
Reduction of Vanadium(V) by Iron(II)-Bearing Minerals



Abstract
1. Introduction
2. Materials and Methods
2.1. Fe Minerals
2.2. Experimental Setup
2.3. X-ray Absorption Spectroscopy Analysis
2.4. V Standards
3. Results and Discussion
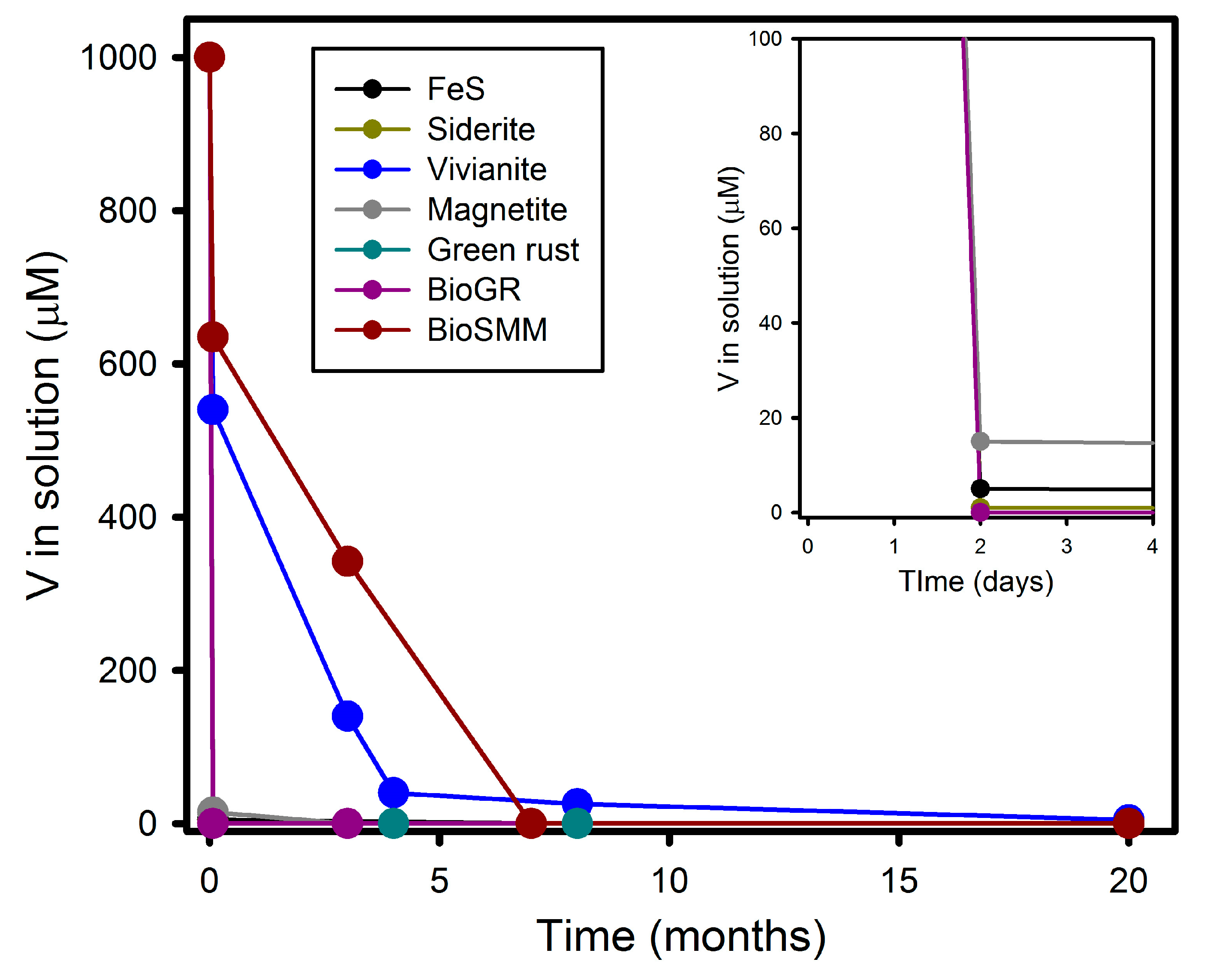
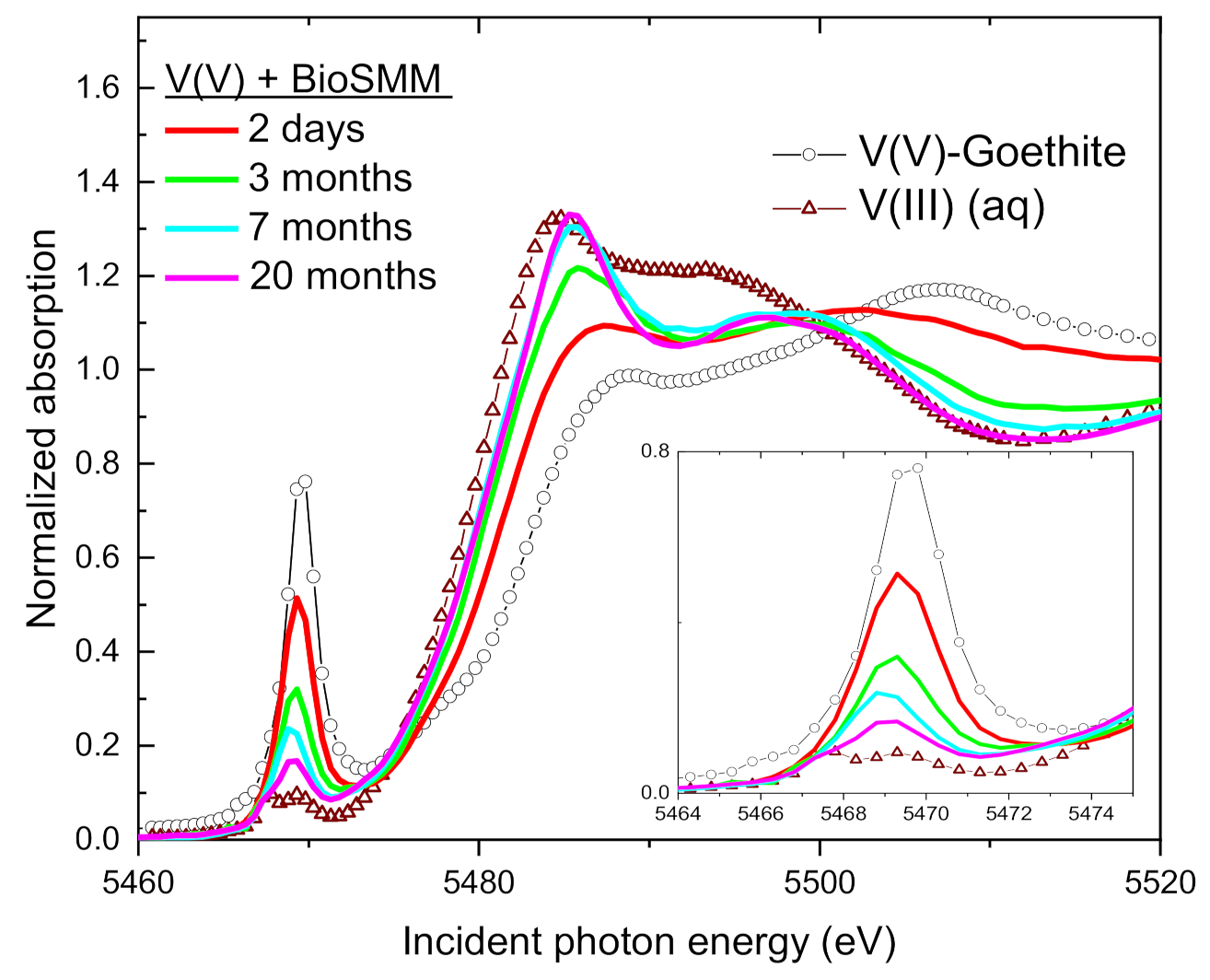
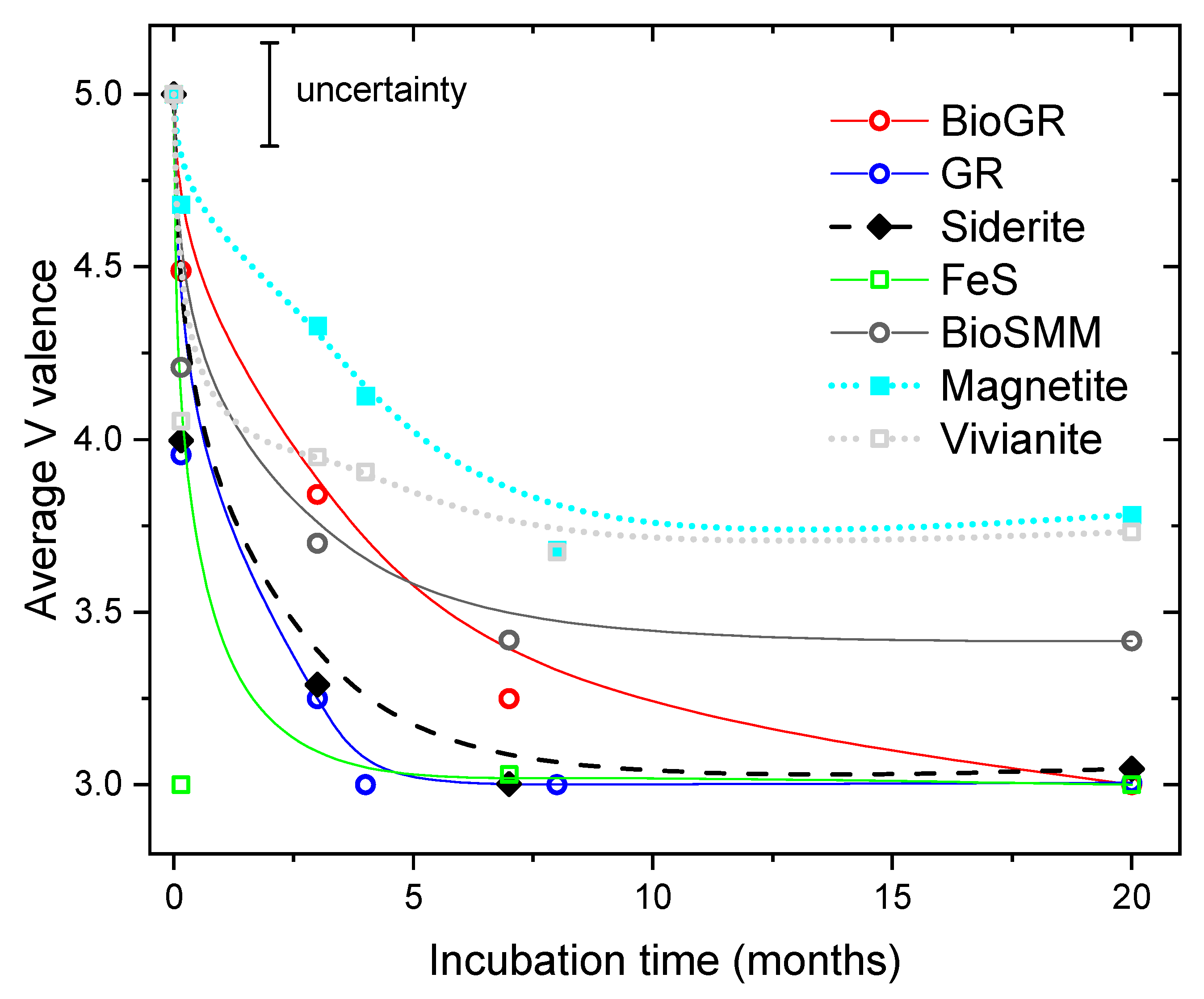
3.1. V(V) Uptake and Extent of Reduction
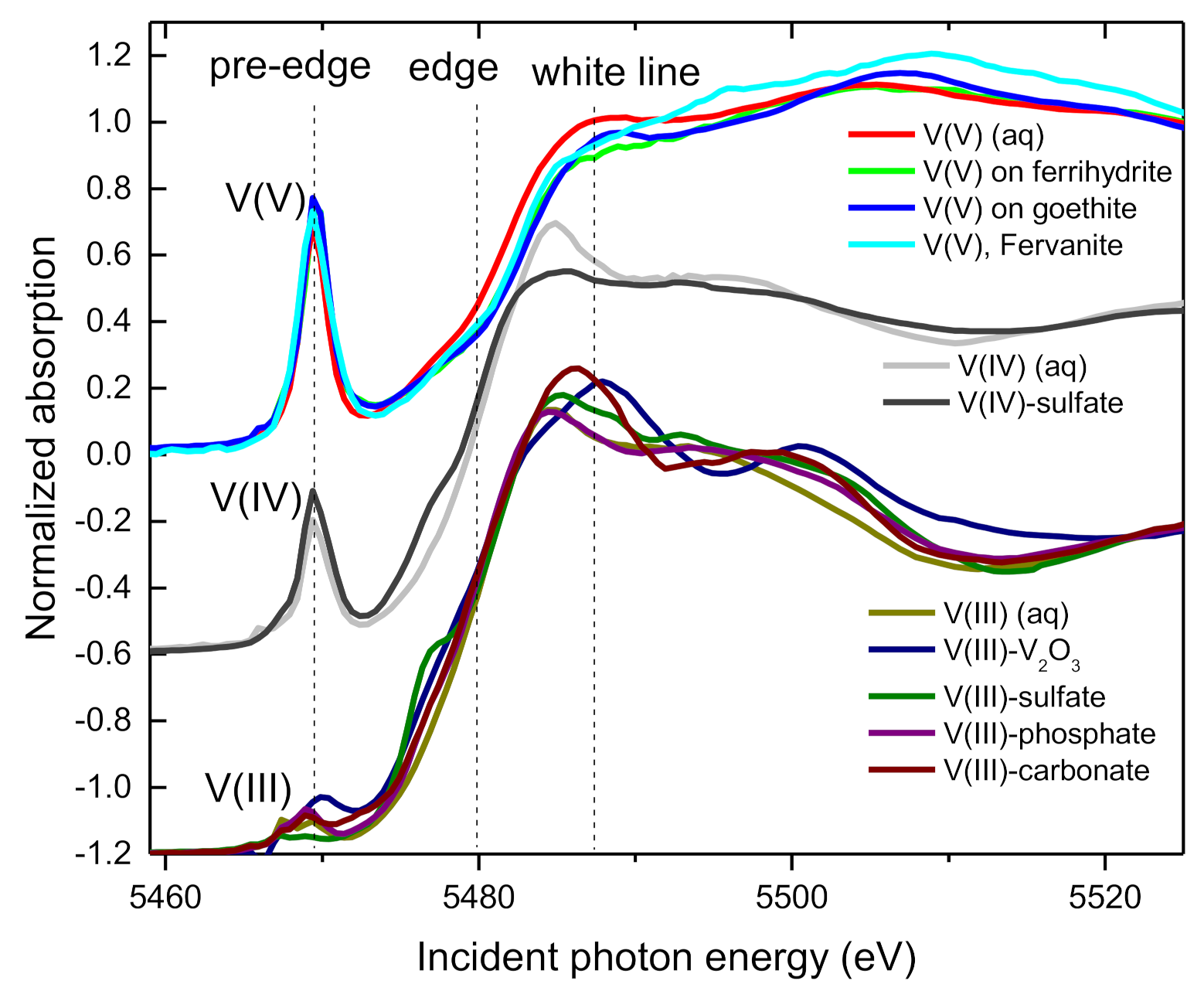
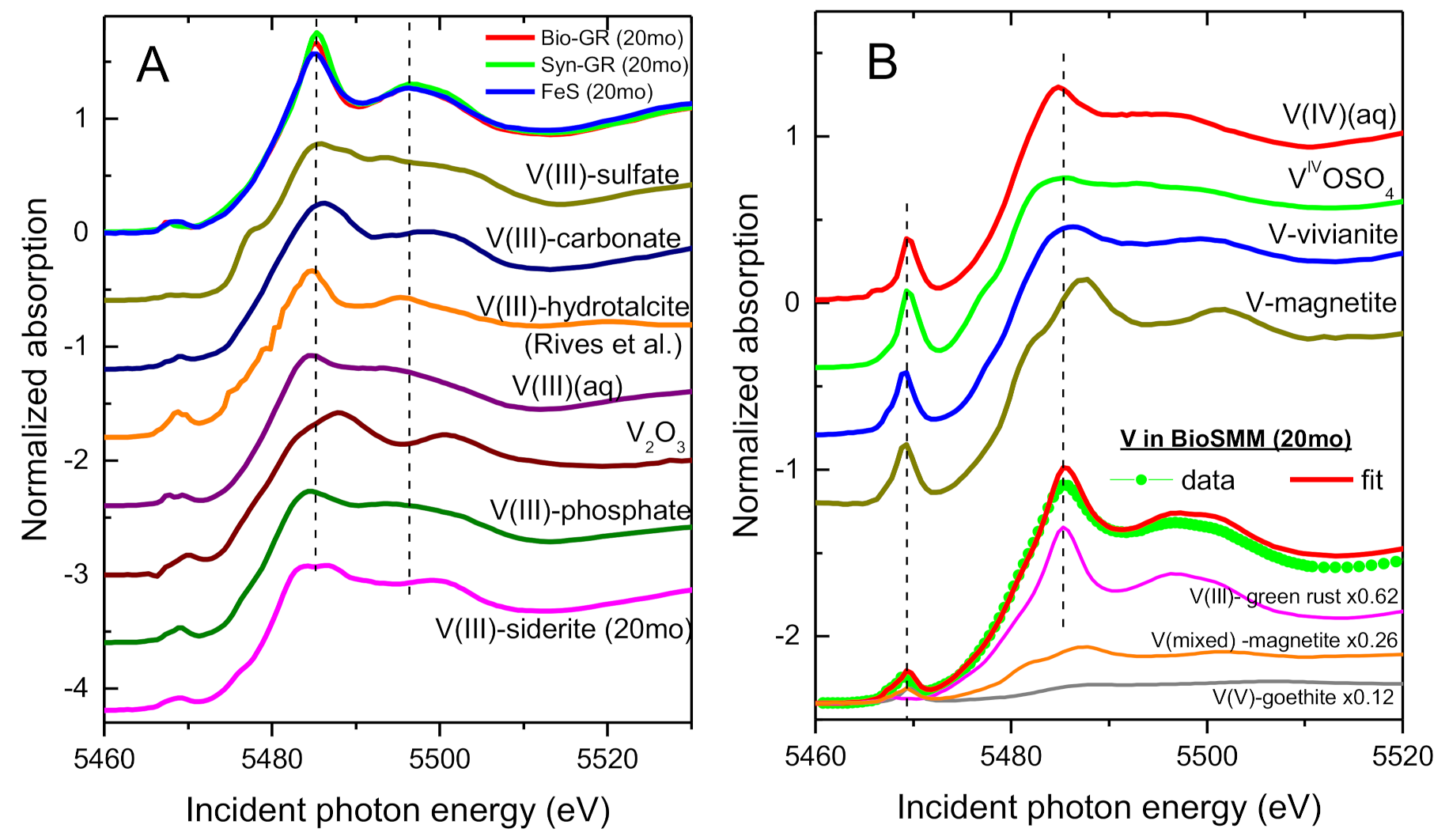
3.2. XANES Spectra of V Standards
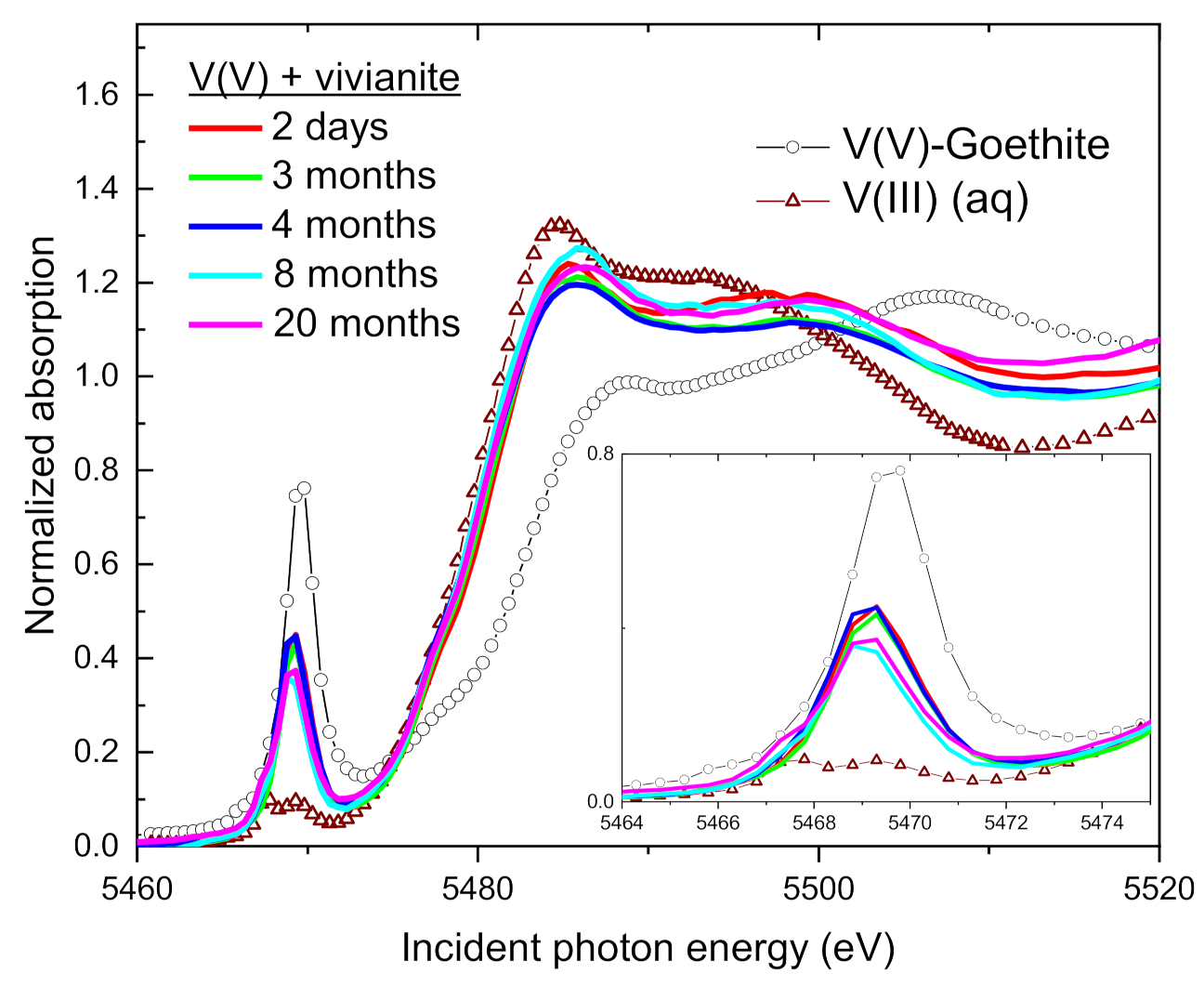
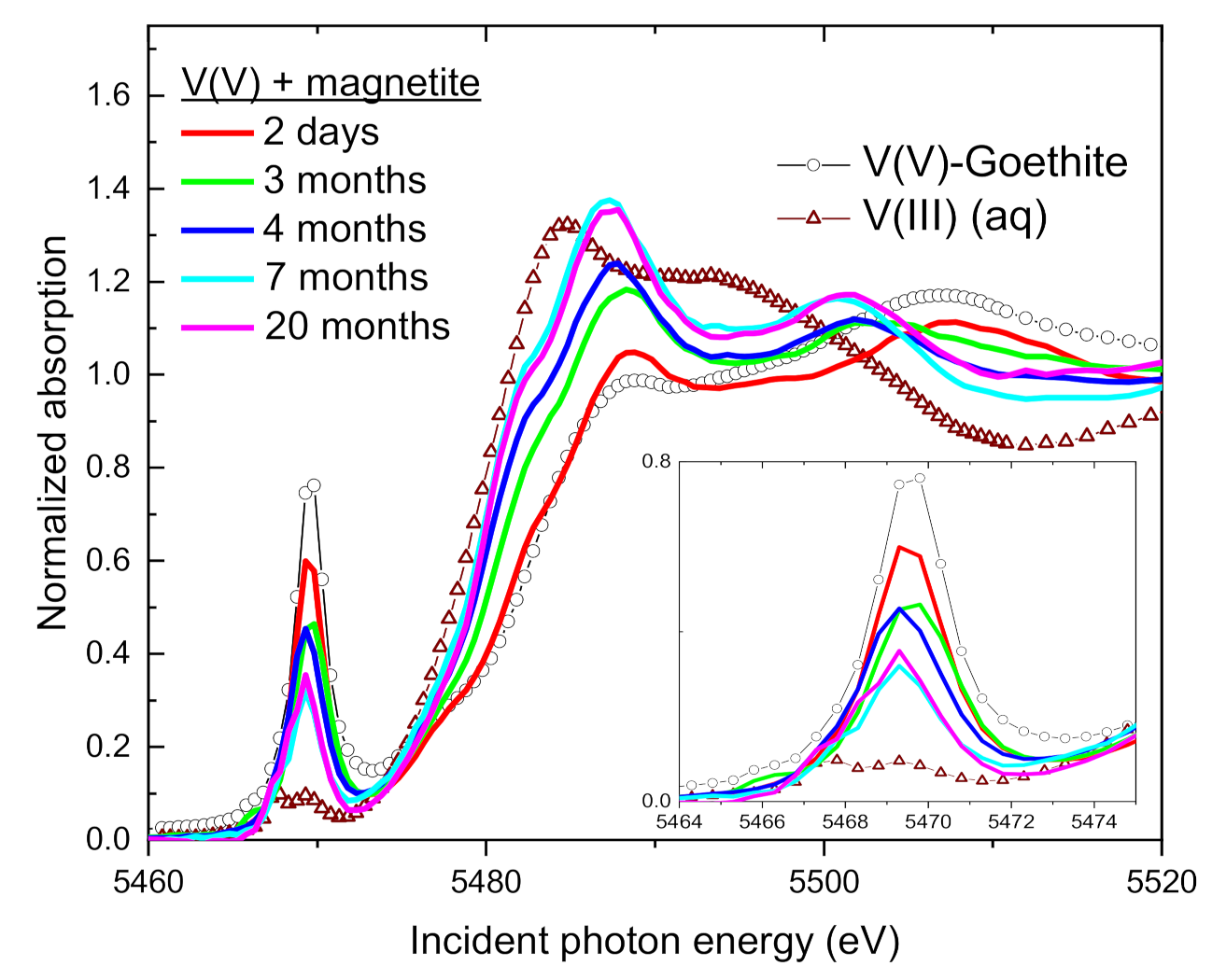
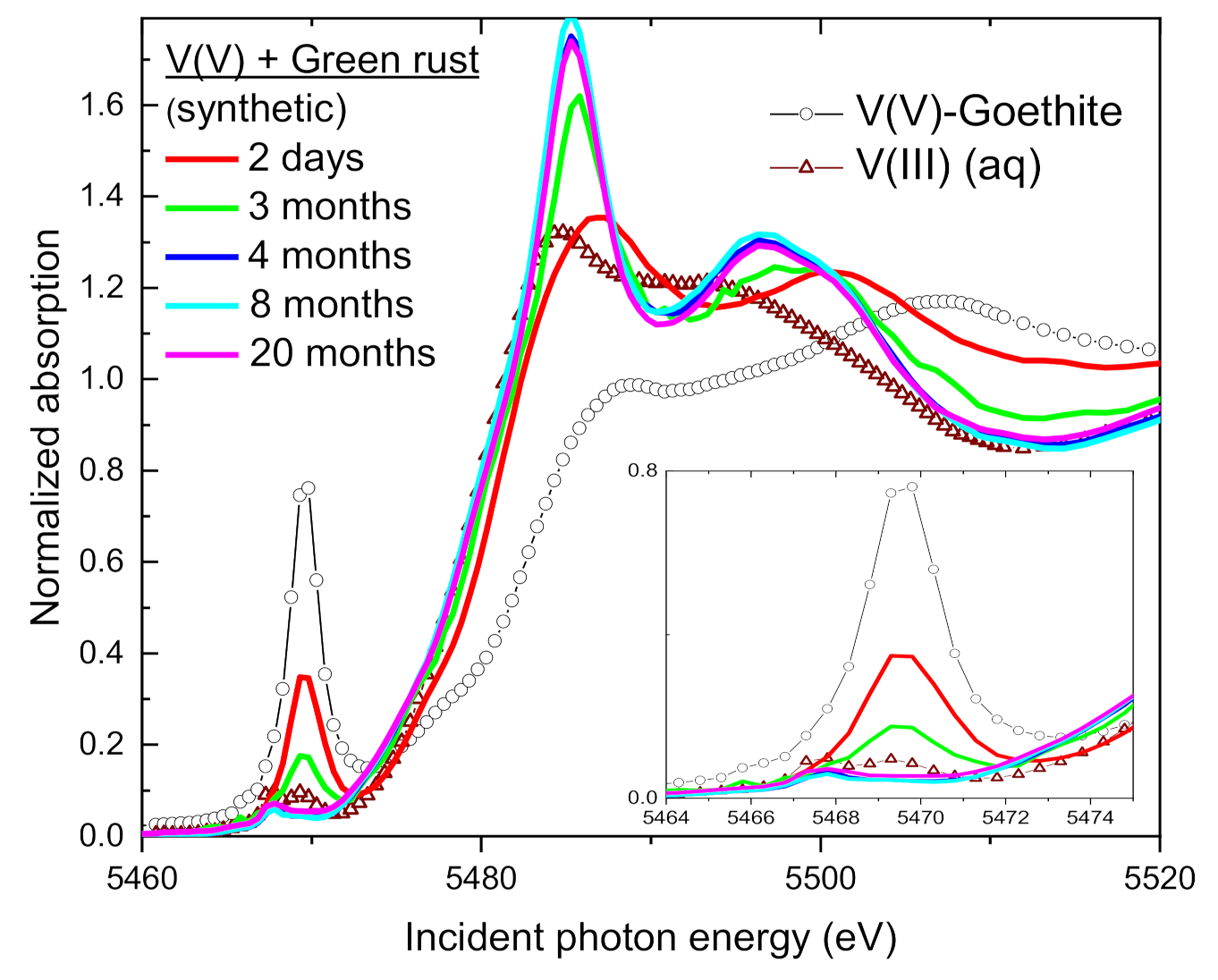
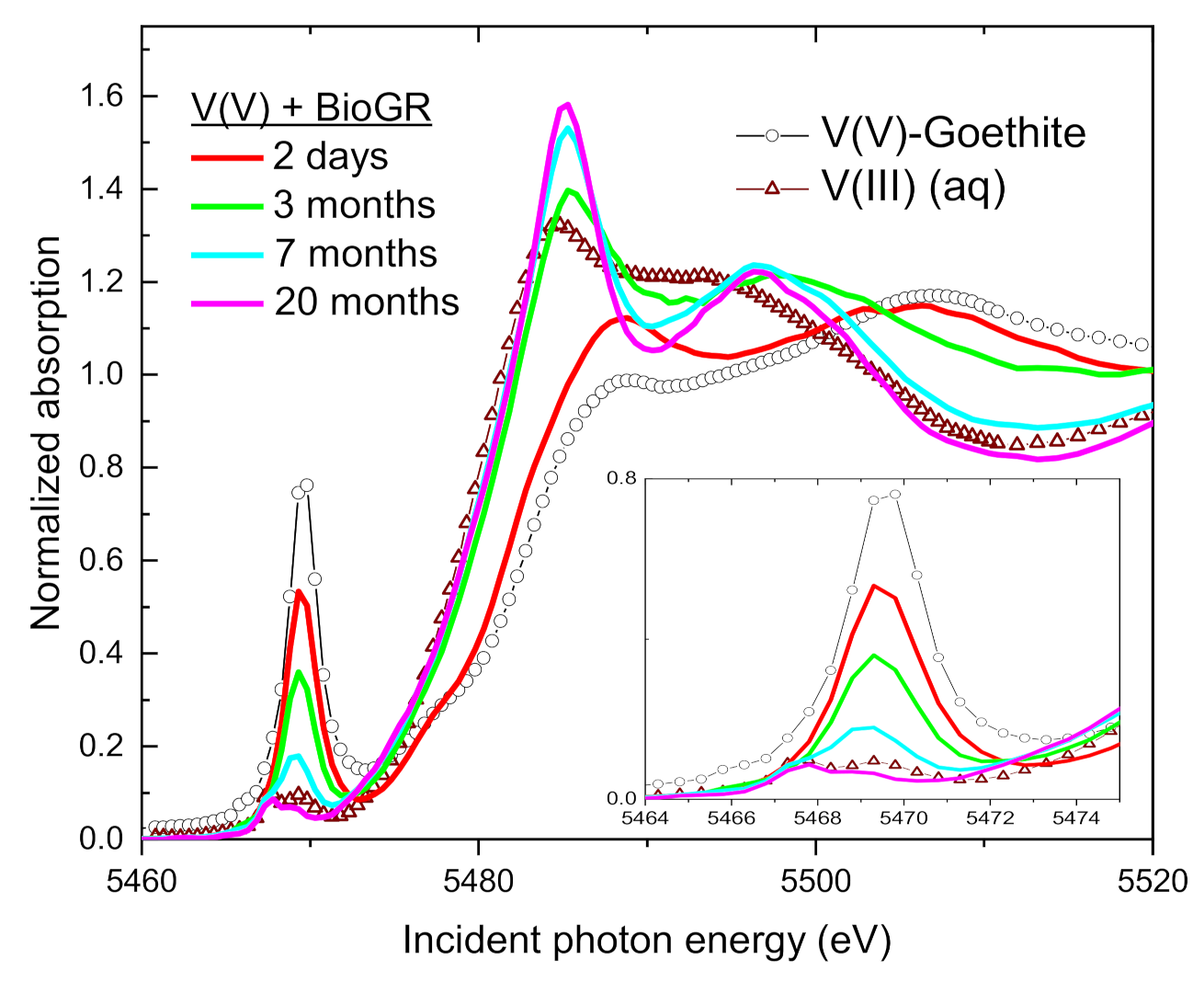
3.3. Change of V Valence with Incubation Time
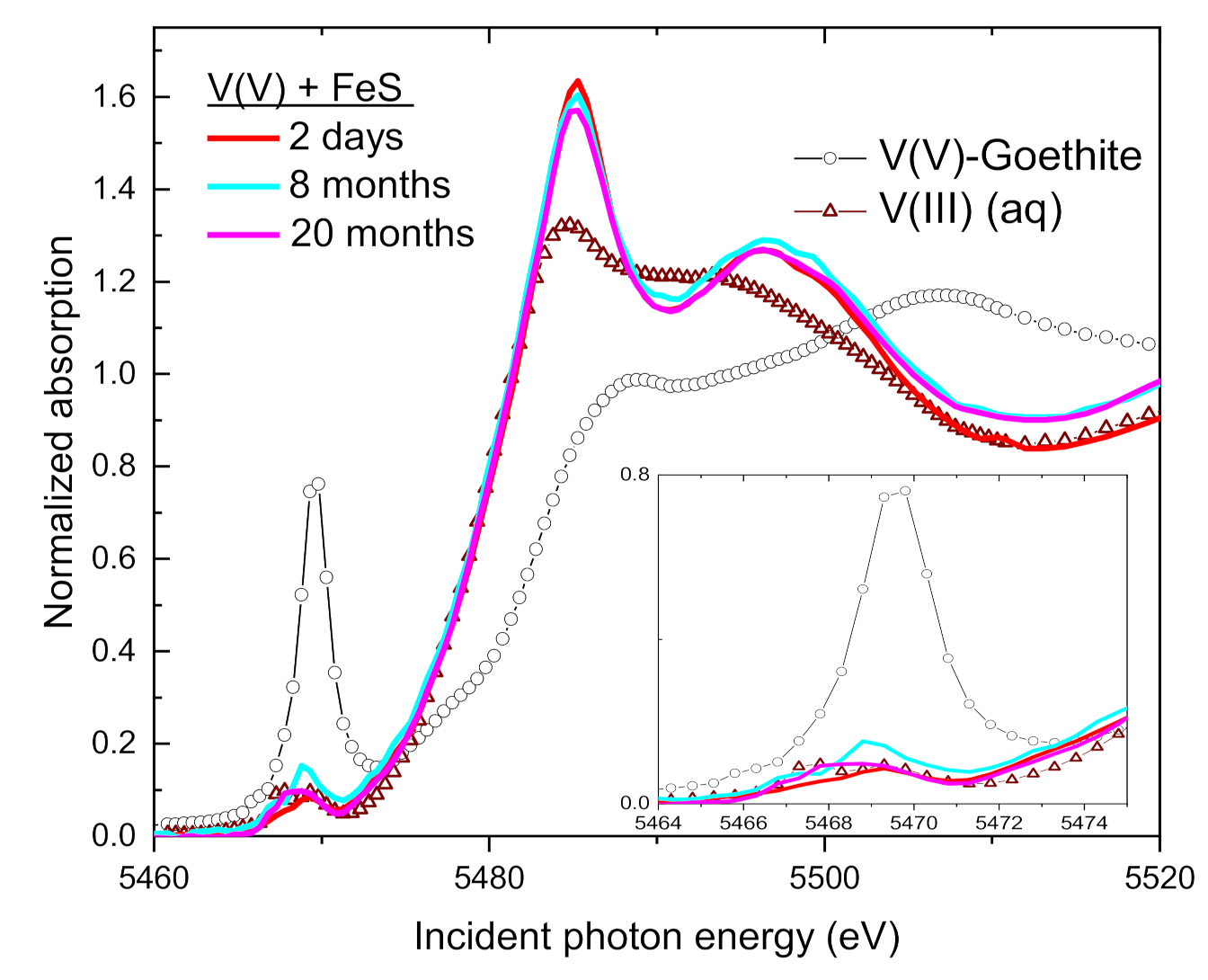
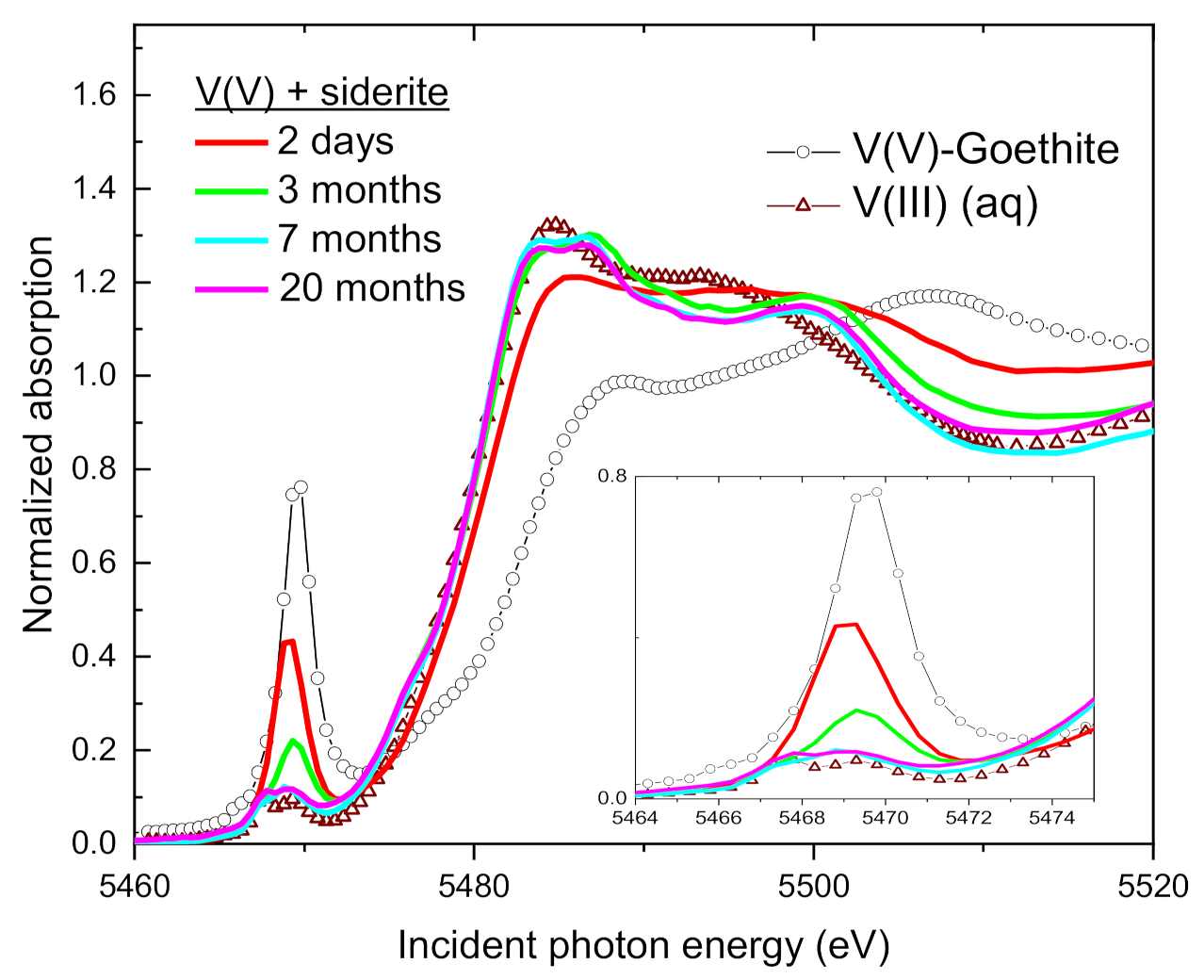
3.4. Speciation of the Reduced V (XANES)
3.5. Environmental Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, J.H.; Huang, F.; Evans, L.; Glasauer, S. Vanadium: Global (bio)geochemistry. Chem. Geol. 2015, 417, 68–89. [Google Scholar] [CrossRef]
- Imtiaz, M.; Rizwan, M.S.; Xiong, S.; Li, H.; Ashraf, M.; Shahzad, S.M.; Shahzad, M.; Rizwan, M.; Tu, S. Vanadium, recent advancements and research prospects: A review. Environ. Int. 2015, 80, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, W.H.; Klein, E.M.; Vengosh, A. Global biogeochemical cycle of vanadium. Proc. Natl. Acad. Sci. USA 2017, 114, E11092–E11100. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.P. Vanadium geochemistry in the biogeosphere –speciation, solid-solution interactions, and ecotoxicity. Appl. Geochem. 2019, 102, 1–25. [Google Scholar] [CrossRef]
- Shaheen, S.M.; Alessi, D.S.; Tack, F.M.G.; Ok, Y.S.; Kim, K.H.; Gustafsson, J.P.; Sparks, D.L.; Rinklebe, J. Redox chemistry of vanadium in soils and sediments: Interactions with colloidal materials, mobilization, speciation, and relevant environmental implications—A review. Adv. Colloid Interface Sci. 2019, 265, 1–13. [Google Scholar] [CrossRef]
- Kabata-Pendias, A. Trace Elements in Soils and Plants, 4th ed.; CRC Press: Boca Raton, FL, USA, 2010; p. 548. [Google Scholar]
- ATSDR. Toxicological Profile for V.; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2012. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp58.pdf (accessed on 12 February 2021).
- Moskalyk, R.R.; Alfantazi, A.M. Processing of vanadium: A review. Miner. Eng. 2003, 16, 793–805. [Google Scholar] [CrossRef]
- Kelley, K.D.; Scott, C.T.; Polyak, D.E.; Kimball, B.E. Vanadium. In Crititcal Mineral Resources of the United States—Economic and Environmental Geology and Prospects for Future Supply: U.S. Geological Survey Professional Paper 1802; Schulz, K.J., DeYoung, J.H., Jr., Seal, R.R., Bradley, D.C., Eds.; United States Geological Survey: Reston, VA, USA, 2017; pp. U1–U36. [Google Scholar] [CrossRef]
- Gardner, C.B.; Carey, A.E.; Lyons, W.B.; Goldsmith, S.T.; McAdams, B.C.; Trierweiler, A.M. Molybdenum, vanadium, and uranium weathering in small mountainous rivers and rivers draining high-standing islands. Geochim. Cosmochim. Acta 2017, 219, 22–43. [Google Scholar] [CrossRef]
- Shiller, A.M.; Mao, L. Dissolved vanadium in rivers: Effects of silicate weathering. Chem. Geol. 2000, 165, 13–22. [Google Scholar] [CrossRef]
- U.S.G.S. Vanadium Mineral Commodity Summary 2020. Available online: https://pubs.usgs.gov/periodicals/mcs2020/mcs2020-vanadium.pdf (accessed on 8 January 2021).
- Watt, J.A.J.; Burke, I.T.; Edwards, R.A.; Malcolm, H.M.; Mayes, W.M.; Olszewska, J.P.; Pan, G.; Graham, M.C.; Heal, K.V.; Rose, N.L.; et al. Vanadium: A re-emerging environmental hazard. Environ. Sci. Technol 2018, 52, 11973–11974. [Google Scholar] [CrossRef]
- Tracey, A.S.; Willsky, G.R.; Takeuchi, E.S. Vanadium: Chemistry, Biochemistry, Pharmacology and Practical Applications; CRC Press: Boca Raton, FL, USA, 2007; p. 250. [Google Scholar] [CrossRef]
- Puttaswamy, N.; Liber, K. Identifying the causes of oil sands coke leachate toxicity to aquatic invertebrates. Environ. Toxicol. Chem. 2011, 30, 2576–2585. [Google Scholar] [CrossRef]
- Larsson, M.A.; Baken, S.; Gustafsson, J.P.; Hadialhejazi, G.; Smolders, E. Vanadium bioavailability and toxicity to soil microorganisms and plants. Environ. Toxicol. Chem. 2013, 32, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Liber, K. Toxicity of aqueous vanadium to zooplankton and phytoplankton species of relevance to the athabasca oil sands region. Ecotoxicol. Environ. Saf. 2017, 137, 1–11. [Google Scholar] [CrossRef]
- Lyalkova, N.N.; Yurkova, N.A. Role of microorganisms in vanadium concentration and dispersion. Geomicrobiol. J. 1992, 10, 15–26. [Google Scholar] [CrossRef]
- Carpentier, W.; Sandra, K.; De Smet, I.; Brigé, A.; De Smet, L.; Van Beeumen, J. Microbial reduction and precipitation of vanadium by Shewanella oneidensis. Appl. Environ. Microbiol. 2003, 69, 3636–3639. [Google Scholar] [CrossRef] [PubMed]
- Bredberg, K.; Karlsson, H.T.; Holst, O. Reduction of vanadium(V) with Acidithiobacillus ferrooxidans and Acidithiobacillus thiooxidans. Bioresour. Technol. 2004, 92, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bernad, I.; Anderson, R.T.; Vrionis, H.A.; Lovley, D.R. Vanadium respiration by Geobacter metallireducens: Novel strategy for in situ removal of vanadium from groundwater. Appl. Environ. Microbiol. 2004, 70, 3091–3095. [Google Scholar] [CrossRef] [PubMed]
- Li, X.S.; Le, X.C. Speciation of vanadium in oilsand coke and bacterial culture by high performance liquid chromatography inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2007, 602, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, Y.; Zou, X.; Luo, X. Study on microbial reduction of vanadium metallurgical waste water. Hydrometallurgy 2009, 99, 13–17. [Google Scholar] [CrossRef]
- van Marwijk, J.; Opperman, D.J.; Piater, L.A.; van Heerden, E. Reduction of vanadium(V) by Enterobacter cloacae EV-SA01 isolated from a South African deep gold mine. Biotechnol. Lett. 2009, 31, 845–849. [Google Scholar] [CrossRef]
- Ogg, C.D.; Patel, B.K. Caloramator mitchellensis sp. nov., a thermoanaerobe isolated from the geothermal waters of the Great Artesian Basin of Australia, and emended description of the genus Caloramator. Int. J. Syst. Evol. Microbiol. 2011, 61, 644–653. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, H.; Zhao, L.; McCarrick, R.; Agrawal, A. Microbial reduction and precipitation of vanadium by mesophilic and thermophilic methanogens. Chem. Geol. 2014, 370, 29–39. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, B.; Li, S.; Yang, M.; Yin, C. Simultaneous microbial reduction of vanadium (V) and chromium (VI) by Shewanella loihica PV-4. Bioresour. Technol. 2017, 227, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Coker, V.S.; van der Laan, G.; Telling, N.D.; Lloyd, J.R.; Byrne, J.M.; Arenholz, E.; Pattrick, R.A.D. Bacterial production of vanadium ferrite spinel (Fe,V)3O4 nanoparticles. Mineral. Mag. 2020, 84, 554–562. [Google Scholar] [CrossRef]
- Sun, X.; Qiu, L.; Kolton, M.; Haggblom, M.; Xu, R.; Kong, T.; Gao, P.; Li, B.; Jiang, C.; Sun, W. V(V) reduction by Polaromonas spp. in vanadium mine tailings. Environ. Sci. Technol. 2020, 54, 14442–14454. [Google Scholar] [CrossRef]
- Yelton, A.P.; Williams, K.H.; Fournelle, J.; Wrighton, K.C.; Handley, K.M.; Banfield, J.F. Vanadate and acetate biostimulation of contaminated sediments decreases diversity, selects for specific taxa, and decreases aqueous V5+ concentration. Environ. Sci. Technol. 2013, 47, 6500–6509. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xia, S.; Zhou, L.; Zhang, Z.; Rittmann, B.E. Bioreduction of vanadium (V) in groundwater by autohydrogentrophic bacteria: Mechanisms and microorganisms. J. Environ. Sci. 2015, 30, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hao, L.; Tian, C.; Yuan, S.; Feng, C.; Ni, J.; Borthwick, A.G. Microbial reduction and precipitation of vanadium (V) in groundwater by immobilized mixed anaerobic culture. Bioresour. Technol. 2015, 192, 410–417. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, B.; Yuan, H.; Cheng, Y.; Wang, S.; He, Z. Microbial reduction of vanadium (V) in groundwater: Interactions with coexisting common electron acceptors and analysis of microbial community. Environ. Pollut. 2017, 231, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhang, B.; Li, J.; Lv, Q.; Wang, S.; Gu, Q. Enhanced vanadium (V) reduction and bioelectricity generation in microbial fuel cells with biocathode. J. Power Sources 2017, 359, 379–383. [Google Scholar] [CrossRef]
- Lai, C.Y.; Dong, Q.Y.; Chen, J.X.; Zhu, Q.S.; Yang, X.; Chen, W.D.; Zhao, H.P.; Zhu, L. Role of extracellular polymeric substances in a methane based membrane biofilm reactor reducing vanadate. Environ. Sci. Technol. 2018, 52, 10680–10688. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, B.; Diao, M.; Shi, J.; Jiang, Y.; Cheng, Y.; Liu, H. Enhancement of synchronous bio-reductions of vanadium (V) and chromium (VI) by mixed anaerobic culture. Environ. Pollut. 2018, 242, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qiu, R.; Lu, L.; Chen, X.; He, C.; Lu, J.; Ren, Z.J. Autotrophic vanadium(V) bioreduction in groundwater by elemental sulfur and zerovalent iron. Environ. Sci. Technol. 2018, 52, 7434–7442. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, S.; Diao, M.; Fu, J.; Xie, M.; Shi, J.; Liu, Z.; Jiang, Y.; Cao, X.; Borthwick, A.G.L. Microbial community responses to vanadium distributions in mining geological environments and bioremediation assessment. J. Geophys. Res. Biogeosci. 2019, 124, 601–615. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, Y.; Zuo, K.; He, C.; Dai, Y.; Ren, Z.J. Microbial vanadate and nitrate reductions coupled with anaerobic methane oxidation in groundwater. J. Hazard. Mater. 2020, 382, 121228. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, B.; Lu, J.; Qiu, R. A newly discovered function of nitrate reductase in chemoautotrophic vanadate transformation by natural mackinawite in aquifer. Water Res. 2021, 189, 116664. [Google Scholar] [CrossRef]
- Lovley, D.R.; Stolz, J.F.; Nord, G.L., Jr.; Phillips, E.J.P. Anaerobic production of magnetite by a dissimilatory iron-reducing microorganism. Nature 1987, 330, 252–254. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Zachara, J.M.; Kennedy, D.W.; Dong, H.; Onstott, T.C.; Hinman, N.W.; Li, S.-M. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim. Cosmochim. Acta 1998, 62, 3239–3257. [Google Scholar] [CrossRef]
- Ona-Nguema, G.; Abdelmoula, M.; Jorand, F.; Benali, O.; Géhin, A.; Block, J.-C.; Génin, J.-M.R. Iron(II,III) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction. Environ. Sci. Technol. 2002, 36, 16–20. [Google Scholar] [CrossRef]
- Glasauer, S.; Weidler, P.G.; Langley, S.; Beveridge, T.J. Controls on Fe reduction and mineral formation by a subsurface bacterium. Geochim. Cosmochim. Acta 2003, 67, 1277–1288. [Google Scholar] [CrossRef]
- Roh, Y.; Zhang, C.-L.; Vali, H.; Lauf, R.J.; Zhou, J.; Phelps, T.J. Biogeochemical and environmental factors in Fe biomineralization: Magnetite and siderite formation. Clays Clay Miner. 2003, 51, 83–95. [Google Scholar] [CrossRef]
- Kukkadapu, R.K.; Zachara, J.M.; Fredrickson, J.K.; Kennedy, D.W.; Dohnalkova, A.C.; Mccready, D.E. Ferrous hydroxy carbonate is a stable transformation product of biogenic magnetite. Am. Mineral. 2005, 90, 510–515. [Google Scholar] [CrossRef][Green Version]
- Behrends, T.; Van Cappellen, P. Transformation of hematite into magnetite during dissimilatory iron reduction-conditions and mechanisms. Geomicrobiol. J. 2007, 24, 403–416. [Google Scholar] [CrossRef]
- Boyanov, M.I.; O’Loughlin, E.J.; Kemner, K.M. Iron phase transformations resulting from the respiration of Shewanella putrefaciens on a mixed mineral phase. J. Phys. Conf. Ser. 2009, 190, 1–4. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Gorski, C.A.; Flynn, T.M.; Scherer, M.M. Electron donor utilization and secondary mineral formation during the bioreduction of lepidocrocite by Shewanella putrefaciens CN32. Minerals 2019, 9, 434. [Google Scholar] [CrossRef]
- Dong, Y.; Sanford, R.A.; Boyanov, M.I.; Flynn, T.M.; O’Loughlin, E.J.; Kemner, K.M.; George, S.; Fouke, K.E.; Li, S.; Huang, D.; et al. Controls on iron reduction and biomineralization over broad environmental conditions as suggested by the Firmicutes Orenia metallireducens strain Z6. Environ. Sci. Technol. 2020, 54, 10128–10140. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Gorski, C.A.; Scherer, M.M.; Kemner, K.M. Effects of Fe(III) oxide mineralogy and phosphate on Fe(II) secondary mineral formation during microbial iron reduction. Minerals 2021, 11, 149. [Google Scholar] [CrossRef]
- Neal, A.L.; Techkarnjanaruk, S.; Dohnalkova, A.; Mccready, D.E.; Peyton, B.M.; Geesey, G.G. Iron sulfides and sulfur species produced at hematite surfaces in the presence of sulfate-reducing bacteria. Geochim. Cosmochim. Acta 2001, 65, 223–235. [Google Scholar] [CrossRef]
- Vaughan, D.J.; Lennie, A.R. The iron sulfide minerals: Their chemistry and role in nature. Sci. Prog. 1991, 75, 371–388. [Google Scholar]
- Herbert, R.B., Jr.; Benner, S.G.; Pratt, A.R.; Blowes, D.W. Surface chemistry and morphology of poorly crystalline iron sulfides precipitated in media containing sulfate-reducing bacteria. Chem. Geol. 1998, 144, 87–97. [Google Scholar] [CrossRef]
- Benning, L.G.; Wilkin, R.T.; Konhauser, K.O. Iron monosulfide stability: Experiments with sulfate reducing bacteria. In Geochemistry of the Earth’s Surface; Ármannsson, H., Ed.; Brookfield A. A. Balkema: Rotterdam, The Netherlands, 1999; pp. 429–432. [Google Scholar]
- Donald, R.; Southam, G. Low temperature anaerobic bacterial diagenesis of ferrous monosulfide to pyrite. Geochim. Cosmochim. Acta 1999, 63, 2019–2023. [Google Scholar] [CrossRef]
- Flynn, T.M.; O’Loughlin, E.J.; Mishra, B.; DiChristina, T.J.; Kemner, K.M. Sulfur-mediated electron shuttling during bacterial iron reduction. Science 2014, 344, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Boyanov, M.I.; Antonopoulos, D.A.; Brulc, J.M.; Johnston, E.R.; Skinner, K.A.; Kemner, K.M.; O’Loughlin, E.J. Effects of dissimilatory sulfate reduction on FeIII (hydr)oxide reduction and microbial community development. Geochim. Cosmochim. Acta 2014, 129, 4570–4576. [Google Scholar] [CrossRef]
- White, A.F.; Peterson, M.L. Reduction of aqueous transition metal species on the surfaces of Fe(II)-containing oxides. Geochim. Cosmochim. Acta 1996, 60, 3799–3814. [Google Scholar] [CrossRef]
- Vessey, C.J.; Lindsay, M.B.J. Aqueous vanadate removal by iron(II)-bearing phases under anoxic conditions. Environ. Sci. Technol. 2020, 54, 4006–4015. [Google Scholar] [CrossRef]
- Johnson, C.R.; Antonopoulos, D.A.; Boyanov, M.I.; Flynn, T.M.; Koval, J.C.; Kemner, K.M.; O’Loughlin, E.J. Reduction of Sb(V) by coupled biotic-abiotic processes under sulfidogenic conditions. Heliyon 2021, 7. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Flynn, T.M.; Gorski, C.; Hofmann, S.M.; McCormick, M.L.; Scherer, M.M.; Kemner, K.M. Effects of bound phosphate on the bioreduction of lepidocrocite (γ-FeOOH) and maghemite (γ-Fe2O3) and formation of secondary minerals. Environ. Sci. Technol. 2013, 47, 9157–9166. [Google Scholar] [CrossRef]
- Segre, C.U.; Leyarovska, N.E.; Chapman, L.D.; Lavender, W.M.; Plag, P.W.; King, A.S.; Kropf, A.J.; Bunker, B.A.; Kemner, K.M.; Dutta, P.; et al. The MRCAT insertion device beamline at the Advanced Photon Source. In Synchrotron Radiation Instrumentation: Eleventh U.S. National Conference. Stanford, CA, USA, 13–15 October 1999; Pianetta, P.A., Arthur, J.R., Brennan, S., Eds.; American Institute of Physics: New York, NY, USA, 2000; Volume CP5321, pp. 419–422. [Google Scholar]
- O’Loughlin, E.J.; Kelly, S.D.; Csencsits, R.; Cook, R.E.; Kemner, K.M. Reduction of uranium(VI) by mixed iron(II)/iron(III) hydroxide (green rust): Formation of UO2 nanoparticles. Environ. Sci. Technol. 2003, 37, 721–727. [Google Scholar] [CrossRef]
- Newville, M.; Livinš, P.; Yacoby, Y.; Rehr, J.J.; Stern, E.A. Near-edge x-ray absorption fine structure of Pb: A comparison of theory and experiment. Phys. Rev. B 1993, 47, 14126–14131. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Zatko, D.A.; Kratochvil, B. Vanadium(III) sulfate as a reducing agent for determination of perchlorate. Anal. Chem. 1965, 37, 1560–1562. [Google Scholar] [CrossRef]
- Poizot, P.; Baudrin, E.; Laruelle, S.; Dupont, L.; Touboul, M.; Tarascon, J.M. Low temperature synthesis and electrochemical performance of crystallized FeVO4·1.1H2O. Solid State Ion. 2000, 138, 31–40. [Google Scholar] [CrossRef]
- Peacock, C.L.; Sherman, D.M. Vanadium(V) adsorption onto goethite (α-FeOOH) at pH 1.5 to 12: A surface complexation model based on ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 2004, 68, 1723–1733. [Google Scholar] [CrossRef]
- Chaurand, P.; Rose, J.; Briois, V.; Salome, M.; Proux, O.; Nassif, V.; Olivi, L.; Susini, J.; Hazemann, J.L.; Bottero, J.Y. New methodological approach for the vanadium K-edge X-ray absorption near-edge structure interpretation: Application to the speciation of vanadium in oxide phases from steel slag. J. Phys. Chem. B 2007, 111, 5101–5110. [Google Scholar] [CrossRef]
- Duchesne, M.A.; Nakano, J.; Hu, Y.; MacLennan, A.; Bennett, J.; Nakano, A.; Hughes, R.W. Synchrotron-based X-ray absorption spectroscopy study of vanadium redox speciation during petroleum coke combustion and gasification. Fuel 2018, 227, 279–288. [Google Scholar] [CrossRef]
- Haskel, D.; Islam, Z.; Lang, J.; Kmety, C.; Srajer, G.; Pokhodnya, K.I.; Epstein, A.J.; Miller, J.S. Local structural order in the disordered vanadium tetracyanoethylene room-temperature molecule-based magnet. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Levina, A.; McLeod, A.I.; Lay, P.A. Vanadium speciation by XANES spectroscopy: A three-dimensional approach. Chemistry 2014, 20, 12056–12060. [Google Scholar] [CrossRef]
- Wong, J.; Lytle, F.W.; Messmer, R.P.; Maylotte, D.H. K-edge absorption spectra of selected vanadium compounds. Phys. Rev. B 1984, 30, 5596–5610. [Google Scholar] [CrossRef]
- Bennett, W.W.; Lombi, E.; Burton, E.D.; Johnston, S.G.; Kappen, P.; Howard, D.L.; Canfield, D.E. Synchrotron X-ray spectroscopy for investigating vanadium speciation in marine sediment: Limitations and opportunities. J. Anal. At. Spectrom. 2018, 33, 1689–1699. [Google Scholar] [CrossRef]
- Frank, P.; Carlson, R.M.K.; Carlson, E.J.; Hodgson, K.O. The vanadium environment in blood cells of Ascidia ceratodes is divergent at all organismal levels: An XAS and EPR spectroscopic study. J. Inorg. Biochem. 2003, 94, 59–71. [Google Scholar] [CrossRef]
- Larsson, M.A.; D’Amato, M.; Cubadda, F.; Raggi, A.; Öborn, I.; Kleja, D.B.; Gustafsson, J.P. Long-term fate and transformations of vanadium in a pine forest soil with added converter lime. Geoderma 2015, 259–260, 271–278. [Google Scholar] [CrossRef]
- Wisawapipat, W.; Kretzschmar, R. Solid phase speciation and solubility of vanadium in highly weathered soils. Environ. Sci. Technol. 2017, 51, 8254–8262. [Google Scholar] [CrossRef] [PubMed]
- Nedrich, S.M.; Chappaz, A.; Hudson, M.L.; Brown, S.S.; Burton, G.A., Jr. Biogeochemical controls on the speciation and aquatic toxicity of vanadium and other metals in sediments from a river reservoir. Sci. Total Environ. 2018, 612, 313–320. [Google Scholar] [CrossRef]
- Gerke, T.L.; Scheckel, K.G.; Schock, M.R. Identification and distribution of vanadinite (Pb5(V5+O4)3Cl) in lead pipe corrosion by-products. Environ. Sci. Technol. 2009, 43, 4412–4418. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, J.A.; Lindsay, M.B.J. Vanadium geochemistry of oil sands fluid petroleum coke. Environ. Sci. Technol. 2017, 51, 3102–3109. [Google Scholar] [CrossRef]
- Larsson, M.A.; Persson, I.; Sjöstedt, C.; Gustafsson, J.P. Vanadate complexation to ferrihydrite: X-ray absorption spectroscopy and CD-MUSIC modelling. Environ. Chem. 2017, 14. [Google Scholar] [CrossRef]
- Schindler, M.; Hawthorne, F.C. Schubnelite, [Fe3+(V5+O4)(H2O)], a novel heteropolyhedral framework mineral. Am. Mineral. 1999, 84, 665–668. [Google Scholar] [CrossRef]
- Hawthorne, F.C.; Schindler, M.; Grice, J.D.; Haynes, P. Orthominasragrite, V4+O (SO4) (H2O)5, a new mineral species from Temple Mountain, Emery County, Utah, U.S.A. Can. Mineral. 2001, 39, 1325–1331. [Google Scholar] [CrossRef]
- Wanty, R.B.; Goldhaber, M.B. Thermodynamics and kinetics of reactions involving vanadium in natural systems: Accumulation of vanadium in sedimentary rocks. Geochim. Cosmochim. Acta 1992, 56, 1471–1483. [Google Scholar] [CrossRef]
- Chen, G.; Liu, H. Understanding the reduction kinetics of aqueous vanadium(V) and transformation products using rotating ring-disk electrodes. Environ. Sci. Technol. 2017, 51, 11643–11651. [Google Scholar] [CrossRef]
- Lee, W.; Batchelor, B. Reductive capacity of natural reductants. Environ. Sci. Technol. 2003, 37, 535–541. [Google Scholar] [CrossRef]
- Christiansen, B.C.; Geckeis, H.; Marquardt, C.M.; Bauer, A.; Römer, J.; Wiss, T.; Schild, D.; Stipp, S.L.S. Neptunyl (NpO2+) interaction with green rust, GRNa,SO4. Geochim. Cosmochim. Acta 2011, 75, 1216–1226. [Google Scholar] [CrossRef]
- Heasman, D.M.; Sherman, D.M.; Ragnarsdottir, K.V. The reduction of aqueous Au3+ by sulfide minerals and green rust phases. Am. Mineral. 2003, 88, 725–738. [Google Scholar] [CrossRef]
- Myneni, S.C.B.; Tokunaga, T.K.; Brown, G.E., Jr. Abiotic selenium redox transformations in the presence of Fe(II,III) oxides. Science 1997, 278, 1106–1109. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Kelly, S.D.; Kemner, K.M.; Csencsits, R.; Cook, R.E. Reduction of AgI, AuIII, CuII, and HgII by FeII/FeIII hydroxysulfate green rust. Chemosphere 2003, 53, 437–446. [Google Scholar] [CrossRef]
- Pepper, S.E.; Bunker, D.J.; Bryan, N.D.; Livens, F.R.; Charnock, J.M.; Pattrick, R.A.D.; Collison, D. Treatment of radioactive wastes: An X-ray adsorption spectroscopy study of the treatment of technetium with green rust. J. Colloid Interface Sci. 2003, 268, 408–412. [Google Scholar] [CrossRef]
- Refait, P.; Simon, L.; Génin, J.-M.R. Reduction of SeO42- anions and anoxic formation of iron(II)-iron(III) hydroxy-selenate green rust. Environ. Sci. Technol. 2000, 34, 819–825. [Google Scholar] [CrossRef]
- Williams, A.G.B.; Scherer, M.M. Kinetics of Cr(VI) reduction by carbonate green rust. Environ. Sci. Technol. 2001, 35, 3488–3494. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.J.; Kelly, S.D.; Kemner, K.M. XAFS investigation of the interactions of UVI with secondary mineralization products from the bioreduction of FeIII oxides. Environ. Sci. Technol. 2010, 44, 1656–1661. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Guldberg, S.; Erbs, M.; Bender Koch, C. Kinetics of nitrate reduction by green rusts—Effects of interlayer anion and Fe(II):Fe(III) ratio. Appl. Clay Sci. 2001, 18, 81–91. [Google Scholar] [CrossRef]
- Bond, D.L.; Fendorf, S. Kinetics and structural constraints of chromate reduction by green rusts. Environ. Sci. Technol. 2003, 37, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Latta, D.E.; Boyanov, M.I.; Kemner, K.M.; O’Loughlin, E.J.; Scherer, M.M. Reaction of uranium(VI) with green rusts: Effect of interlayer anion. Curr. Inorg. Chem. 2015, 5, 156–168. [Google Scholar] [CrossRef]
- Zegeye, A.; Etique, M.; Carteret, C.; Ruby, C.; Schaaf, P.; Francius, G. Origin of the differential nanoscale reactivity of biologically and chemically formed green rust crystals investigated by chemical force spectroscopy. J. Phys. Chem. C 2014, 118, 5978–5987. [Google Scholar] [CrossRef]
- Remy, P.P.; Etique, M.; Hazotte, A.A.; Sergent, A.S.; Estrade, N.; Cloquet, C.; Hanna, K.; Jorand, F.P. Pseudo-first-order reaction of chemically and biologically formed green rusts with Hg and CHNO: Effects of pH and stabilizing agents (phosphate, silicate, polyacrylic acid, and bacterial cells). Water Res. 2015, 70C, 266–278. [Google Scholar] [CrossRef]
- Yan, S.; Boyanov, M.I.; Mishra, B.; Kemner, K.M.; O’Loughlin, E.J. U(VI) reduction by biogenic and abiotic hydroxycarbonate green rusts: Impacts on U(IV) speciation and stability over time. Environ. Sci. Technol. 2018, 52, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- Gorski, C.A.; Nurmi, J.T.; Tratnyek, P.G.; Hofstetter, T.B.; Scherer, M.M. Redox behavior of magnetite: Implications for contaminant reduction. Environ. Sci. Technol. 2010, 44, 55–60. [Google Scholar] [CrossRef]
- Latta, D.E.; Gorski, C.A.; Boyanov, M.I.; O’Loughlin, E.J.; Kemner, K.M.; Scherer, M.M. Influence of magnetite stoichiometry on UVI reduction. Environ. Sci. Technol. 2012, 46, 778–786. [Google Scholar] [CrossRef]
- Pasakarnis, T.S.; Boyanov, M.I.; Kemner, K.M.; Mishra, B.; O’Loughlin, E.J.; Parkin, G.; Scherer, M.M. Influence of chloride and Fe(II) content on the reduction of Hg(II) by magnetite. Environ. Sci. Technol. 2013, 47, 6987–6994. [Google Scholar] [CrossRef]
- Veeramani, H.; Alessi, D.S.; Suvorova, E.I.; Lezama-Pacheco, J.S.; Stubbs, J.E.; Sharp, J.O.; Dippon, U.; Kappler, A.; Bargar, J.R.; Bernier-Latmani, R. Products of abiotic U(VI) reduction by biogenic magnetite and vivianite. Geochim. Cosmochim. Acta 2011, 75, 2512–2528. [Google Scholar] [CrossRef]
- Bae, S.; Sihn, Y.; Kyung, D.; Yoon, S.; Eom, T.; Kaplan, U.; Kim, H.; Schafer, T.; Han, S.; Lee, W. Molecular identification of Cr(VI) removal mechanism on vivianite surface. Environ. Sci. Technol. 2018, 52, 10647–10656. [Google Scholar] [CrossRef]
- Etique, M.; Bouchet, S.; Byrne, J.M.; ThomasArrigo, L.K.; Kaegi, R.; Kretzschmar, R. Mercury reduction by nanoparticulate vivianite. Environ. Sci. Technol. 2021. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.L.; Kim, H.S.; Bouwer, E.J.; Adriaens, P. Abiotic transformation of chlorinated solvents as a consequence of microbial iron reduction: An investigation of the role of biogenic magnetite in mediating reductive dechlorination. In Proceedings of Thirtieth Mid-Atlantic Industrial and Hazardous Waste Conference—Hazardous and Industrial Wastes; Suri, R.P.S., Christensen, G.L., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 339–348. [Google Scholar]
- Rives, V.; Labajos, F.M.; Ulibarri, M.A.; Malet, P. A new hydrotalcite-like compound containing V3+ ions in the layers. Inorg. Chem. 1993, 32, 5000–5001. [Google Scholar] [CrossRef]
- Labajos, F.M.; Rives, V.; Malet, P.; Centeno, M.A.; Ulibarri, M.A. Synthesis and characterization of hydrotalcite-like compounds containing V(3+) in the layers and of their calcination products. Inorg Chem. 1996, 35, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Hering, J.G.; Stumm, W. Oxidative and reductive dissolution of minerals. In Mineral-Water Interface Geochemistry; Hochella, M.F.J., White, A.F., Eds.; American Mineralogical Society: Washington, DC, USA, 1990; Volume 23, pp. 427–465. [Google Scholar]
- Heron, G.; Christensen, T.H. Impact of sediment-bound iron on redox buffering in a landfill leachate polluted aquifer (Vejen, Denmark). Environ. Sci. Technol. 1995, 29, 187–192. [Google Scholar] [CrossRef]
- Rügge, K.; Hofstetter, T.B.; Haderlein, S.B.; Bjerg, P.L.; Knudsen, S.; Zraunig, C.; Mosbæk, H.; Christensen, T.H. Characterization of predominant reductants in an anaerobic leachate-contaminated aquifer by nitroaromatic probe compounds. Environ. Sci. Technol. 1998, 32, 23–31. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Time | V(V) | V(IV) | V(III) | Average V Valence b | R-Factor c |
|---|---|---|---|---|---|---|
| FeS (mackinawite) | 2 days | 0 | 0 | 100 | 3.0 | 0.008 |
| 8 months | 0 | 3 | 97 | 3.0 | 0.013 | |
| 20 months | 0 | 0 | 100 | 3.0 | 0.014 | |
| Siderite | 2 days | 14 | 72 | 14 | 4.0 | 0.019 |
| 3 months | 0 | 29 | 71 | 3.3 | 0.010 | |
| 7 months | 0 | 0 | 100 | 3.0 | 0.005 | |
| 20 months | 2 | 0 | 100 | 3.0 | 0.004 | |
| Vivianite | 2 days | 13 | 80 | 8 | 4.1 | 0.014 |
| 3 months | 12 | 70 | 18 | 3.9 | 0.014 | |
| 4 months | 13 | 65 | 22 | 3.9 | 0.028 | |
| 8 months | 4 | 60 | 36 | 3.7 | 0.019 | |
| 20 months | 18 | 36 | 45 | 3.7 | 0.011 | |
| Magnetite | 2 days | 84 | 0 | 16 | 4.7 | 0.005 |
| 3 months | 53 | 26 | 21 | 4.3 | 0.005 | |
| 4 months | 35 | 43 | 22 | 4.1 | 0.013 | |
| 7 months | 17 | 34 | 49 | 3.7 | 0.006 | |
| 20 months | 20 | 78 | 22 | 3.8 | 0.012 | |
| Green Rust | 2 days | 14 | 67 | 19 | 4.0 | 0.004 |
| 3 months | 13 | 0 | 87 | 3.3 | 0.005 | |
| 4 months | 0 | 0 | 100 | 3.0 | 0.011 | |
| 8 months | 0 | 0 | 100 | 3.0 | 0.014 | |
| 20 months | 1 | 0 | 99 | 3.0 | 0.010 | |
| Biogenic Green Rust | 2 days | 70 | 8 | 22 | 4.5 | 0.005 |
| (BioGR) | 3 months | 36 | 47 | 17 | 4.2 | 0.010 |
| 7 months | 5 | 15 | 80 | 3.2 | 0.006 | |
| 20 months | 0 | 0 | 100 | 3.0 | 0.012 | |
| Biogenic | 2 days | 52 | 32 | 16 | 4.4 | 0.008 |
| Sid/mack/mag | 3 months | 34 | 5 | 61 | 3.7 | 0.004 |
| (BioSMM) | 7 months | 21 | 0 | 79 | 3.4 | 0.004 |
| 20 months | 22 | 0 | 78 | 3.4 | 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Loughlin, E.J.; Boyanov, M.I.; Kemner, K.M. Reduction of Vanadium(V) by Iron(II)-Bearing Minerals. Minerals 2021, 11, 316. https://doi.org/10.3390/min11030316
O’Loughlin EJ, Boyanov MI, Kemner KM. Reduction of Vanadium(V) by Iron(II)-Bearing Minerals. Minerals. 2021; 11(3):316. https://doi.org/10.3390/min11030316
Chicago/Turabian StyleO’Loughlin, Edward J., Maxim I. Boyanov, and Kenneth M. Kemner. 2021. "Reduction of Vanadium(V) by Iron(II)-Bearing Minerals" Minerals 11, no. 3: 316. https://doi.org/10.3390/min11030316
APA StyleO’Loughlin, E. J., Boyanov, M. I., & Kemner, K. M. (2021). Reduction of Vanadium(V) by Iron(II)-Bearing Minerals. Minerals, 11(3), 316. https://doi.org/10.3390/min11030316