
Water in the Alluaudite Type-Compounds: Synthesis, Crystal Structure and Magnetic Properties of Co3(AsO4)0.5+x(HAsO4)2−x(H2AsO4)0.5+x[(H,□)0.5(H2O,H3O)0.5]2x+



Abstract
1. Introduction
2. Materials and Methods
2.1. Single Crystal X-ray Diffraction Analysis
2.2. Chemical Composition and Micromorphology
2.3. Vibrational Spectroscopy
2.4. Magnetic Properties
3. Results and Discussion
3.1. Description of the Crystal Structure
3.2. Infrared and Raman Spectra of CoAsAllu
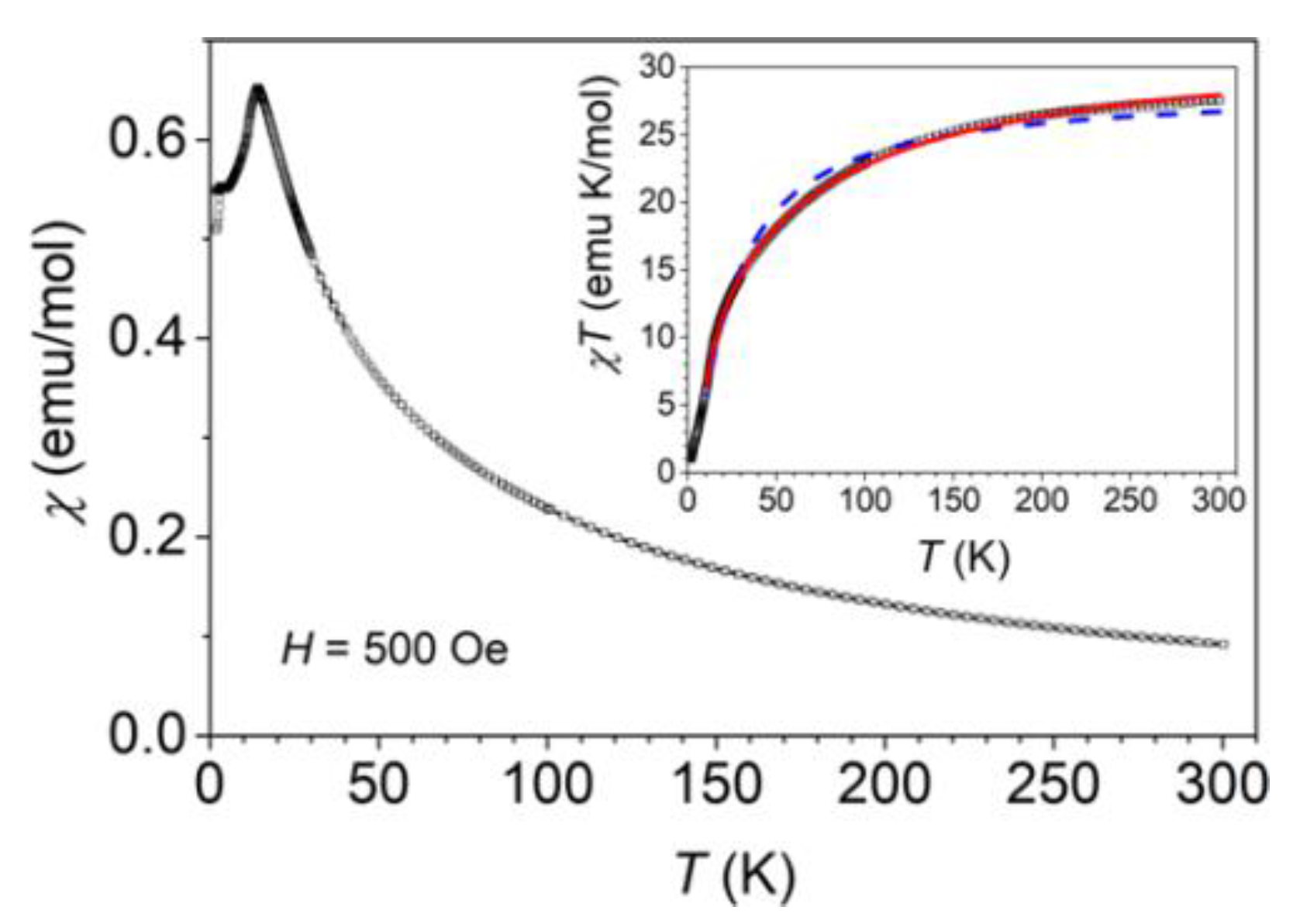
3.3. Magnetic Properties
4. Summery
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowagiel, M.; Samsel, M.J.; Pietrzak, T.K. Towards the high phase purity of nanostructured alluaudite-type glass-ceramics cathode materials for sodium ion batteries. Materials 2021, 14, 4997. [Google Scholar] [CrossRef]
- Buzlukov, A.L.; Medvedeva, N.I.; Suetin, D.V.; Serdtsev, A.V.; Baklanova, Y.V.; Solodovnikov, S.F.; Tyutyunnik, A.P.; Denisova, T.A.; Gulyaeva, O.A. Revealing sodium-ion diffusion in alluaudite-type Na4–2xM1+x(MoO4)3 (M = Mg, Zn, Cd) from 23Na MAS NMR and ab initio studies. J. Solid State Chem. 2021, 293, 121800. [Google Scholar] [CrossRef]
- Korzenski, M.B.; Schimek, G.L.; Kolis, J.W.; Long, G.J. Hydrothermal synthesis, structure, and characterization of a mixed-valent iron (II/III) phosphate, NaFe3.67(PO4)3: A new variation of the alluaudite structure type. J. Solid State Chem. 1998, 139, 152–160. [Google Scholar] [CrossRef]
- Richardson, T.J. Phosphate-stabilized lithium intercalation compounds. J. Pow. Sources 2003, 119–121, 262–265. [Google Scholar] [CrossRef]
- Kacimi, M.; Ziyad, M.; Hatert, F. Structural features of AgCaCdMg2(PO4)3 and AgCd2Mg2(PO4)3, two new compounds with the alluaudite-type structure, and their catalytic activity in butan-2-ol conversion. Mater. Res. Bull. 2005, 40, 682–693. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Park, I.; Park, Y.-U.; Yoo, Y.-K.; Park, K.-J.; Lee, S.; Kang, K. LiFePO4 with an alluaudite crystal structure for lithium ion batteries. Energy Environ. Sci. 2013, 6, 830–834. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Park, K.Y.; Park, Y.U.; Lee, S.; Kwon, H.S.; Yoo, H.L.; Kang, K. Alluaudite LiMnPO4: A new Mn-based positive electrode for Li rechargeable batteries. J. Mater. Chem. 2014, A2, 8632–8636. [Google Scholar] [CrossRef]
- Aksenov, S.M.; Yamnova, N.A.; Kabanova, N.A.; Volkov, A.S.; Gurbanova, O.A.; Deyneko, D.V.; Dimitrova, O.V.; Krivovichev, S.V. Topological features of the alluaudite-type framework and its derivatives: Synthesis and crystal structure of NaMnNi2(H2/3PO4)3. Crystals 2021, 11, 237. [Google Scholar] [CrossRef]
- Tait, K.T.; Hawthorne, F.C.; Halden, N. Alluaudite-group phosphate and arsenate minerals. Can. Mineral. 2021, 59, 243–263. [Google Scholar] [CrossRef]
- Hatert, F.; Keller, P.; Lissner, F.; Antenucci, D.; Fransolet, A.-M. First experimental evidence of alluaudite-like phosphates with high Li-content: The (Na1-xLix)MnFe2(PO4)3 series (x = 0 to 1). Eur. J. Mineral. 2000, 12, 847–857. [Google Scholar] [CrossRef]
- Hatert, F. A new nomenclature scheme for the alluaudite supergroup. Eur. J. Mineral. 2019, 31, 807–822. [Google Scholar] [CrossRef]
- Đorđević, T.; Wittwer, A.; Krivovichev, S.V. Three new alluaudite-like protonated arsenates: NaMg3(AsO4)(AsO3OH)2, NaZn3(AsO4)(AsO3OH)2 and Na(Na0.6Zn0.4)Zn2(H0.6AsO4)(AsO3OH)2. Eur. J. Mineral. 2015, 27, 559–573. [Google Scholar] [CrossRef]
- Stojanović, J.; Đorđević, T.; Karanović, L. Structural features of two novel alluaudite-like arsenates Cd1.16Zn2.34(AsO4)1.5(HAsO4)(H2AsO4)0.5 and Cd0.74Mg2.76(AsO4)1.5(HAsO4)(H2AsO4)0.5. J. Alloys Compd. 2012, 520, 180–189. [Google Scholar] [CrossRef][Green Version]
- Ben Yahia, H.; Shikano, M.; Tabuchi, M.; Belharouak, I. Synthesis, crystal structure, and properties of the alluaudite-type vanadates Ag2-xNaxMn2Fe(VO4)3. Inorg. Chem. 2016, 55, 4643–4649. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, L.L.; Kendrick, E.; Knight, K.S.; Wright, A.J.; Slater, P.R. Investigation into the dehydration of selenate doped Na2M(SO4)2·2H2O (M = Mn, Fe, Co and Ni): Stabilisation of the high Na content alluaudite phases Na3M1.5(SO4)3-1.5x(SeO4)1.5x (M = Mn, Co and Ni) through selenate incorporation. J. Solid State Chem. 2018, 258, 64–71. [Google Scholar] [CrossRef]
- Solodovnikov, S.F.; Solodovnikova, Z.A.; Zolotova, E.S.; Yudin, V.N.; Gulyaeva, O.A.; Tushinova, Y.L.; Kuchumov, B.M. Nonstoichiometry in the systems Na2MoO4–MMoO4 (M = Co, Cd), crystal structures of Na3.36Co1.32(MoO4)3, Na3.13Mn1.43(MoO4)3 and Na3.72Cd1.14(MoO4)3, crystal chemistry, compositions and ionic conductivity of alluaudite-type double molybdates and tungstates. J. Solid State Chem. 2017, 253, 121–128. [Google Scholar] [CrossRef]
- Pekov, I.V.; Koshlyakova, N.N.; Agakhanov, A.A.; Zubkova, N.V.; Belakovskiy, D.I.; Vigasina, M.F.; Turchkova, A.G.; Sidorov, E.G.; Pushcharovsky, D.Y. New arsenate minerals from the Arsenatnaya fumarole, Tolbachik volcano, Kamchatka, Russia. XIV. Badalovite, NaNaMg(MgFe3+)(AsO4)3, a member of the alluaudite group. Mineral. Mag. 2020, 84, 616–622. [Google Scholar] [CrossRef]
- Kampf, A.R.; Nash, B.P.; Celestian, A.J.; Dini, M.; Molina Donoso, A.A. Camanchacaite, chinchorroite, espadaite, magnesiofluckite, picaite and ríosecoite: Six new hydrogen-arsenate minerals from the Torrecillas mine, Iquique Province, Chile. Mineral. Mag. 2019, 83, 655–671. [Google Scholar] [CrossRef]
- Pekov, I.V.; Koshlyakova, N.N.; Agakhanov, A.A.; Zubkova, N.V.; Belakovskiy, D.I.; Vigasina, M.F.; Turchkova, A.G.; Sidorov, E.G.; Pushcharovsky, D.Y. New arsenate minerals from the Arsenatnaya fumarole, Tolbachik volcano, Kamchatka, Russia. XV. Calciojohillerite, NaCaMgMg2(AsO4)3, a member of the alluaudite group. Mineral. Mag. 2021, 85, 215–223. [Google Scholar] [CrossRef]
- Đorđević, T.; Kolitsch, U.; Nasdala, L. A single-crystal X-ray and Raman spectroscopic study of hydrothermally synthesized arsenates and vanadates with the descloizite and adelite structure types. Am. Mineral. 2016, 101, 1135–1149. [Google Scholar] [CrossRef]
- Đorđević, T.; Gerger, S.; Karanović, L. Hydrothermal and ionothermal synthesis of mineral-related arsenates in the system CdO–MO–As2O5 (M2+ = Mg, Co, Ni, Cu, Zn) and their crystal structures. Eur. J. Mineral. 2017, 29, 73–89. [Google Scholar] [CrossRef]
- Đorđević, T.; Karanović, L. An update on the mineral-like Sr-containing transition metal arsenates. Mineral. Mag. 2021, 85, 416–430. [Google Scholar] [CrossRef]
- Đorđević, T.; Wittwer, A.; Jagličić, Z.; Djerdj, I. Hydrothermal synthesis of single crystal CoAs2O4 and NiAs2O4 compounds and their magnetic properties. RSC Adv. 2015, 5, 18270–18278. [Google Scholar] [CrossRef]
- Đorđević, T.; Karanović, L.; Jagličić, Z. A new copper(II) arsenate, Na2Cu3(AsO3OH)4·4H2O containing discrete [Cu3O12]18-units: Synthesis, crystal structure and magnetic properties. J. Solid State Chem. 2018, 265, 55–63. [Google Scholar] [CrossRef]
- STOE. X-AREA and X-RED32 Software; Stoe & Cie: Darmstadt, Germany, 2013. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement viaSIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Wills, A.S.; Brown, I.D. VaList- Bond Valence Software for Windows; CEA: Bruyères-le-Châtel, France, 1999; Available online: http://www.CCP14.ac.uk (accessed on 20 October 2021).
- Dowty, E. ATOMS 6.1, a Computer Program; Shape Software, Inc.: Kingsport, TN, USA, 1999. [Google Scholar]
- Kahn, O. Molecular Magnetism; Wiley VCH Publishing: Hoboken, NJ, USA, 1993. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. 1991, B47, 192–197. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Chervonnyi, A.D. ‘IR Spectra of Minerals and Related Compounds, and Reference Samples’ Data. In Infrared Spectroscopy of Minerals and Related Compounds; Springer Mineralogy, Springer: Cham, Switzerland, 2010. [Google Scholar]
- Đorđević, T.; Karanović, L. A new polymorph of Ba(AsO3OH): Synthesis, crystal structure and vibrational spectra. J. Solid State Chem. 2010, 183, 2835–2844. [Google Scholar] [CrossRef]
- Mabbs, F.E.; Machin, D.J. Magnetism and Transition Metal Complexes; Dover Publications, Inc.: Mineola, NY, USA, 1973. [Google Scholar]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Saunders College Publishing: Philadelphia, PA, USA, 1976. [Google Scholar]
- Jones, L.F.; Kilner, C.A.; Halcrow, M.A. A comparison of different methods for fitting susceptibility data of cobalt(II) coordination polymers in a new cobalt(II)/sulfate 1-D chain. New J. Chem. 2007, 31, 1530–1534. [Google Scholar] [CrossRef]
- Rueff, M.; Masciocchi, N.; Rabu, P.; Sironi, A.; Skoulios, A. Structure and magnetism of a polycrystalline transition metal soap - CoII[OOC(CH2)10COO](H2O)2. Eur. J. Inorg. Chem. 2001, 2001, 2843–2848. [Google Scholar] [CrossRef]
- Carlin, R.L. Magnetochemistry; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Yakubovich, O.V.; Kiryukhina, G.V.; Dimitrova, O.V. Crystal chemistry of KCuMn3(VO4)3 in the context of detailed systematics of the alluaudite family. Crystallogr. Rep. 2016, 61, 556–575. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
| Chemical formula | As6H8Co6O25 |
| Molecular mass, Mr | 1211.16 |
| Crystal system, space group | Monoclinic, C2/c |
| Temperature (K) | 293 |
| a, b, c (Å) | 11.6978(8), 12.5713(7), 6.7705(5) |
| β (°) | 113.255(5) |
| V (Å3) | 914.76 (11) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 16.222 |
| Crystal size (mm) | 0.08 × 0.02 × 0.02 |
| Data Collection | |
| Diffractometer | STOE StadiVari |
| Absorption correction | Multi-scan |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 8740, 1639, 1414 |
| Rint | 0.041 |
| (sin θ/λ)max (Å−1) | 0.757 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.020, 0.043, 1.00 |
| No. of reflections | 1639 |
| No. of parameters | 88 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (eÅ−3) | 0.67, −1.49 |
| Atom | x | y | z | Uiso*/Ueq | Occupancy (<1) |
|---|---|---|---|---|---|
| As1 | 0.500000 | 0.81531(2) | 1.250000 | 0.00713(6) | |
| As2 | 0.29008(2) | 0.61716(2) | 0.39402(3) | 0.00896(6) | |
| Co1 | 0.500000 | 0.78694(3) | 0.750000 | 0.00777(8) | |
| Co2 | 0.30090(3) | 0.66037(2) | 0.89043(4) | 0.00849(6) | |
| O1 | 0.46638(14) | 0.74458(12) | 1.0241(2) | 0.0084(2) | |
| O2 | 0.62013(14) | 0.89752(13) | 1.2730(3) | 0.0111(3) | |
| H2 | 0.604032 | 0.958135 | 1.298744 | 0.013* | 0.5 |
| O3 | 0.35017(15) | 0.68027(13) | 0.6308(2) | 0.0098(3) | |
| O4 | 0.13697(15) | 0.59005(13) | 0.3394(3) | 0.0122(3) | |
| H4 | 0.132130 | 0.554871 | 0.437751 | 0.015* | 0.5 |
| O5 | 0.29295(15) | 0.68858(12) | 0.1877(2) | 0.0093(3) | |
| O6 | 0.36646(16) | 0.49889(13) | 0.4159(3) | 0.0149(3) | |
| H6 | 0.439143 | 0.510404 | 0.435550 | 0.018* | 0.5 |
| O7 | 0.500000 | 0.5129(8) | 0.750000 | 0.048(2) | 0.5 |
| Atom | U11 | U22 | U33 | U12 | U13 | U23 |
|---|---|---|---|---|---|---|
| As1 | 0.00900(13) | 0.00658(12) | 0.00501(11) | 0.000 | 0.00190(9) | 0.000 |
| As2 | 0.01160(10) | 0.00900(10) | 0.00620(8) | −0.00325(7) | 0.00345(7) | −0.00085 (6) |
| Co1 | 0.00725(16) | 0.00874(16) | 0.00791(15) | 0.000 | 0.00362(12) | 0.000 |
| Co2 | 0.00752(13) | 0.01095(13) | 0.00756(11) | −0.00178(9) | 0.00357(9) | −0.00083(8) |
| O1 | 0.0077(6) | 0.0119(6) | 0.0057(5) | −0.0030(5) | 0.0027(4) | −0.0024(5) |
| O2 | 0.0078(6) | 0.0082(6) | 0.0172(6) | −0.0007(5) | 0.0048(5) | −0.0001(5) |
| O3 | 0.0105(6) | 0.0129(7) | 0.0063(5) | −0.0040(5) | 0.0036(5) | −0.0023(5) |
| O4 | 0.0088(6) | 0.0095(6) | 0.0185(7) | −0.0007(5) | 0.0057(5) | −0.0002(5) |
| O5 | 0.0107(6) | 0.0114(6) | 0.0069(5) | 0.0007(5) | 0.0045(5) | 0.0018(5) |
| O6 | 0.0131(7) | 0.0075(6) | 0.0241(8) | 0.0000(5) | 0.0073(6) | −0.0019(6) |
| O7 | 0.036(4) | 0.076(6) | 0.028(3) | 0.000 | 0.010(3) | 0.000 |
| Atom1-Atom2 | Bond lengths (Å) | Bond Valences (v.u.) | Atom1-Atom2 | Bond Lengths (Å) | Bond Valences (v.u.) |
|---|---|---|---|---|---|
| As1—O1 | 1.6763(14) | 1.279 | As2—O5 | 1.6718(15) | 1.293 |
| As1—O1i | 1.6763(14) | 1.279 | As2—O3 | 1.6742(15) | 1.286 |
| As1—O2 | 1.7012(16) | 1.195 | As2—O6 | 1.7106(16) | 1.163 |
| As1—O2i | 1.7012(16) | 1.195 | As2—O4 | 1.7139(16) | 1.154 |
| Average/Σνij | <1.689> | 4.948(1) | <1.693> | 4.896(2) | |
| Co1—O3 | 2.0987(15) | 0.333 | Co2—O3 | 2.0702(15) | 0.360 |
| Co1—O3iii | 2.0987(15) | 0.333 | Co2—O1 | 2.0745(15) | 0.355 |
| Co1—O1iii | 2.1114(14) | 0.322 | Co2—O2vi | 2.0750(16) | 0.355 |
| Co1—O1 | 2.1115(14) | 0.322 | Co2—O5ii | 2.0818(15) | 0.349 |
| Co1—O4vii | 2.1352(17) | 0.302 | Co2—O6v | 2.1265(16) | 0.309 |
| Co1—O4iv | 2.1352(17) | 0.302 | Co2—O5iv | 2.1523(15) | 0.288 |
| average/Σνij | <2.115> | 1.914 (4) | <2.097> | 2.016(1) |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H2⋯O4viii | 0.82 | 1.70 | 2.455(2) | 152 |
| O4—H4⋯O2vi | 0.82 | 2.40 | 3.023(2) | 133 |
| O6—H6⋯O6ix | 0.82 | 2.10 | 2.872(4) | 158 |
| O6—H6⋯O7ix | 0.82 | 1.70 | 2.261(2) | 124 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Đorđević, T.; Karanović, L.; Jagodič, M.; Jagličić, Z. Water in the Alluaudite Type-Compounds: Synthesis, Crystal Structure and Magnetic Properties of Co3(AsO4)0.5+x(HAsO4)2−x(H2AsO4)0.5+x[(H,□)0.5(H2O,H3O)0.5]2x+. Minerals 2021, 11, 1372. https://doi.org/10.3390/min11121372
Đorđević T, Karanović L, Jagodič M, Jagličić Z. Water in the Alluaudite Type-Compounds: Synthesis, Crystal Structure and Magnetic Properties of Co3(AsO4)0.5+x(HAsO4)2−x(H2AsO4)0.5+x[(H,□)0.5(H2O,H3O)0.5]2x+. Minerals. 2021; 11(12):1372. https://doi.org/10.3390/min11121372
Chicago/Turabian StyleĐorđević, Tamara, Ljiljana Karanović, Marko Jagodič, and Zvonko Jagličić. 2021. "Water in the Alluaudite Type-Compounds: Synthesis, Crystal Structure and Magnetic Properties of Co3(AsO4)0.5+x(HAsO4)2−x(H2AsO4)0.5+x[(H,□)0.5(H2O,H3O)0.5]2x+" Minerals 11, no. 12: 1372. https://doi.org/10.3390/min11121372
APA StyleĐorđević, T., Karanović, L., Jagodič, M., & Jagličić, Z. (2021). Water in the Alluaudite Type-Compounds: Synthesis, Crystal Structure and Magnetic Properties of Co3(AsO4)0.5+x(HAsO4)2−x(H2AsO4)0.5+x[(H,□)0.5(H2O,H3O)0.5]2x+. Minerals, 11(12), 1372. https://doi.org/10.3390/min11121372