examples) | Assignment of Absorption | Lower Limit (nm) | Upper Limit (nm) | Literature | Intensity Script | Wavelength Script |
|---|---|---|---|---|---|---|---|
| all hydroxylated minerals | νOH | 2667 | 5000 | [23,65] | - | - | |
| di-oct. sheet silicate | kaolin group | νOH | 2705, 2725, 2738, 2761 | [41] | 2830D | 2830W | |
| di-oct. sheet silicate | white mica | νOH | 2677, 2762, 2775, 2799, 2807 | [58] | |||
| tri-oct. sheet silicate | dark mica | νOH | 2668, 2677, 2680 | [68] | |||
| di-/tri-oct. sheet silicate | white and dark micas | νOH | 2810 (Mg-rich) | 2853 (Fe2+-rich) | [69,70] | ||
| double chain silicate | amphibole | νOH | 2723 (Mg-rich) | 2759 (Fe2+-rich) | [67,71] | ||
| cyclosilicate | tourmaline | νOH | 2675 to 2890 | [49,72] | |||
| iron oxides | goethite | νH-O-H | 2899 | 2903 | [66] | - | - |
| di-oct. sheet silicate | Al-smectites | νH-O-H | 2946 (Mg-rich) | 2987 (K-rich) | [73] | - | - |
| all H2O-bearing minerals | νH-O-H | 3000 | [74] | 3000D | - | ||
| iron oxides | νOH | 3113 | 3131 | [66] | - | - | |
| cyclosilicate | tourmaline | 2νBO3 | 3704 | unpublished | - | - | |
| cyclosilicate | tourmaline | 2νBO3 | 3811 | unpublished | 3800D | 3800W | |
| carbonate | carbonate | 2νCO3, ν + νCO3 | 3930 | 4150 | unpublished | 4000D | 4000W |
| tectosilicate | quartz | 2νSiO | 4470 | unpublished (inferred from [75]) | 4470D | - | |
| tectosilicate | quartz | 2νSiO | 4670 | unpublished (inferred from [75]) | - | - | |
| phosphate | 2ν3(PO4) | 4805 | unpublished | - | - | ||
| tectosilicate | quartz | 2νSiO | 4920 | unpublished | - | - | |
| sulphate | jarosite | 2ν3(SO4)2−, 2d (OH) | 4960 | [35] | 4500D | 4500W | |
| sulphate | jarosite | 2ν3(SO4)2−, 2δ(OH) | 5099 | [35] | - | - | |
| tectosilicate | quartz | 2νSiO | 5330 | unpublished | 5330D | - | |
| di-oct. sheet silicate | kaolin group | 2νSiO | 5480 | unpublished | 5480D | - | |
| Mineral Group | Mineral Species (Examples) | Assignment of Absorption | Lower Limit (nm) | Upper Limit (nm) | Literature | Intensity Script | Wavelength Script |
|---|---|---|---|---|---|---|---|
| carbonate | ankerite, calcite, dolomite, magnesite, siderite | ν3 asymmetric stretch CO3 | 6405R to 6598R | [76,85] | 6500P | - | |
| NH4 (multiple minerals) | NH bending | 6993* to 7400* | [86] | - | - | ||
| cyclosilicate | tourmaline (schorl) | νas(BO3) | 7391* | [87] | 7400P | - | |
| cyclosilicate | tourmaline (schorl) | νas(BO3) | 7968* (7880 to 7995) | [87] | - | - | |
| sulphate | alunite, jarosite | ν3SO4 | 7899E, 8197E | [88] | - | - | |
| sulphate | jarosite, barite, alunite, anhydrite | ν3SO4 | all*: 8439, 8480 to 8540, 8576, 8650 | [35,88,89] | - | - | |
| sulphate | anhydrite, barite, alunite, jarosite | ν3SO4 | all*: 8920, 8930 to 8980, 8969, 8993 | [88,89] | - | - | |
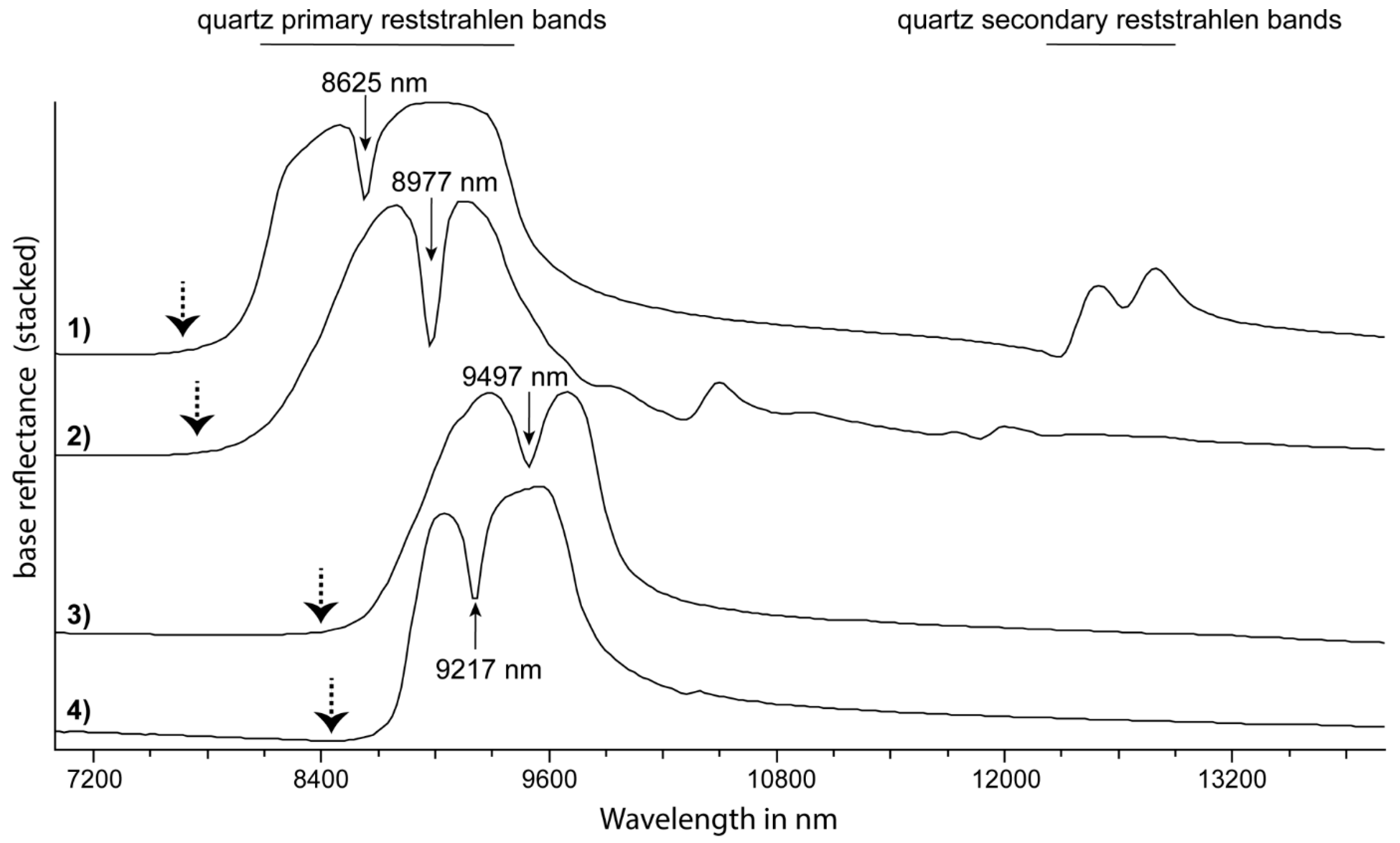
| tectosilicate | quartz | νSiO | 8496R (8598*) | [80,85] | R8250/R7400 | - | |
| tectosilicate | quartz | νSiO | 9074R (9328*) | [80,85] | - | - | |
| tectosilicate | quartz | “quartz RSC feature” | 8625R | [85] | 8625D | - | |
| tectosilicate | K-feldspar (orthoclase) | νSiO | 8739R | [90] | - | - | |
| single chain silicate | pyroxene (diopside) | νSiO | 8960 to 9100 | [91,92] | 9000P | 9000PW | |
| phosphate | apatite | νas(PO4) | 9050R (9160*) | [93,94,95] | - | ||
| phosphate | apatite | “apatite RSC feature” | 9210R | unpublished | 9200D | - | |
| phosphate | apatite | νas(PO4) | 9550R (9620*) | [93,94,95] | |||
| tri-oct. sheet silicate | talc | νSiO | 9280R | [8,93] | |||
| tri-oct. sheet silicate | talc | “talc RSC feature” | 9470R | unpublished | 9470D | - | |
| tri-oct. sheet silicate | talc | νSiO | 9680R | [8,93] | |||
| di-oct. sheet silicate | muscovite | νSiO | 9130R | [31] | - | - | |
| tri-oct. sheet silicate | biotite, phlogopite | νSiO | 9200R | [31] | - | - | |
| tri-oct. sheet silicate | ripidolite (clinochlore) | νSiO | 9217* to 9225* | [96] | - | - | |
| single chain silicate | pyroxene (diopside) | νSiO | 9290 to 9390 | [92] | minor | minor | |
| nesosilicates | olivine (Fo#91 to Fo#0) | νSiO | 9337 to 9852 | [97] | tbd | tbd | |
| sulphate | jarosite, barite | ν3SO4 | all*: 9217, 9230 to 9260 | [35,89] | - | - | |
| sulphate | alunite, jarosite, barite | ν1SO4 | all*: 9737, 9940, 10150 to 10180 | [88,89] | - | - | |
| di-oct. sheet silicate | muscovite | νSiO | 9600R | [31] | - | - | |
| di-oct. sheet silicate | montmorillonite (+- mica) | νSiO | 9600* to 9710* | [98] | |||
| tri-oct. sheet silicate | biotite, phlogopite | νSiO | 9780R to 9800R | [31] | |||
| di-oct. sheet silicate | kaolinite | νSiO | 9074*, 9681*, 9891* | [98] | |||
| tectosilicate | K-feldspar (orthoclase) | νSiO | 9558R | [90] | |||
| tectosilicate | plagioclase feldspars | νSiO | 9660R | [85] | 9660P | 9660P/9920P | |
| tectosilicate | plagioclase feldspars | νSiO | 9920R | [85] | 9920P | ||
| nesosilicates | vesuvianite | νSiO in Si2O7 group | 9900R | [91,99] | 9900P | - | |
| nesosilicates | olivine (Fo#91 to Fo#0) | νSiO | 10,121R to 10,537R | [97] | minor | minor | |
| nesosilicates | olivine (Fo#91 to Fo#0) | nSiO | 10,515R to 11,099R | [97] | tbd | tbd | |
| di-oct. sheet silicate | kaolin group | δOH | 10,661* | [98] | - | - | |
| single chain silicate | pyroxene (diopside) | νSiO | 10,850R | [92] | minor | minor | |
| di-oct. sheet silicate | kaolin group | δOH | 10,929* | [98] | - | - | |
| nesosilicates | grandite series | νSi-O in SiO4 group (“B”) | 10,953R to 11,368R | [91,100] | 11,100P | 11,100PW | |
| carbonate | ankerite, calcite, dolomite, magnesite, siderite | ν2 out-of-plane bend CO3 | 11,058R to 11,372R | [76,85] | - | 11,300PW | |
| nesosilicates | grandite series | grandite feature “T” | 11,250R to 11,600R | [91] | 11,300D | 11,300DW | |
| single chain silicate | pyroxene (enstatite) | νSiO | 11,470R to 11,570R | [92] | - | 11,500PW | |
| carbonate | ankerite, calcite, dolomite, magnesite, siderite | ν4 in-plane bend CO3 (trough!) | 13,200R to 14,000R | [76] | - | 14,000DW | |
| sulphate | anhydrite | n4SO4 | 14,780* | [89] | - | - | |
| Depth Script | Wavelength Script | wvl Range of Mineral-Related Absorptions | wvl Range for Continuum Removal | Literature Examples for Applying the Respective Scripts (or Using the Respective Absorption Features) to Investigate Respective Geological Environments | Applicable to Mineral Group | ||
|---|---|---|---|---|---|---|---|
| - | - | from | to | from | to | - | - |
| 1400D | 1400W | 1387 | 1445 | 1350 | 1450 | [116] | OH-bearing |
| 1480D | 1480W | 1471 | 1491 | 1440 | 1520 | [32,37,117] | sulphate, prehnite |
| 1550D | 1550W | 1520 | 1563 | 1510 | 1610 | [33] | epidote |
| 1760D | 1760W | 1751 | 1764 | 1730 | 1790 | [117] | sulphate |
| 1850D | - | 1849 | 1820 | 1880 | [47] | NH4 | |
| 2080D | - | 2080 | 2060 | 2100 | [45] | talc | |
| 2160D | - | 2159 | 2166 | 2138 | 2179 | [107] | kaolin, pyrophyllite |
| 2200D | 2200W | 2185 | 2215 | 2120 | 2245 | [106] | Al-bearing sheetsilicate |
| 2250D | 2250W | 2248 | 2268 | 2230 | 2280 | [17,105] | biotite, chlorite, epidote |
| 2290D | 2290W | 2279 | 2338 | 2270 | 2320 | [108] | FeMg-clays, amphibole, talc |
| 2320D | 2320W | 2300 | 2340 | 2295 | 2345 | [45] | amphibole, carbonate |
| 2350D | 2350W | 2320 | 2366 | 2310 | 2370 | [18] | chlorite, micas, epidote |
| 2390D | 2390W | 2377 | 2406 | 2375 | 2435 | [45] | amphibole, annite, talc |
| 2830D | 2830W | 2677 | 2890 | 2770 | 2890 | this contribution | OH-bearing |
| 3000D | - | 3000 | 2650 | 3900 | this contribution | H2O | |
| 4000D | 4000W | 3930 | 4150 | 3800 | 4200 | this contribution | carbonate |
| 4470D | - | 4470 | 4350 | 4550 | this contribution | quartz | |
| 4500D | 4500W | 4570 | 4850 | 4090 | 5040 | this contribution | sulphate |
| 4500D | 4500W | 4625 | 4770 | 4090 | 5040 | this contribution | carbonate |
| 5330D | - | 5330 | 5210 | 5450 | this contribution | quartz | |
| 5480D | - | 5480 | 5380 | 5580 | this contribution | kaolin | |
| Depth Script | Wavelength Script | wvl Range of Mineral-Related Absorptions | wvl Range for Continuum Removal | Literature Examples for Applying the Respective Scripts (or Using the Respective Absorption Features) to Investigate Respective Geological Environments | Applicable to Mineral Group | ||
|---|---|---|---|---|---|---|---|
| - | - | from | to | from | to | - | - |
| 6500P | - | 6405 | 6598 | 6300 | 6700 | [76,109] | carbonate |
| 7400P | - | 7391 | 7200 | 7550 | this contribution | tourmaline | |
| R8250/R7400* | - | - | 7350 | 8270 | [85] | quartz | |
| 8625D | - | 8625 | 8565 | 8705 | [85,109] | quartz | |
| 9000P | 9000PW | 9337 | 9852 | 8650 | 9350 | [91] | pyroxene, pyroxenoid |
| 9660P | - | 9660 | 9400 | 9780 | [85] | feldspar | |
| 9920P | - | 9920 | 9800 | 10,150 | [85] | feldspar | |
| 9660P/9920P^ | - | - | - | 10,150 | [85] | feldspar | |
| 9900P | - | 9900 | 9500 | 10,200 | [91] | vesuvianite | |
| 11100P | 11,100PW | 10,953 | 11,368 | 10,850 | 11,300 | [91] | garnet |
| - | 11,300PW | 11,058 | 11,372 | 10,900 | 11,800 | [76,91,109] | carbonate |
| - | 11,300DW | 11,250 | 11,600 | 11,200 | 11,600 | [91] | garnet |
| - | 11,500PW | 11,470 | 11,570 | 9800 | 11,650 | [92] | pyroxene |
| - | 14,000DW | 13,200 | 14,000 | 13,000 | 14,000 | [62,73,76,91,99,109] | carbonate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laukamp, C.; Rodger, A.; LeGras, M.; Lampinen, H.; Lau, I.C.; Pejcic, B.; Stromberg, J.; Francis, N.; Ramanaidou, E. Mineral Physicochemistry Underlying Feature-Based Extraction of Mineral Abundance and Composition from Shortwave, Mid and Thermal Infrared Reflectance Spectra. Minerals 2021, 11, 347. https://doi.org/10.3390/min11040347
Laukamp C, Rodger A, LeGras M, Lampinen H, Lau IC, Pejcic B, Stromberg J, Francis N, Ramanaidou E. Mineral Physicochemistry Underlying Feature-Based Extraction of Mineral Abundance and Composition from Shortwave, Mid and Thermal Infrared Reflectance Spectra. Minerals. 2021; 11(4):347. https://doi.org/10.3390/min11040347
Chicago/Turabian StyleLaukamp, Carsten, Andrew Rodger, Monica LeGras, Heta Lampinen, Ian C. Lau, Bobby Pejcic, Jessica Stromberg, Neil Francis, and Erick Ramanaidou. 2021. "Mineral Physicochemistry Underlying Feature-Based Extraction of Mineral Abundance and Composition from Shortwave, Mid and Thermal Infrared Reflectance Spectra" Minerals 11, no. 4: 347. https://doi.org/10.3390/min11040347
APA StyleLaukamp, C., Rodger, A., LeGras, M., Lampinen, H., Lau, I. C., Pejcic, B., Stromberg, J., Francis, N., & Ramanaidou, E. (2021). Mineral Physicochemistry Underlying Feature-Based Extraction of Mineral Abundance and Composition from Shortwave, Mid and Thermal Infrared Reflectance Spectra. Minerals, 11(4), 347. https://doi.org/10.3390/min11040347