
Secondary Sulfates from the Monte Arsiccio Mine (Apuan Alps, Tuscany, Italy): Trace-Element Budget and Role in the Formation of Acid Mine Drainage



Abstract
1. Introduction
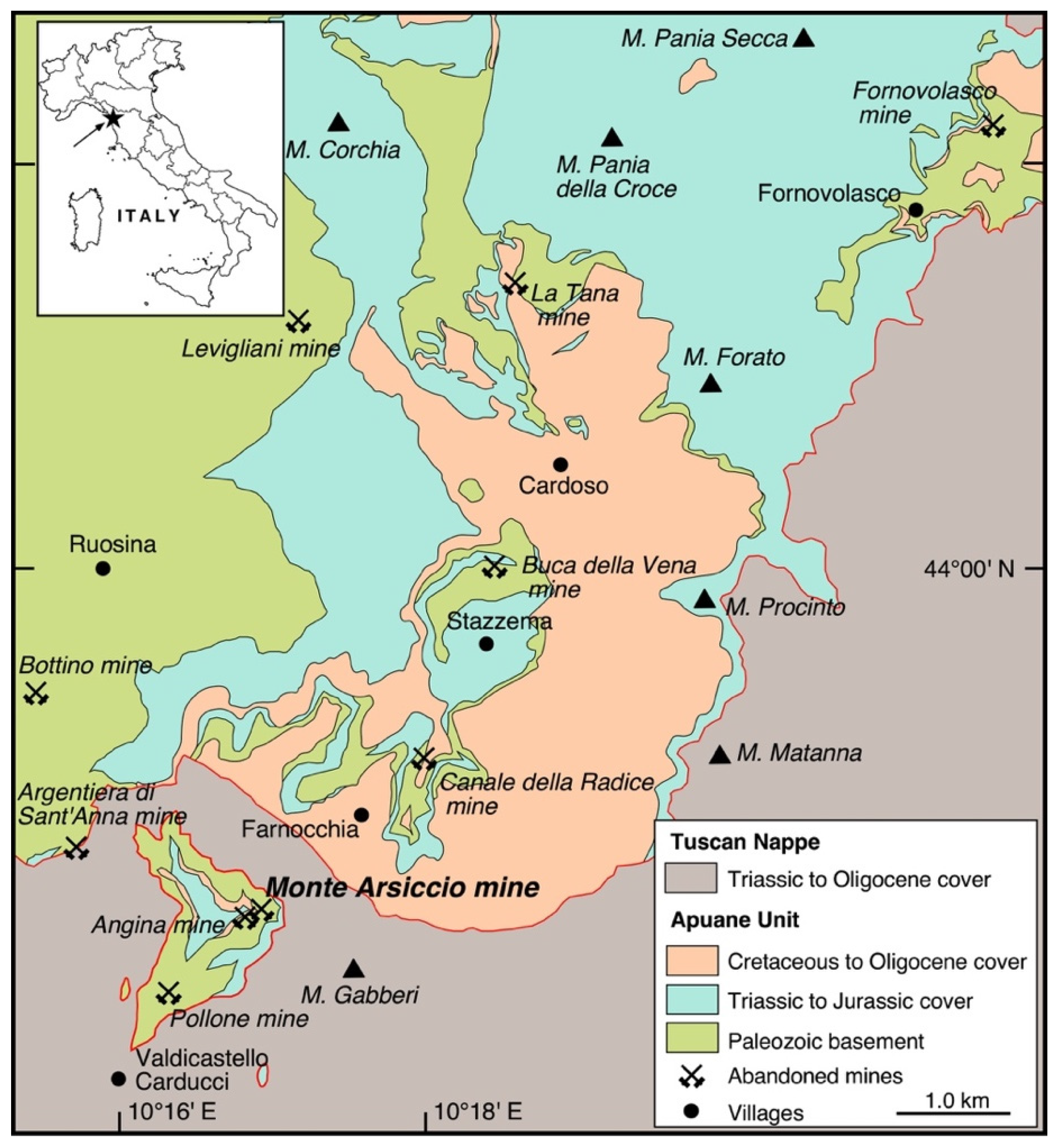
2. Geological Background
3. The Monte Arsiccio Sulfate Assemblage
- (1)
- The widespread occurrence of K-sulfates: it is worth noting that three new K-Fe3+ sulfate minerals were found in this assemblage (scordariite, magnanelliite, and giacovazzoite [23,24,25]), along with all the other currently known species in the K2O-Fe2O3-SO3-H2O system, with the only exception being represented by yavapaiite not yet identified at this locality:
- (2)
- The occurrence of the uncommon fluo-sulfate khademite [26];
- (3)
4. Experimental
4.1. Samples
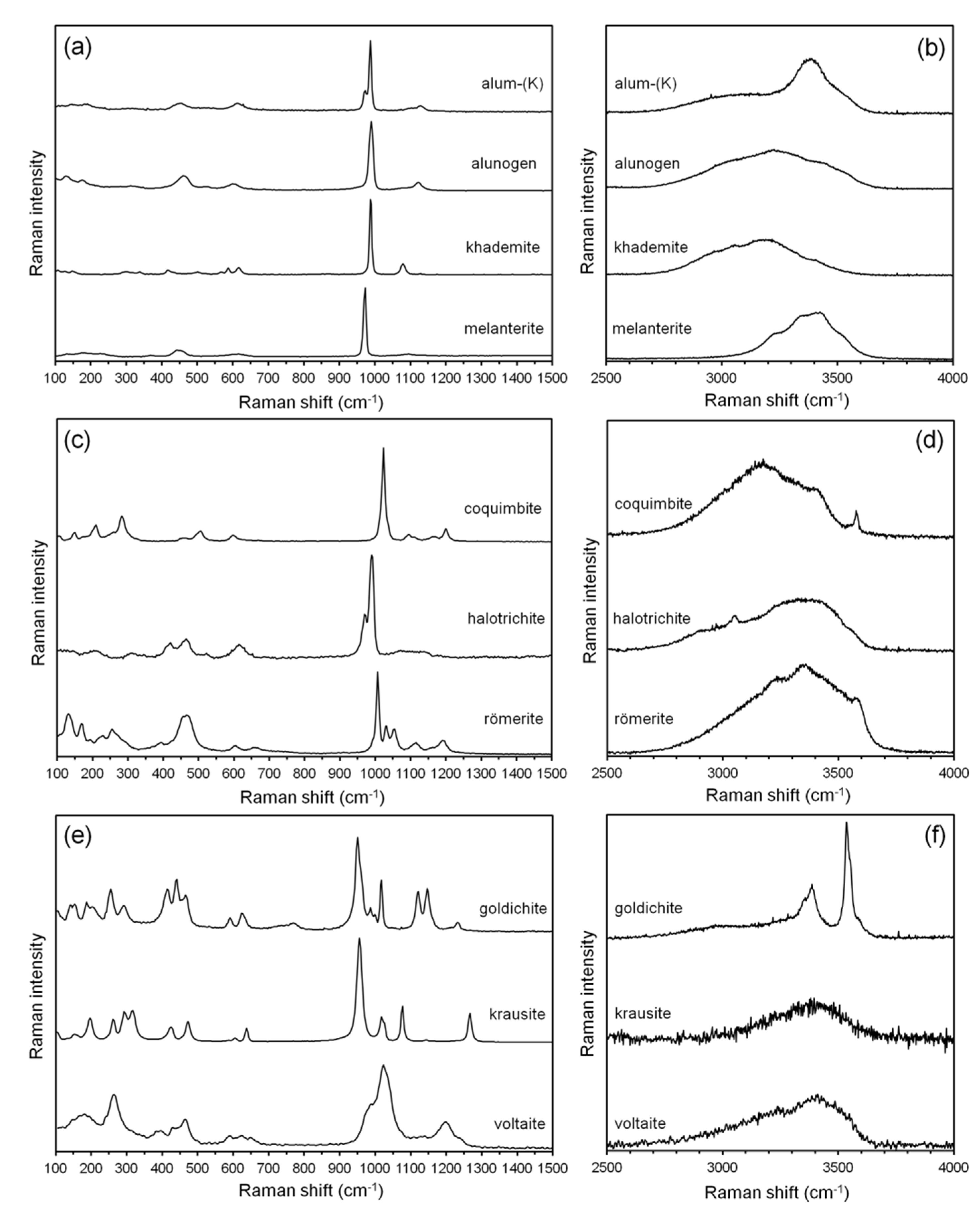
4.2. X-ray Diffraction and Micro-Raman Spectroscopy
4.3. Inductively Coupled Plasma Mass Spectrometry
4.4. Dissolution Experiments
5. Results
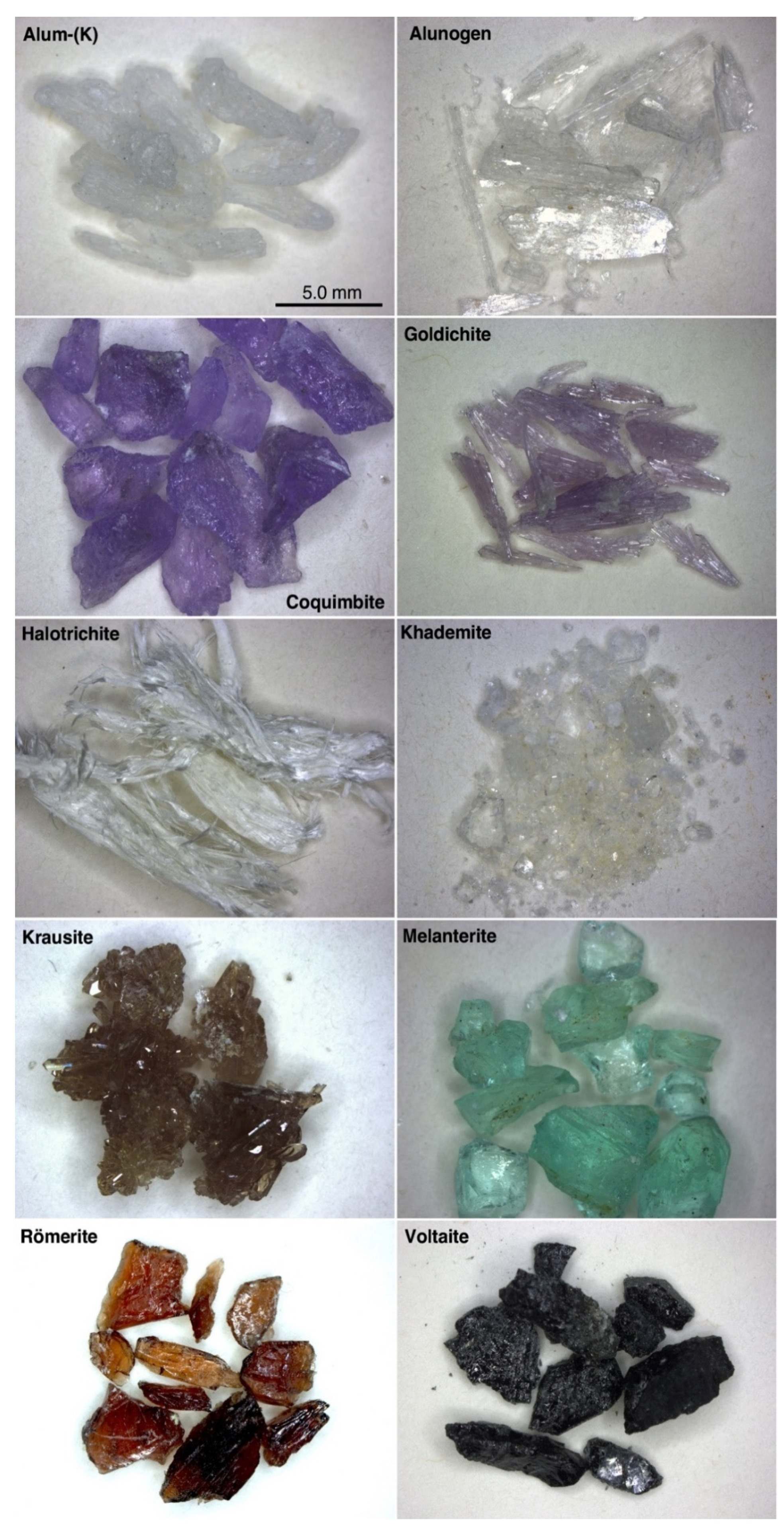
5.1. Mineralogy of the Studied Samples
- (1)
- Sulfates characterized by unconnected SO4 tetrahedra: alum-(K), alunogen, khademite, and melanterite (Figure 4a,b);
- (2)
- Sulfates with finite clusters of polyhedra: coquimbite, halotrichite, and rӧmerite (Figure 4c,d);
- (3)
- Sulfates with infinite chains: krausite (Figure 4e,f);
- (4)
- Sulfates with infinite sheets: goldichite (Figure 4e,f);
- (5)
- Sulfates with three-dimensional framework: voltaite (Figure 4e,f).
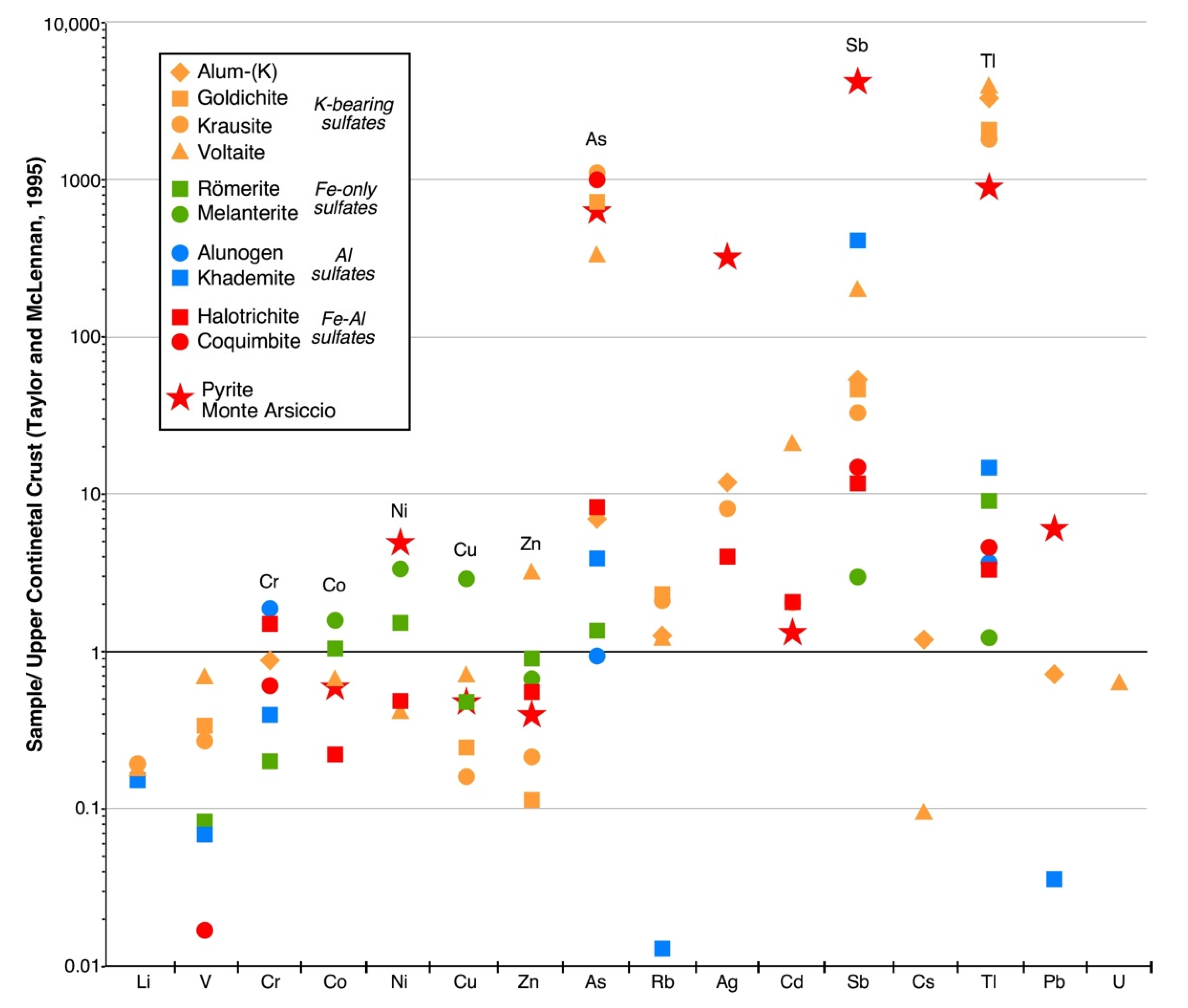
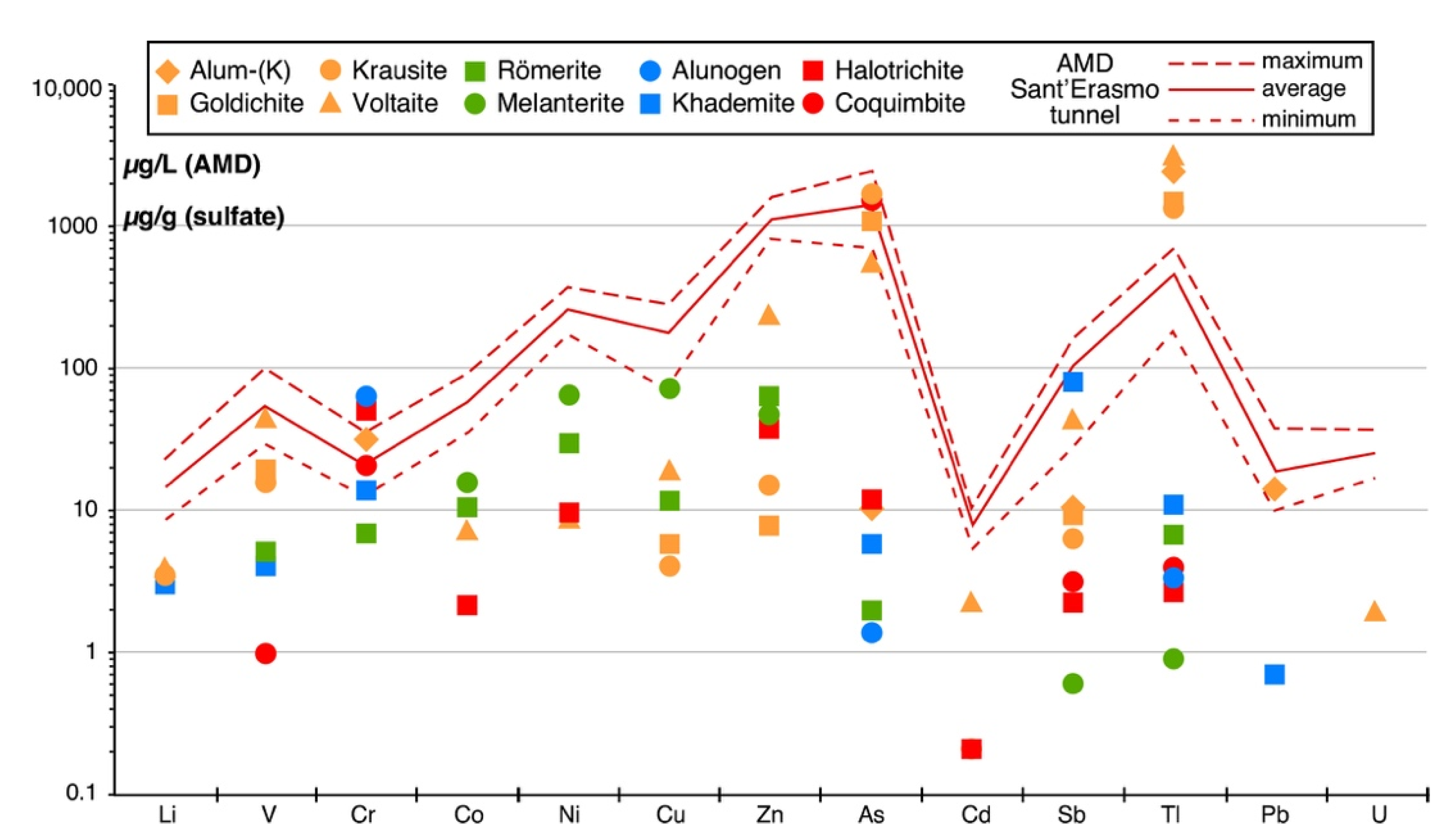
5.2. Trace-Element Content in Sulfate Minerals
- (1)
- Tl (1370–2988 µg/g) and Rb (140–252 µg/g) in the four K-bearing sulfates alum-(K), goldichite, krausite, and voltaite;
- (2)
- As (505–1680 µg/g) in coquimbite, goldichite, krausite, and voltaite;
- (3)
- Sb (41–81 µg/g) in khademite and voltaite;
- (4)
- Co, Ni, Cu and Zn in melanterite; Co, Ni, and Zn in römerite;
- (5)
- Cr (14–65 µg/g) in alum-(K), alunogen, coquimbite, halotrichite, and khademite.
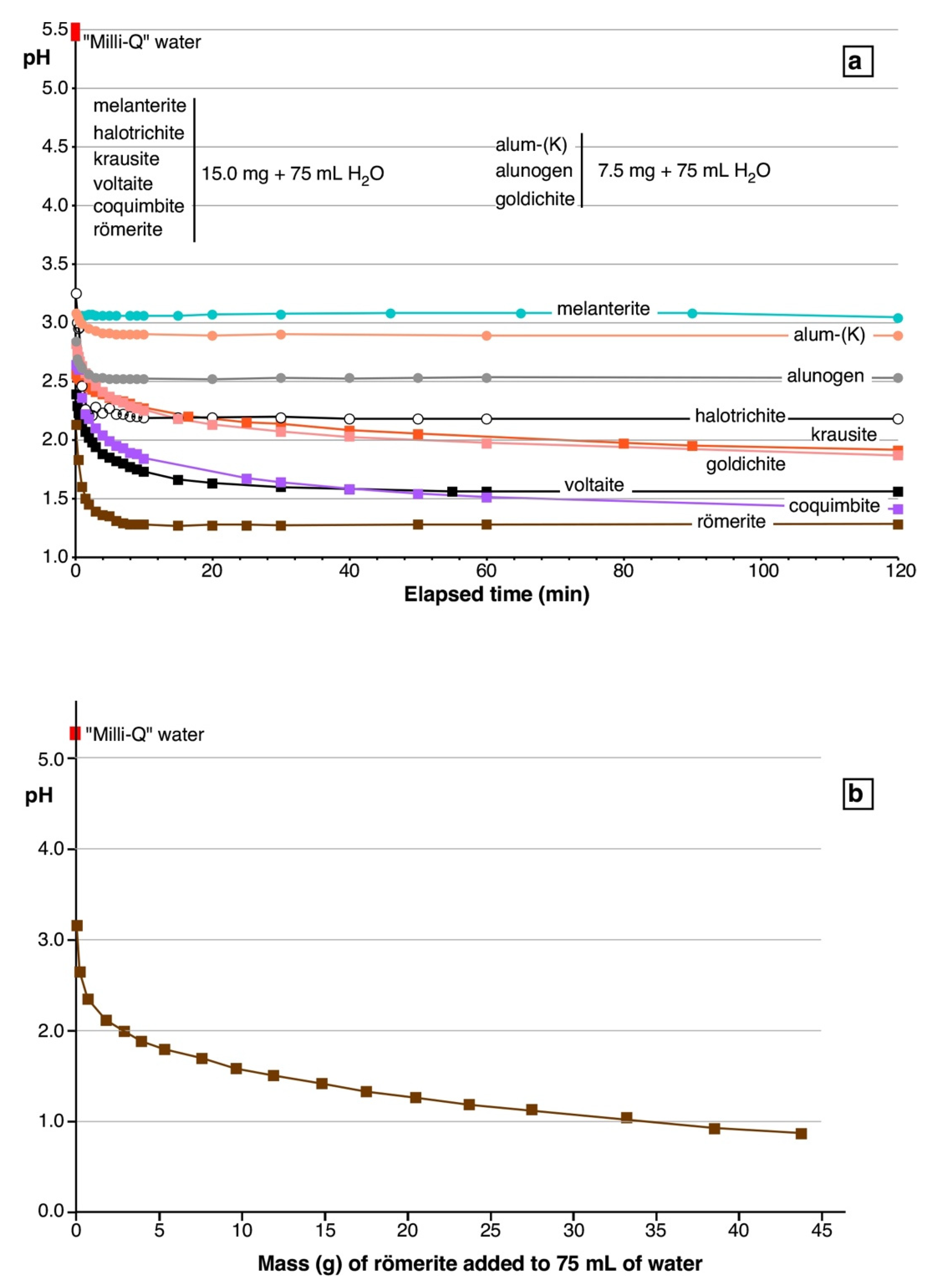
5.3. Dissolution Experiments
6. Discussion
6.1. From Geochemistry to Crystal-Chemistry: The Role of Sulfate Minerals in Sequestering PTE
6.2. Sulfate Dissolution and Acidity Production
6.3. Sulfate Mineralogy and AMD Geochemistry
7. Conclusions
- The studied secondary sulfates show a widely variable ability to store PTE in accordance with their widely variable crystal-chemistry.
- The four K-bearing sulfates store a significant quantity of Tl (and Rb), whereas coquimbite, goldichite, krausite, and voltaite are a repository of As. The Al-rich sulfates alunogen, halotrichite, alum-(K), coquimbite, and khademite show an enrichment in Cr. The Fe-only sulfates melanterite and römerite are characterized by a modest enrichment in the base metals Cu, Zn, Co, and Ni. Finally, khademite and, less so, voltaite exhibit moderately high concentrations of Sb.
- Dissolution experiments demonstrate that the dissolution of the studied sulfates always produces acidity. The acidity is ultimately generated by Fe3+ hydrolysis. Even the secondary sulfates not containing Fe3+ in their ideal chemical formula (alum-(K), alunogen, halotrichite, and melanterite) generate acidity upon their dissolution in water due to the measured occurrence of minor amounts of Fe3+. The highly soluble secondary sulfates containing high proportions of Fe3+ (e.g., römerite, coquimbite, and voltaite) can easily induce a decrease of the pH of the solutions down to values <1.0.
- The geochemistry of the AMD flowing out of the Monte Arsiccio mine is the result of a very complex series of processes occurring inside the mineralized area. However, the comparison between the PTE concentrations of the AMD and those measured in the studied secondary sulfates suggests that the dissolution of the latter may exert a strong control over the concentrations of dissolved Tl, As, Sb, and Cr.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rimstidt, J.D.; Vaughan, D.J. Pyrite oxidation: A state-of-the-art assessment of the reaction mechanism. Geochim. Cosmochim. Acta 2003, 67, 873–880. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Alpers, C.N. Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California. Proc. Natl. Acad. Sci. USA 1999, 96, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Jambor, J.L.; Nordstrom, K.D.; Alpers, C.N. Metal-sulfates salts from sulfide minerals oxidation. In. Sulfate minerals—crystallography, geochemistry and environmental significance. Rev. Mineral. Geochem. 2000, 40, 305–340. [Google Scholar] [CrossRef]
- Dimitrova, D.; Mladenova, V.; Hecht, L. Efflorescent sulfate crystallization on fractured and polished colloform pyrite surfaces: A migration pathway of trace elements. Minerals 2020, 10, 12. [Google Scholar] [CrossRef]
- Cravotta, C.A., III. Secondary iron-sulfate minerals as sources of sulfate and acidity. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society: Washington, DC, USA, 1994; pp. 345–364. [Google Scholar]
- Williamson, M.A.; Rimstidt, J.D. The kinetics and electrochemical rate-determining step of aqueous pyrite oxidation. Geochim. Cosmochim. Acta 1994, 58, 5443–5454. [Google Scholar] [CrossRef]
- Biagioni, C.; D’Orazio, M.; Vezzoni, S.; Dini, A.; Orlandi, P. Mobilization of Tl-Hg-As-Sb-(Ag,Cu)-Pb sulfosalt melts during low-grade metamorphism in the Alpi Apuane (Tuscany, Italy). Geology 2013, 41, 747–750. [Google Scholar] [CrossRef]
- D’Orazio, M.; Biagioni, C.; Dini, A.; Vezzoni, S. Thallium-rich pyrite ores from the Apuan Alps, Tuscany, Italy: Constraints for their origin and environmental concerns. Miner. Deposita 2017, 52, 687–707. [Google Scholar] [CrossRef]
- George, L.L.; Biagioni, C.; D’Orazio, M.; Cook, N.J. Textural and trace element evolution of pyrite during greenschist facies metamorphic recrystallization in the southern Apuan Alps (Tuscany, Italy): Influence on the formation of Tl-rich sulfosalt melt. Ore Geol. Rev. 2018, 102, 59–105. [Google Scholar] [CrossRef]
- Perotti, M.; Petrini, R.; D’Orazio, M.; Ghezzi, L.; Giannecchini, R.; Vezzoni, S. Thallium and other potentially toxic elements in the Baccatoio Stream Catchment (Northern Tuscany, Italy) receiving drainages from abandoned mines. Mine Water Environ. 2018, 37, 431–441. [Google Scholar] [CrossRef]
- Ghezzi, L.; D’Orazio, M.; Doveri, M.; Lelli, M.; Petrini, R.; Giannecchini, R. Groundwater and potentially toxic elements in dismissed mining area: Thallium contamination of drinking spring water in the Apuan Alps (Tuscany, Italy). J. Geochem. Expl. 2019, 197, 84–92. [Google Scholar] [CrossRef]
- D’Orazio, M.; Campanella, B.; Bramanti, E.; Onor, M.; Vianello, G.; Vittori-Antisari, L.; Petrini, R. Thallium pollution in water, soils and plants from a past-mining site of Tuscany: Sources, transfer processes and toxicity. J. Geochem. Expl. 2020, 209, 106434. [Google Scholar] [CrossRef]
- Giannecchini, R.; D’Amato Avanzi, G. Historical research as a tool in estimating hydrogeological hazard in a typical small alpine-like area: The example of the Versilia River basin (Apuan Alps, Italy). Phys. Chem. Earth 2012, Parts A/B/C 49, 32–43. [Google Scholar] [CrossRef]
- Piccini, L.; Pranzini, G.; Tedici, L.; Forti, P. Le risorse idriche dei complessi carbonatici del comprensorio apuo-versiliese. Quaderni di Geologia Applicata 1999, 6, 61–78. [Google Scholar]
- D’Achiardi, A. Mineralogia della Toscana; Tipografia Nistri: Pisa, Italy, 1872; Volume 1, p. 276. [Google Scholar]
- Biagioni, C.; Mauro, D.; Pasero, M. Sulfates from the pyrite ore deposits of the Apuan Alps (Tuscany, Italy): A review. Minerals 2020, 10, 1092. [Google Scholar] [CrossRef]
- Frau, F. The formation-dissolution-precipitation cycle of melanterite at the abandoned pyrite mine of Genna Luas in Sardinia, Italy: Environmental implications. Mineral. Mag. 2000, 64, 995–1006. [Google Scholar] [CrossRef]
- Valente, T.M.; Gomes, C.L. Occurrence, properties and pollution potential of environmental minerals in acid mine drainage. Sci. Tot. Environ. 2009, 407, 1135–1152. [Google Scholar] [CrossRef]
- Jerz, J.K.; Rimstidt, J.D. Efflorescent iron sulfate minerals: Paragenesis, relative stability, and environmental impact. Am. Mineral. 2003, 88, 1919–1932. [Google Scholar] [CrossRef]
- Hammarstrom, J.M.; Seall II, R.R.; Meier, A.L.; Kornfeld, J.M. Secondary sulfate minerals associated with acid drainage in the eastern US: Recycling of metals and acidity in surficial environments. Chem. Geol. 2005, 215, 407–431. [Google Scholar] [CrossRef]
- Carmignani, L.; Dessau, G.; Duchi, G. I giacimenti a barite, pirite ed ossidi di ferro delle Alpi Apuane. Studio minerogenetico e strutturale. Boll. Soc. Geol. It. 1976, 95, 1009–1061. [Google Scholar]
- Biagioni, C.; D’Orazio, M.; Fulignati, P.; George, L.L.; Mauro, D.; Zaccarini, F. Sulfide melts in ore deposits from low-grade metamorphic settings: Insights from fluid and Tl-rich sulfosalt microinclusions from the Monte Arsiccio mine (Apuan Alps, Tuscany, Italy). Ore Geol. Rev. 2020, 123, 103589. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Mauro, D.; Pasero, M. Crystal-chemistry of sulfates from the Apuan Alps (Tuscany, Italy). IV. Giacovazzoite, K5Fe3+3O(SO4)6(H2O)9·H2O, the natural analogue of the β-Maus’s Salt and its dehydration product. Phys. Chem. Miner. 2020, 47, 7. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Mauro, D.; Hålenius, U. Crystal chemistry of sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A new metavoltine-related mineral. Minerals 2019, 9, 702. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Kampf, A.R. Crystal chemistry of sulfates from the Apuan Alps (Tuscany, Italy). VII. Magnanelliite, K3Fe3+2(SO4)4(OH)(H2O)2, a new sulfate from the Monte Arsiccio mine. Minerals 2019, 9, 779. [Google Scholar] [CrossRef]
- Mauro, D.; Biagioni, C.; Pasero, M.; Zaccarini, F. Crystal-chemistry of sulfates from the Apuan Alps, Tuscany, Italy. VIII. New data on khademite, Al(SO4)F(H2O)5. Mineral. Mag. 2020, 84, 540–546. [Google Scholar] [CrossRef]
- Mauro, D.; Biagioni, C.; Pasero, M.; Skogby, H.; Zaccarini, F. Redefinition of coquimbite, AlFe3+3(SO4)6(H2O)12·6H2O. Mineral. Mag. 2020, 84, 275–282. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Hawthorne, F.C.; Krivovichev, S.V.; Burns, P.C. The crystal chemistry of sulfate minerals. Sulfate Minerals-Crystallography, geochemistry and environmental significance. Rev. Mineral. Geochem. 2000, 40, 1–101. [Google Scholar] [CrossRef]
- Frost, R.L.; Kloprogge, J.T. Raman microscopy study of kalinite, tschermigite and lonecreekite at 298 and 77 K. N. Jahr. Mineral. Monat. 2001, 2001, 27–40. [Google Scholar]
- Biagioni, C.; Mauro, D.; Pasero, M.; Bonaccorsi, E.; Lepore, G.O.; Zaccarini, F.; Skogby, H. Crystal-chemistry of sulfates from the Apuan Alps (Tuscany, Italy). VI. Tl-bearing alum-(K) and voltaite from the Fornovolasco mining complex. Am. Mineral. 2020, 105, 1088–1098. [Google Scholar] [CrossRef]
- Frost, R.L.; Žigovečki Gobac, Ž.; López, A.; Xi, Y.; Scholz, R.; Lana, C.; Fernandes Lima, R.M. Characterization of the sulphate mineral coquimbite, a secondary iron sulphate from Javier Ortega mine, Lucanas Province, Peru—Using infrared, Raman spectroscopy and thermogravimetry. J. Mol. Struct. 2014, 1063, 251–258. [Google Scholar] [CrossRef]
- Locke, A.J.; Martens, W.N.; Frost, R.L. Natural halotrichites—An EDX and Raman spectroscopic study. J. Raman Spectr. 2007, 38, 1429–1435. [Google Scholar] [CrossRef]
- Mauro, D.; Biagioni, C.; Pasero, M.; Skogby, H. Crystal-chemistry of sulfates from the Apuan Alps (Tuscany, Italy). III. Mg-rich sulfate assemblages from the Fornovolasco mining complex. Atti Soc. Tosc. Sci. Nat. Mem. 2019, 126, 33–44. [Google Scholar]
- Mauro, D.; Biagioni, C.; Pasero, M. Crystal-chemistry of sulfates from Apuan Alps (Tuscany, Italy). I. Crystal structure and hydrogen bond system of melanterite, Fe(H2O)6(SO4)·H2O. Period. Mineral. 2018, 87, 89–96. [Google Scholar]
- Frost, R.L.; Palmer, S.J.; Čejka, J.; Sejkora, J.; Plášil, J.; Jebavá, I.; Keeffe, E.C. A Raman spectroscopic study of M2+M3+ sulphate minerals, römerite Fe2+Fe3+2(SO4)4·14H2O and botryogen Mg2+Fe3+(SO4)2(OH)·7H2O. J. Raman Spectr. 2011, 42, 825–830. [Google Scholar] [CrossRef]
- Mauro, D.; Biagioni, C.; Pasero, M.; Zaccarini, F. Crystal-chemistry of sulfates from Apuan Alps (Tuscany, Italy). II. Crystal structure and hydrogen bonding system of römerite, Fe2+Fe3+2(SO4)4(H2O)14. Atti Soc. Tosc. Sci. Nat. Mem. 2018, 125, 5–11. [Google Scholar]
- Košek, F.; Edwards, H.G.M.; Jehlička, J. Raman spectroscopic vibrational analysis of the complex ion sulfates clairite, metavoltine, and voltaite from the burning coal dump Anna I, Alsdorf, Germany. J. Raman Spectr. 2020, 51, 1454–1461. [Google Scholar] [CrossRef]
- Košek, F.; Culca, A.; Žáček, V.; Laufek, F.; Škoda, R.; Jehlička, J. Native alunogen: A Raman spectroscopic study of a well-described specimen. J. Mol. Struct. 2018, 1157, 191–200. [Google Scholar] [CrossRef]
- Taylor, S.R.; McLennan, S.M. The geochemical evolution of the continental crust. Rev. Geophys. 1995, 33, 241–265. [Google Scholar] [CrossRef]
- Vezzoni, S.; Pieruccioni, D.; Galanti, Y.; Biagioni, C.; Dini, A. Permian hydrothermal alteration preserved in polymetamorphic basement and constraints for ore-genesis (Alpi Apuane, Italy). Geosciences 2020, 10, 399. [Google Scholar] [CrossRef]
- Romero, A.; González, I.; Galán, E. The role of efflorescent sulfates in the storage of trace elements in stream waters polluted by acid mine-drainage: The case of Peña del Hierro, southwestern Spain. Can. Mineral. 2006, 44, 1431–1446. [Google Scholar] [CrossRef]
- Nieva, N.E.; Garcia, M.G.; Borgnino, L.; Borda, L.G. The role of efflorescent salts associated with sulfide-rich mine wastes in the short-term cycling of arsenic: Insights from XRD, XAS, and μ-XRF studies. J. Hazard. Mater. 2021, 404, 124158. [Google Scholar] [CrossRef] [PubMed]
- Rutstein, M.S. Nickeloan melanterite from Sudbury Basin. Am. Mineral. 1980, 65, 968–969. [Google Scholar]
- Li, W.; Chen, G. Lishizhenite—A new zinc sulphate mineral. Acta Mineral. Sin. 1990, 10, 299–305. [Google Scholar]
- Li, W.; Chen, G.; Sun, S. Zincovoltaite—A new sulphate mineral. Acta Mineral. Sin. 1987, 7, 307–312. [Google Scholar]
- Nyburg, S.C.; Steed, J.W.; Aleksovska, S.; Petruševski, V.M. Structure of the alums. I. On the sulfate group disorder in the α-alums. Acta Crystallogr. 2000, B56, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, H.E. New minerals from California. Mineral. Rec. 1971, 2, 152–161. [Google Scholar]
- Biagioni, C.; Orlandi, P.; Pasero, M. Ankangite from the Monte Arsiccio mine (Apuan Alps, Tuscany, Italy): Occurrence, crystal structure, and classification problems in cryptomelane group minerals. Period. Mineral. 2009, 78, 3–11. [Google Scholar]
- Biagioni, C.; Orlandi, P.; Pasero, M.; Nestola, F.; Bindi, L. Mapiquiroite, (Sr,Pb)(U,Y)Fe2(Ti,Fe3+)18O38, a new member of the crichtonite group from the Apuan Alps, Tuscany, Italy. Eur. J. Mineral. 2014, 26, 427–437. [Google Scholar] [CrossRef]
- Biagioni, C.; Bonaccorsi, E.; Perchiazzi, N.; Hålenius, U.; Zaccarini, F. Derbylite and graeserite from the Monte Arsiccio mine, Apuan Alps, Tuscany, Italy: Occurrence and crystal-chemistry. Mineral. Mag. 2020, 84, 766–777. [Google Scholar] [CrossRef]
- Hawkes, S.J. All positive ions give acid solutions in water. J. Chem. Educ. 1996, 73, 516–517. [Google Scholar] [CrossRef]
- Hurowitz, J.A.; Tosca, N.J.; Dyar, M.D. Acid production by FeSO4·nH2O dissolution and implications for terrestrial and martian aquatic systems. Am. Mineral. 2009, 94, 409–414. [Google Scholar] [CrossRef]
- Giles, D.E.; Mohapatra, M.; Issa, T.B.; Anand, S.; Singh, P. Iron and aluminium based adsorption strategies for removing arsenic from water. J. Environ. Manag. 2011, 92, 3011–3022. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Ideal Chemical Formula | Mineral | Ideal Chemical Formula |
|---|---|---|---|
| Alum-(K) | KAl(SO4)2(H2O)12 | Jarosite | KFe3+3(SO4)2(OH)6 |
| Alunogen | Al2(SO4)3(H2O)12·5H2O | Khademite | Al(SO4)F(H2O)5 |
| Anhydrite | Ca(SO4) | Krausite | KFe3+(SO4)2(H2O) |
| Coquimbite | AlFe3+3(SO4)6(H2O)12·6H2O | Magnanelliite | K3Fe3+2(SO4)4(OH)(H2O)2 |
| Epsomite | Mg(SO4)(H2O)6·H2O | Magnesiocopiapite | MgFe3+4(SO4)6(OH)2(H2O)14·6H2O |
| Giacovazzoite | K5Fe3+3O(SO4)6(H2O)9·H2O | Melanterite | Fe2+(SO4)(H2O)6·H2O |
| Goldichite | KFe3+(SO4)2(H2O)4 | Römerite | Fe2+Fe3+2(SO4)4(H2O)14 |
| Gypsum | Ca(SO4)(H2O)2 | Scordariite | K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11 |
| Halotrichite | Fe2+Al2(SO4)4(H2O)17·5H2O | Voltaite | K2Fe2+5Fe3+3Al(SO4)12(H2O)18 |
| Element | Analyte Isotope | Detection Limit |
|---|---|---|
| - | (mass number) | (µg/g) |
| Li | 7 | 0.5 |
| V | 51 | 1 |
| Cr | 53 | 1 |
| Co | 59 | 0.1 |
| Ni | 60 | 0.5 |
| Cu | 63 | 1 |
| Zn | 66 | 1 |
| As | 75 | 0.5 |
| Rb | 85 | 0.5 |
| Sr | 86 | 1 |
| Mo | 98 | 0.5 |
| Ag | 107 | 0.1 |
| Cd | 111 | 0.1 |
| Sb | 121 | 0.5 |
| Cs | 133 | 0.05 |
| Tl | 205 | 0.05 |
| Pb | 206, 207, 208 | 0.5 |
| U | 238 | 0.05 |
| Internal Standards | ||
| Rh | 103 | |
| Re | 187 | |
| Bi | 209 | |
| Mineral | Me–O and Lattice Modes | ν2 (SO4) | ν4 (SO4) | ν1 (SO4) | ν3 (SO4) | ν (H2O) |
|---|---|---|---|---|---|---|
| Alum-(K) | 146, 188, 313 | 450, 515 | 614 | 972, 987 | 1102, 1130 | 2916, 3139, 3382, 3526 |
| Alunogen | 133, 177, 280, 319 | 459, 524 | 602 | 990 | 1087, 1123 | 2979, 3241, 3441, 3526 |
| Coquimbite | 148, 208, 281 | 457, 504 | 598 | 1024 | 1095, 1164, 1200 | 2944, 3180, 3405, 3577 |
| Goldichite | 141, 155, 186, 203, 255, 291 | 414, 441, 467 | 591, 623, 632, 741, 771 | 951, 990 | 1017, 1120, 1147, 1232 | 1637, 3009, 3328, 3382, 3536, 3553, 3558 |
| Halotrichite | 212, 316, 350 | 423, 468 | 620 | 975, 995 | 1075, 1102, 1141 | 2895, 3049, 3264, 3423 |
| Khademite | 127, 150, 300, 337 | 417, 432, 500, 514 | 567, 587, 602, 617 | 987 | 1079, 1129 | 2965, 3049, 3197, 3405 |
| Krausite | 142, 196, 259, 291, 315 | 422, 456, 472 | 635 | 953, 983, 1016, 1076 | 1141, 1219, 1265 | 3390 |
| Melanterite | 140, 197, 243, 381 | 447 | 607 | 980 | 1101, 1142 | 1655, 3243, 3335, 3427, 3521 |
| Römerite | 138, 173, 230, 260 | 463 | 608, 665 | 1011, 1036 | 1058, 1117, 1166, 1196 | 1653, 3249, 3352, 3486 |
| Voltaite | 179, 263, 392 | 433, 464, 589 | 621, 650 | 988, 1025 | 1200, 1240 | 3204, 3437 |
| Alum-(K) | Alunogen | Coquimbite | Goldichite | Halotrichite | Khademite | Krausite | Melanterite | Römerite | Voltaite | |
|---|---|---|---|---|---|---|---|---|---|---|
| Li | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | 3.0 | 3.8 | <0.5 | <0.5 | 3.8 |
| V | <1 | <1 | 1 | 20 | <1 | 4 | 16 | <1 | 5 | 42 |
| Cr | 31 | 65 | 21 | <1 | 52 | 14 | <1 | <1 | 7 | <1 |
| Co | <0.1 | <0.1 | <0.1 | <0.1 | 2.2 | <0.1 | <0.1 | 15.8 | 10.5 | 6.8 |
| Ni | <0.5 | <0.5 | <0.5 | <0.5 | 9.6 | <0.5 | <0.5 | 66 | 30.0 | 8.4 |
| Cu | <1 | <1 | <1 | 6 | <1 | <1 | 4 | 72 | 12 | 18 |
| Zn | <1 | <1 | <1 | 8 | 39 | <1 | 15 | 47 | 64 | 230 |
| As | 10.4 | 1.4 | 1531 | 1094 | 12.3 | 5.8 | 1680 | <0.5 | 2.0 | 505 |
| Rb | 140 | <0.5 | <0.5 | 252 | <0.5 | 1.4 | 236 | <0.5 | <0.5 | 140 |
| Sr | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 |
| Mo | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 |
| Ag | 0.6 | <0.1 | <0.1 | <0.1 | 0.2 | <0.1 | 0.4 | <0.1 | <0.1 | <0.1 |
| Cd | <0.1 | <0.1 | <0.1 | <0.1 | 0.2 | <0.1 | 0.2 | <0.1 | <0.1 | 2.1 |
| Sb | 10.7 | <0.5 | 3.0 | 9.4 | 2.3 | 81 | 6.5 | 0.6 | <0.5 | 41 |
| Cs | 4.5 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | 0.36 |
| Tl | 2501 | 3.5 | 3.9 | 1541 | 2.77 | 10.9 | 1370 | 0.92 | 6.8 | 2988 |
| Pb | 14.3 | <0.5 | <0.5 | <0.5 | <0.5 | 0.7 | <0.5 | <0.5 | <0.5 | <0.5 |
| U | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | 1.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Orazio, M.; Mauro, D.; Valerio, M.; Biagioni, C. Secondary Sulfates from the Monte Arsiccio Mine (Apuan Alps, Tuscany, Italy): Trace-Element Budget and Role in the Formation of Acid Mine Drainage. Minerals 2021, 11, 206. https://doi.org/10.3390/min11020206
D’Orazio M, Mauro D, Valerio M, Biagioni C. Secondary Sulfates from the Monte Arsiccio Mine (Apuan Alps, Tuscany, Italy): Trace-Element Budget and Role in the Formation of Acid Mine Drainage. Minerals. 2021; 11(2):206. https://doi.org/10.3390/min11020206
Chicago/Turabian StyleD’Orazio, Massimo, Daniela Mauro, Marta Valerio, and Cristian Biagioni. 2021. "Secondary Sulfates from the Monte Arsiccio Mine (Apuan Alps, Tuscany, Italy): Trace-Element Budget and Role in the Formation of Acid Mine Drainage" Minerals 11, no. 2: 206. https://doi.org/10.3390/min11020206
APA StyleD’Orazio, M., Mauro, D., Valerio, M., & Biagioni, C. (2021). Secondary Sulfates from the Monte Arsiccio Mine (Apuan Alps, Tuscany, Italy): Trace-Element Budget and Role in the Formation of Acid Mine Drainage. Minerals, 11(2), 206. https://doi.org/10.3390/min11020206