The High Temperature Co-Processing of Nickel Sulfide and Nickel Laterite Sources



Abstract
1. Introduction
- (1)
- high in capital expenditure and, for several operations, there have been significant cost blow outs;
- (2)
- in most cases have required long ramp-up times, often not achieving nameplate capacity;
- (3)
- best suited to limonitic fractions of the nickel laterite profile to ensure low acid consumption;
- (4)
- composed of variable and complex downstream processing operations; and
- (5)
- large generators of waste materials that must be treated and stored appropriately.
- (1)
- The potential to inject a higher pulp density ore/concentrate mix enhancing throughput, as it has been shown that the addition of nickel sulfide-containing material to nickel laterite ore lowers the pulp yield stress [16];
- (2)
- Little or no requirement to burn sulfur to generate sulfuric acid, with the elimination of potential attendant issues for a sulfuric acid plant such as the inadvertent generation of hydrogen;
- (3)
- Less loss of heat from sulfur burning not recovered as steam or hot water that can be used in the process along with potential lower plant use of water for cooling in the sulfuric acid plant;
- (4)
- The in situ generation of heat resulting in reduced requirement for steam injection and associated increase in working pulp density;
- (5)
- Depending upon the sulfidic ore/concentrate source, the generation of a higher metal value concentration leach liquor, e.g., pyrrhotite, which generally contains low but not insignificant amounts of nickel, does not need to be rejected when generating a nickel concentrate;
- (6)
- Greater tolerance for high magnesium content in the nickel concentrate compared with material sent to nickel smelters, though preferably hosted by poorly leached minerals such as talc;
- (7)
- The in situ solubilization of toxic impurities, such as arsenic, which can then be more readily managed via the formation of stable ferric arsenate compounds.
- (1)
- If sulfur is not burned, there needs to be an alternative energy source available to generate steam and/or electricity requirements;
- (2)
- An oxidant is required for in situ generation of the sulfuric acid, which is expected to be oxygen that is generated on site;
- (3)
- The complexity of the equipment will be greater than that required for high pressure acid leaching and, if oxygen is used as the oxidant, some of the materials of construction may be more exotic/expensive.
2. Materials and Methods
3. Results and Discussion
3.1. Feed Materials
Laterite Ore, Nickel Concentrate, and Site Process Water
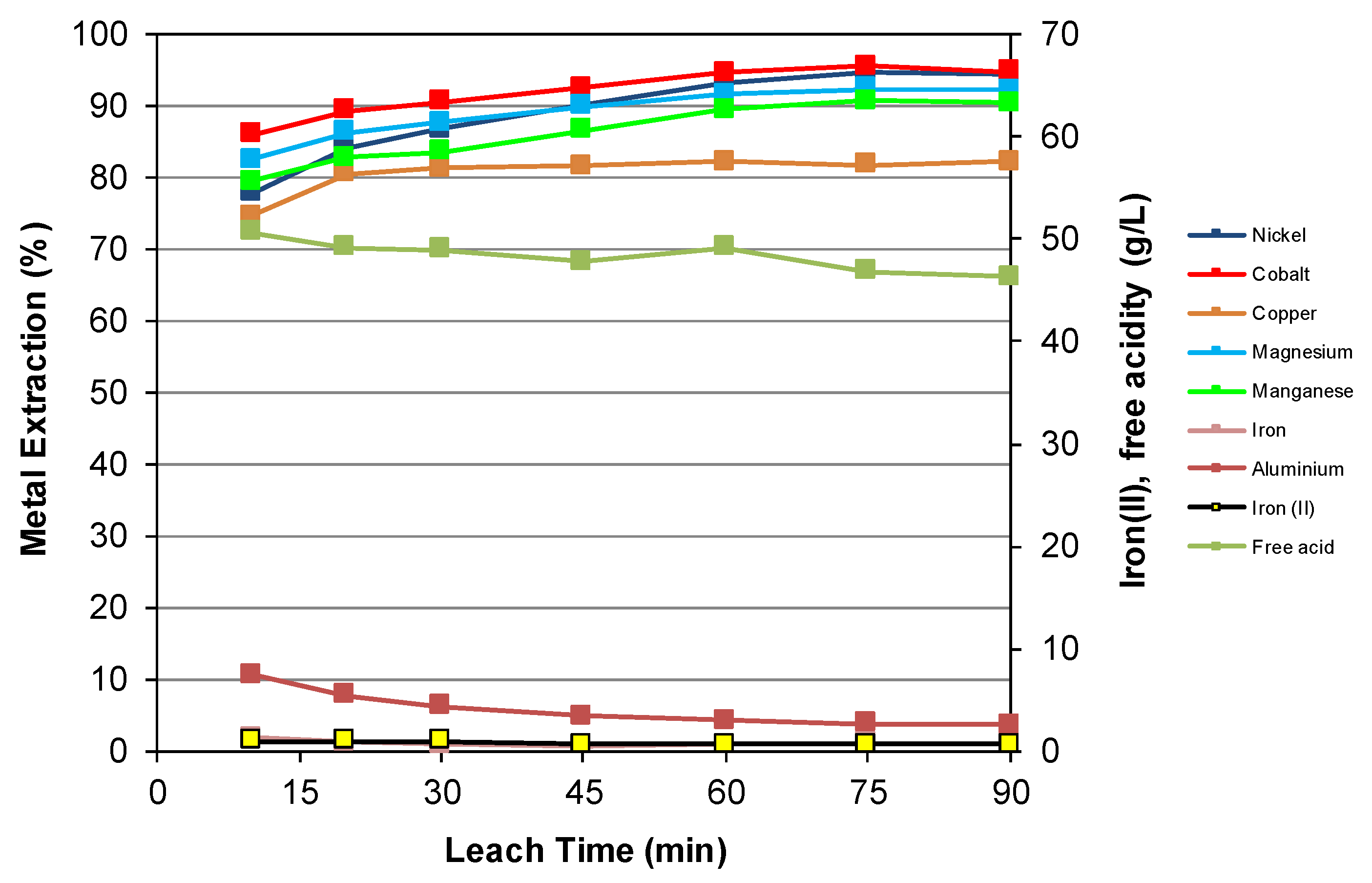
3.2. Nickel Laterite Leaching
Nax+2a(H3O)1-x-2aFey-2bAl3-y+2b(SO4)2(OH)6 + aH2SO4(aq) + bFe2(SO4)3(aq) + aH2O
(a ≤ 1 − x, b ≤ y)
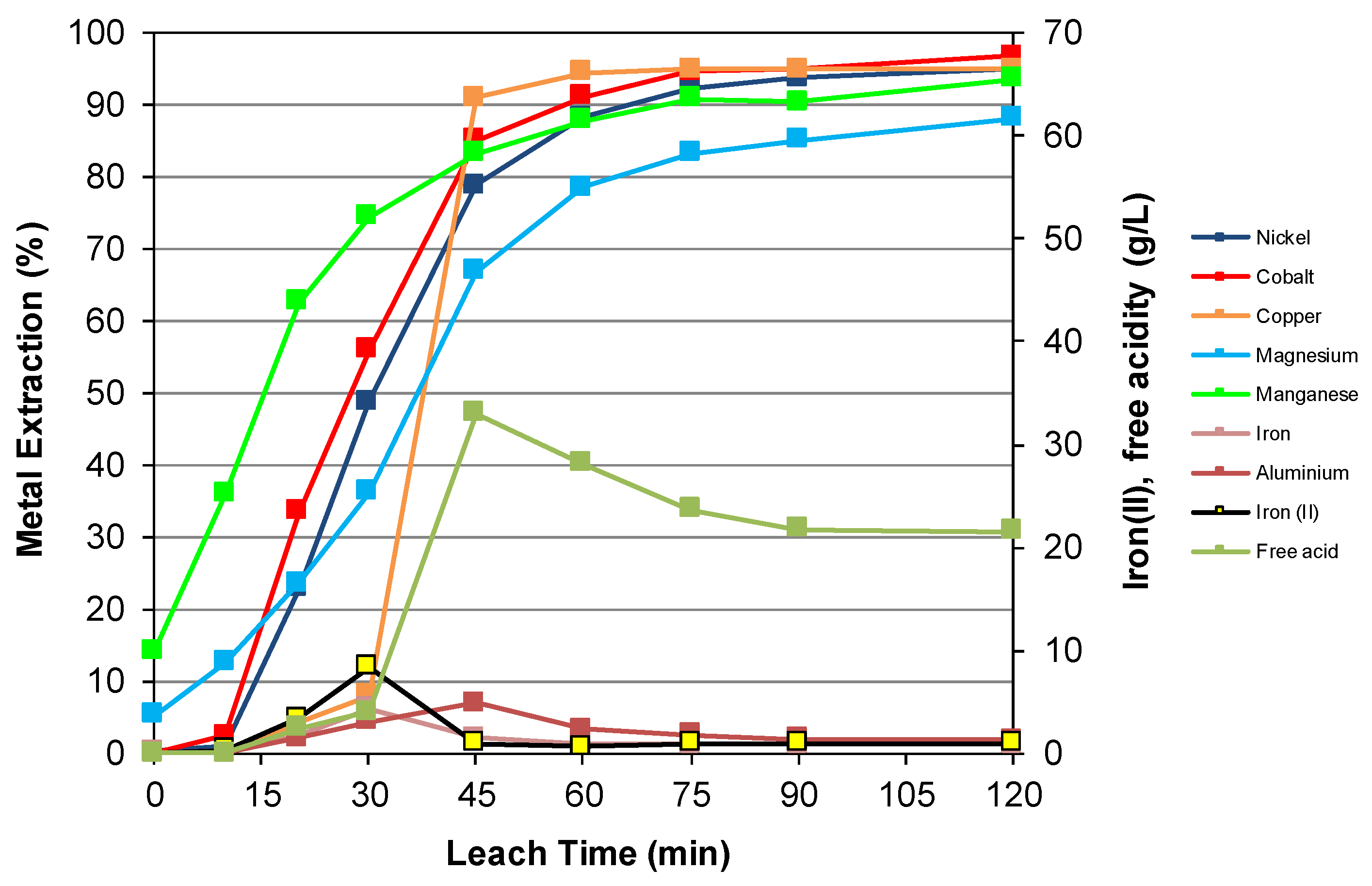
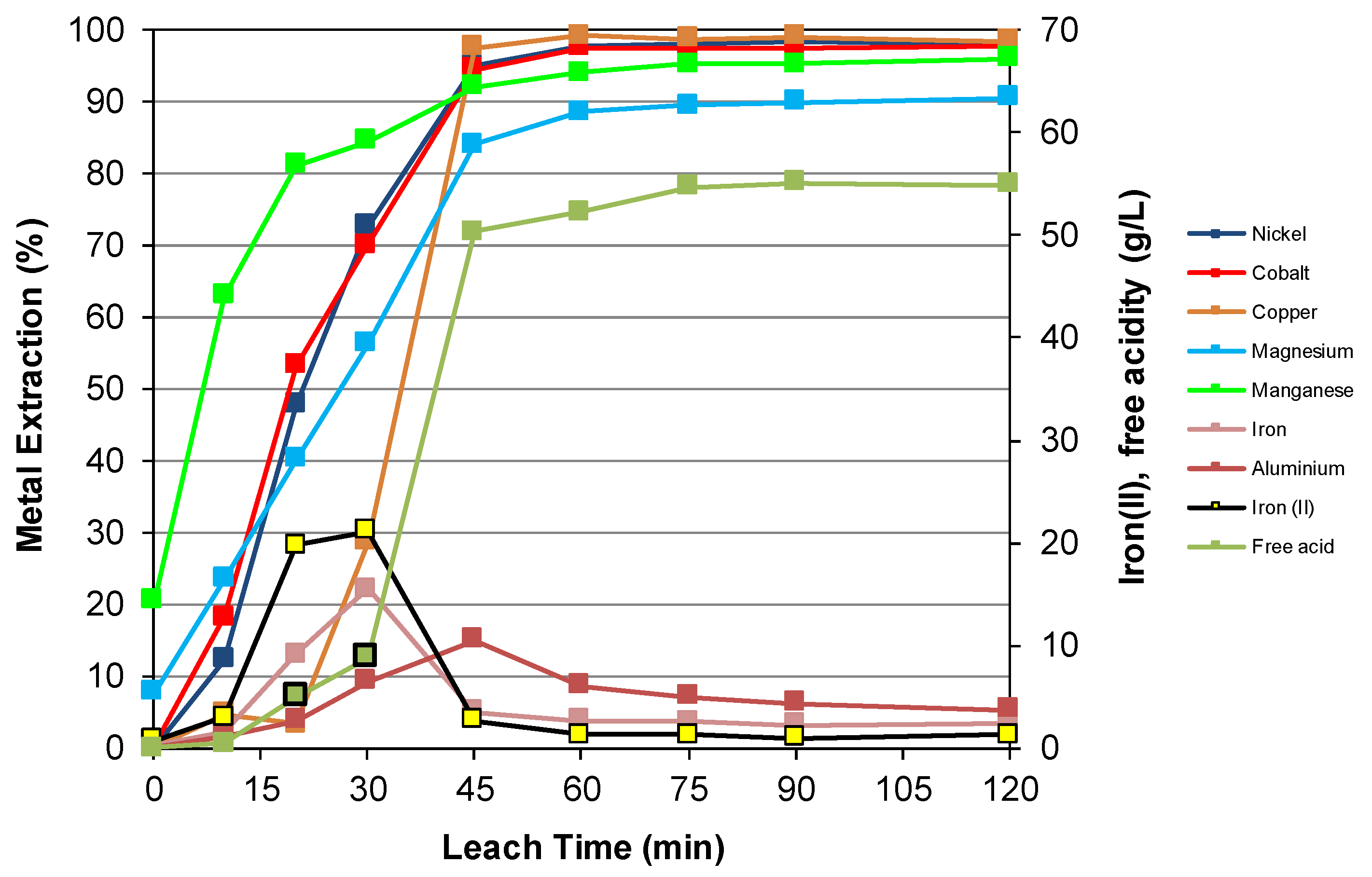
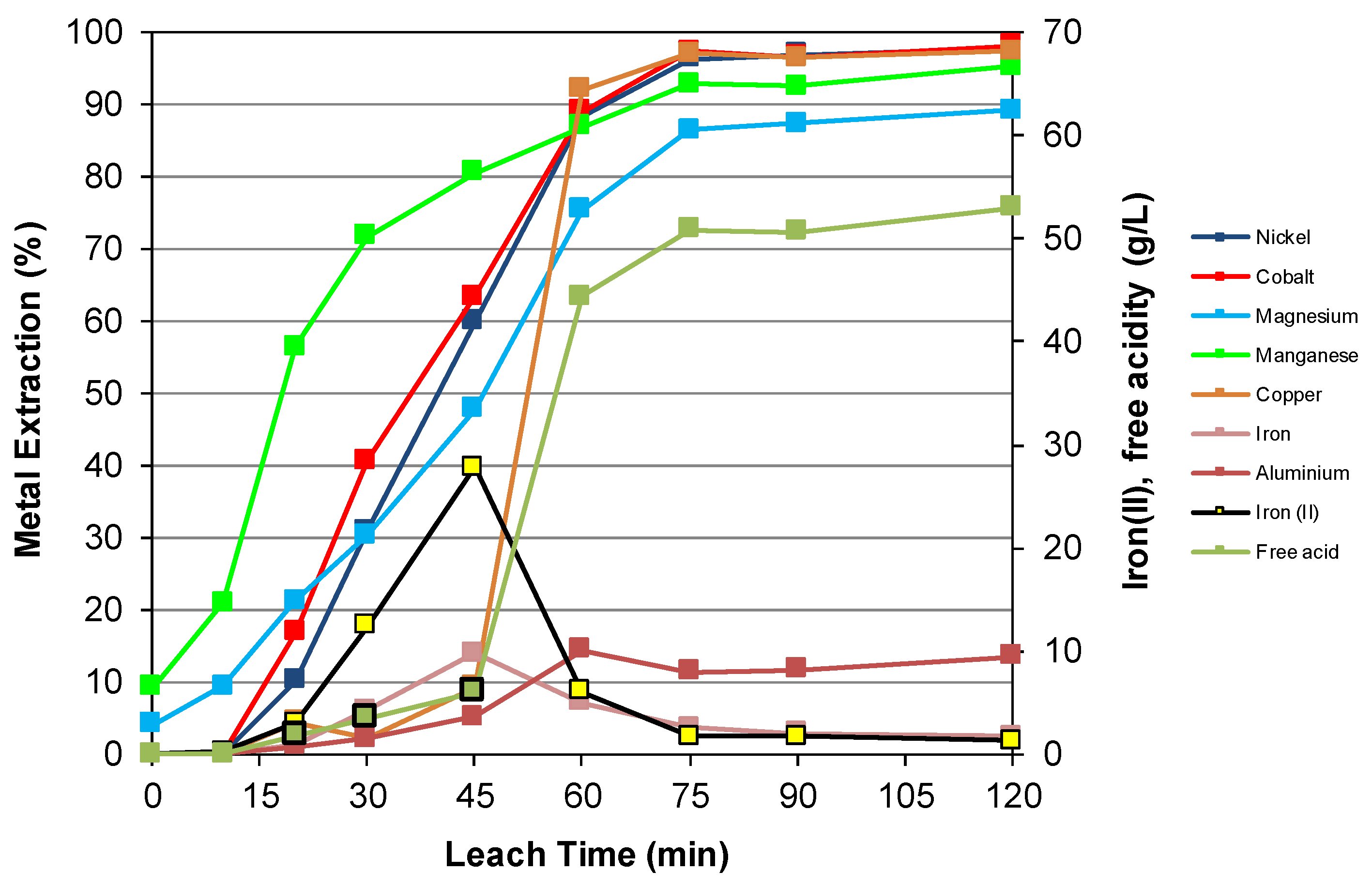
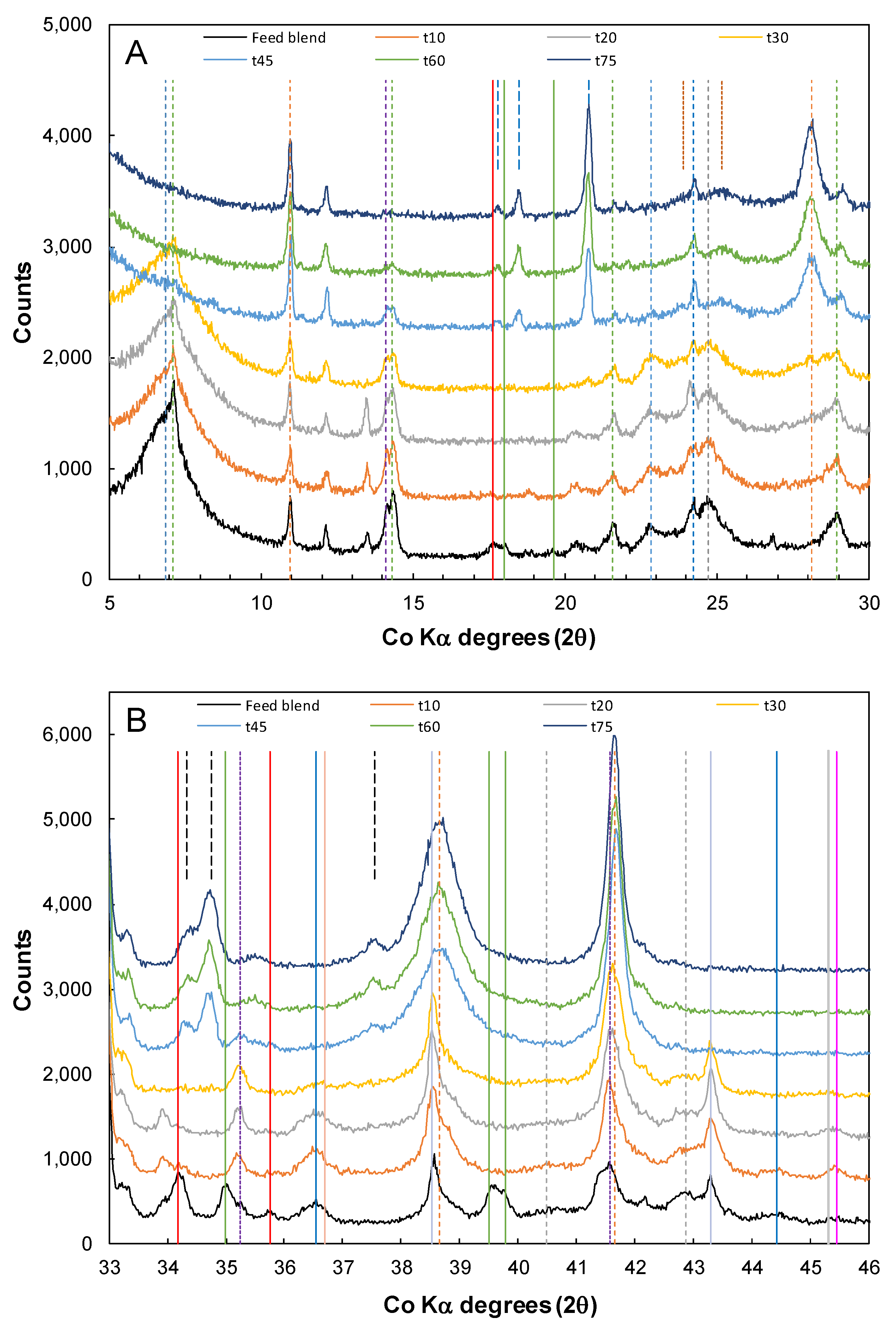
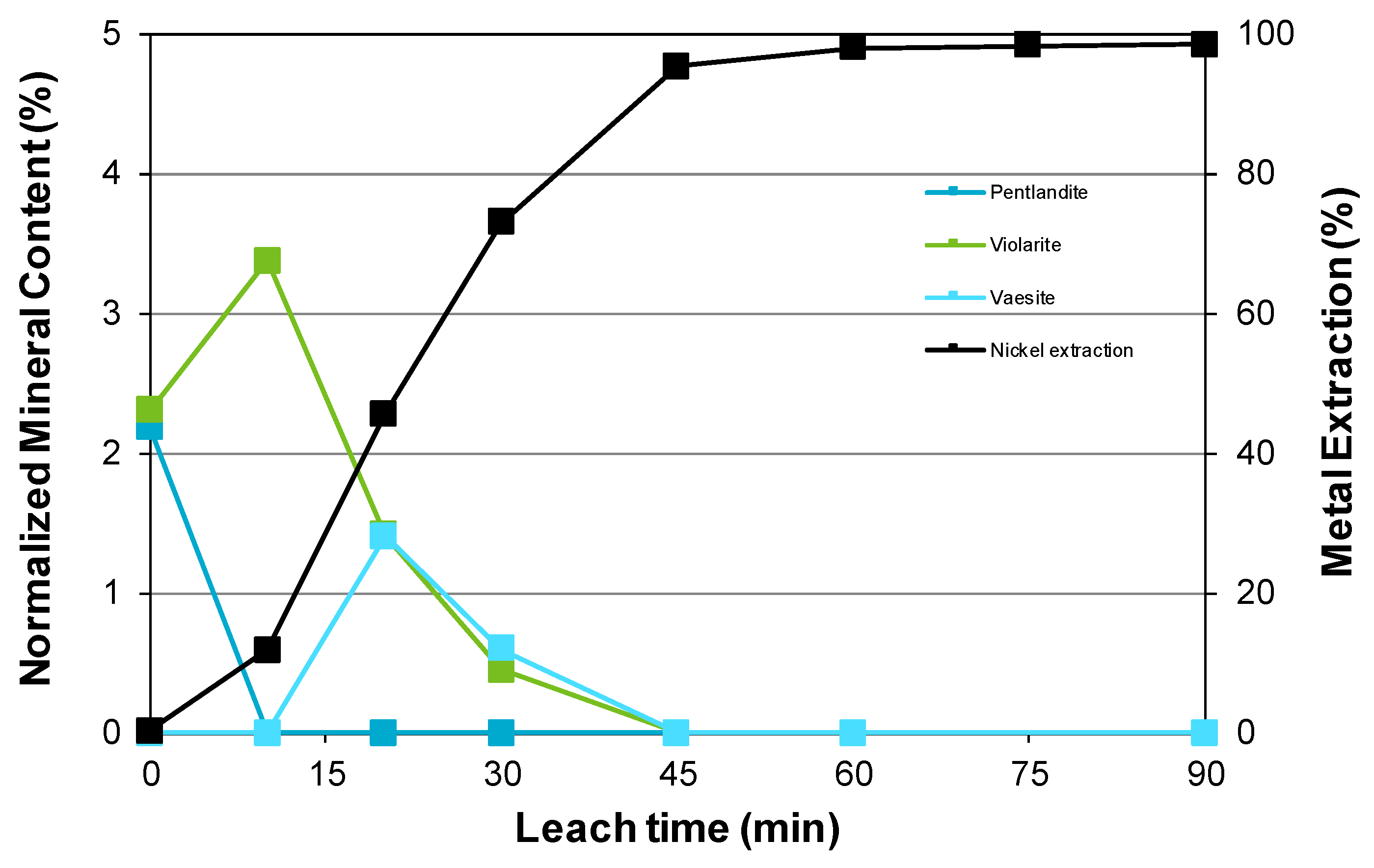
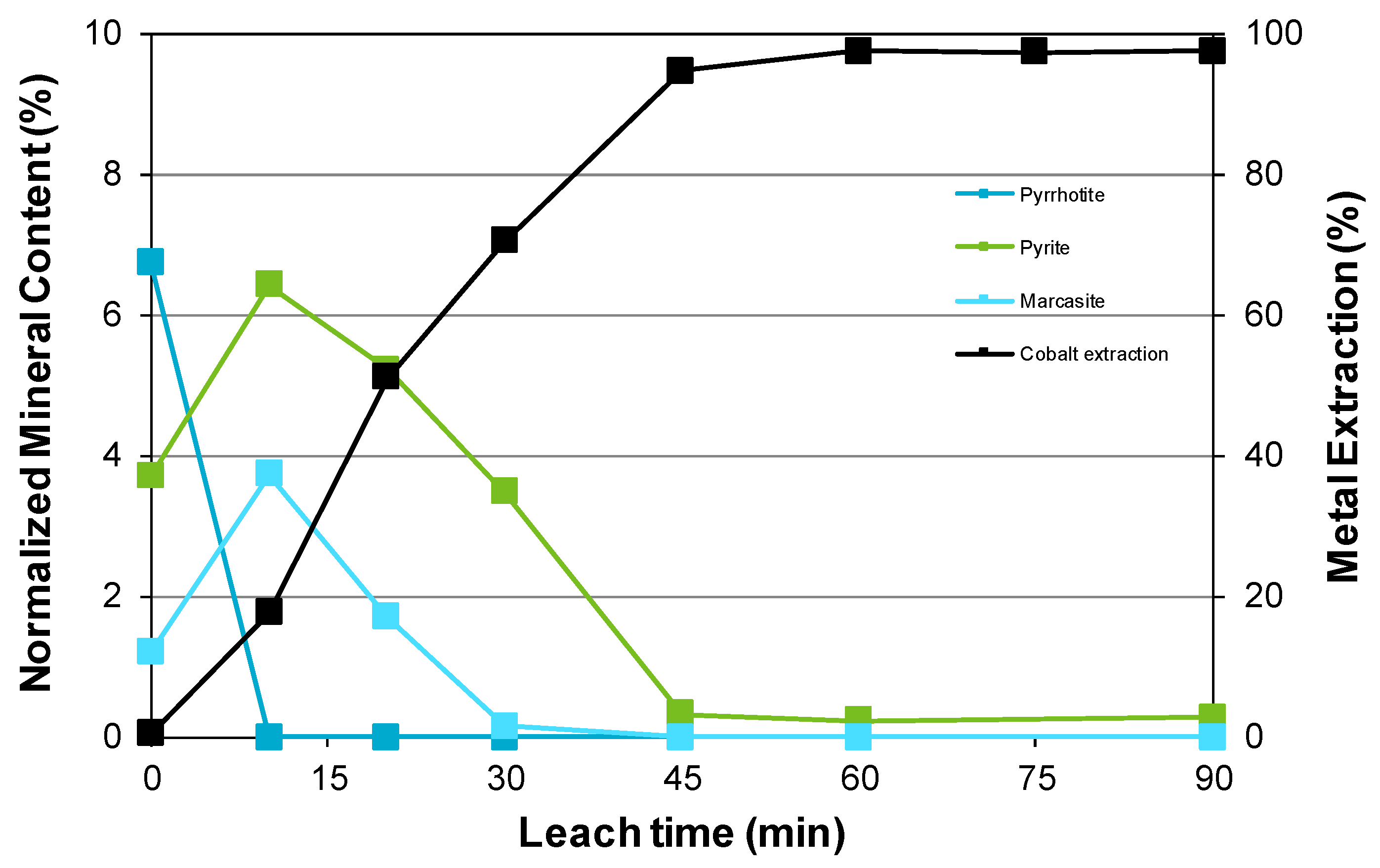
3.3. Nickel Sulfide with Nickel Laterite Co-Processing
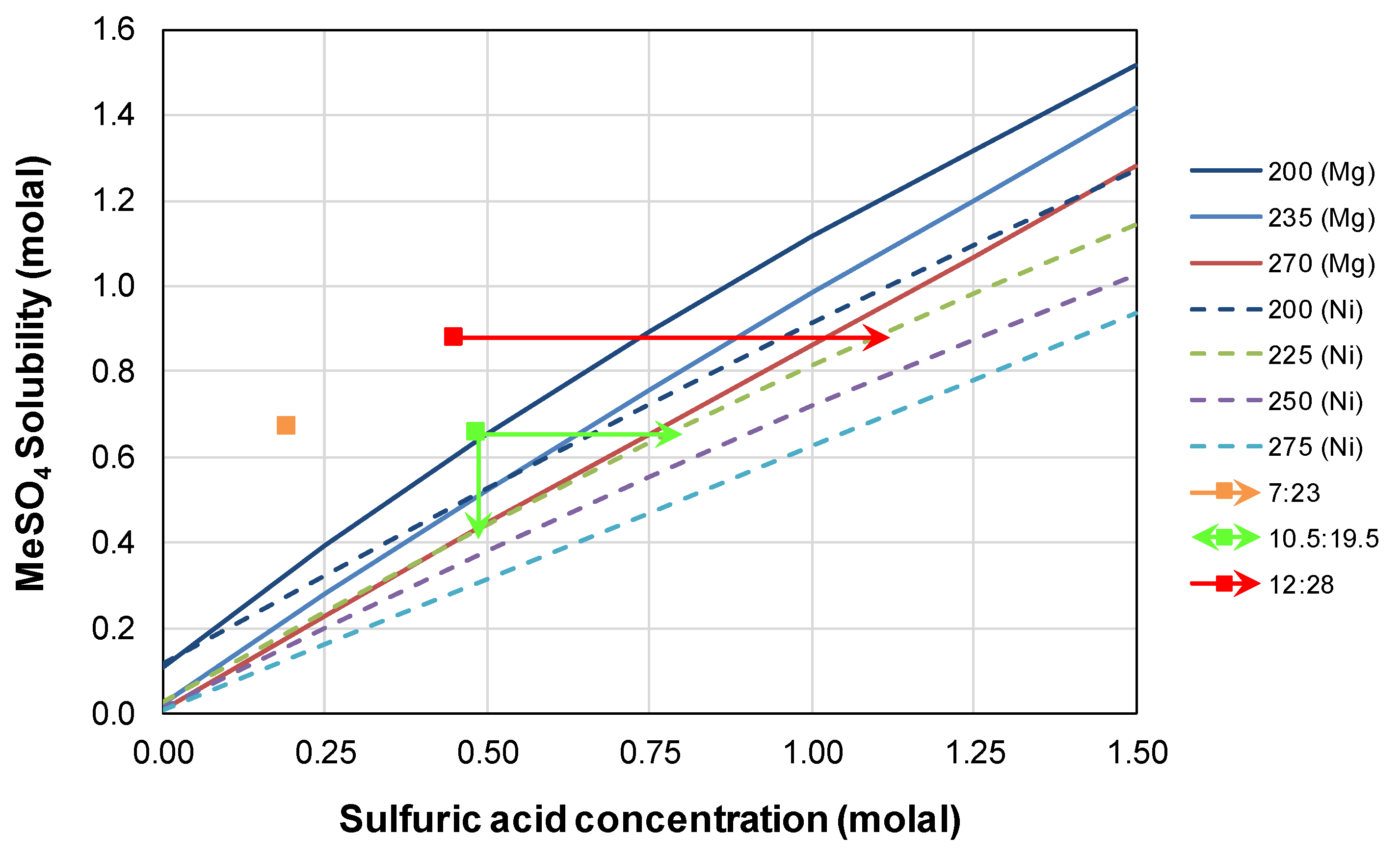
3.4. Divalent Metal Sulfate Solubility
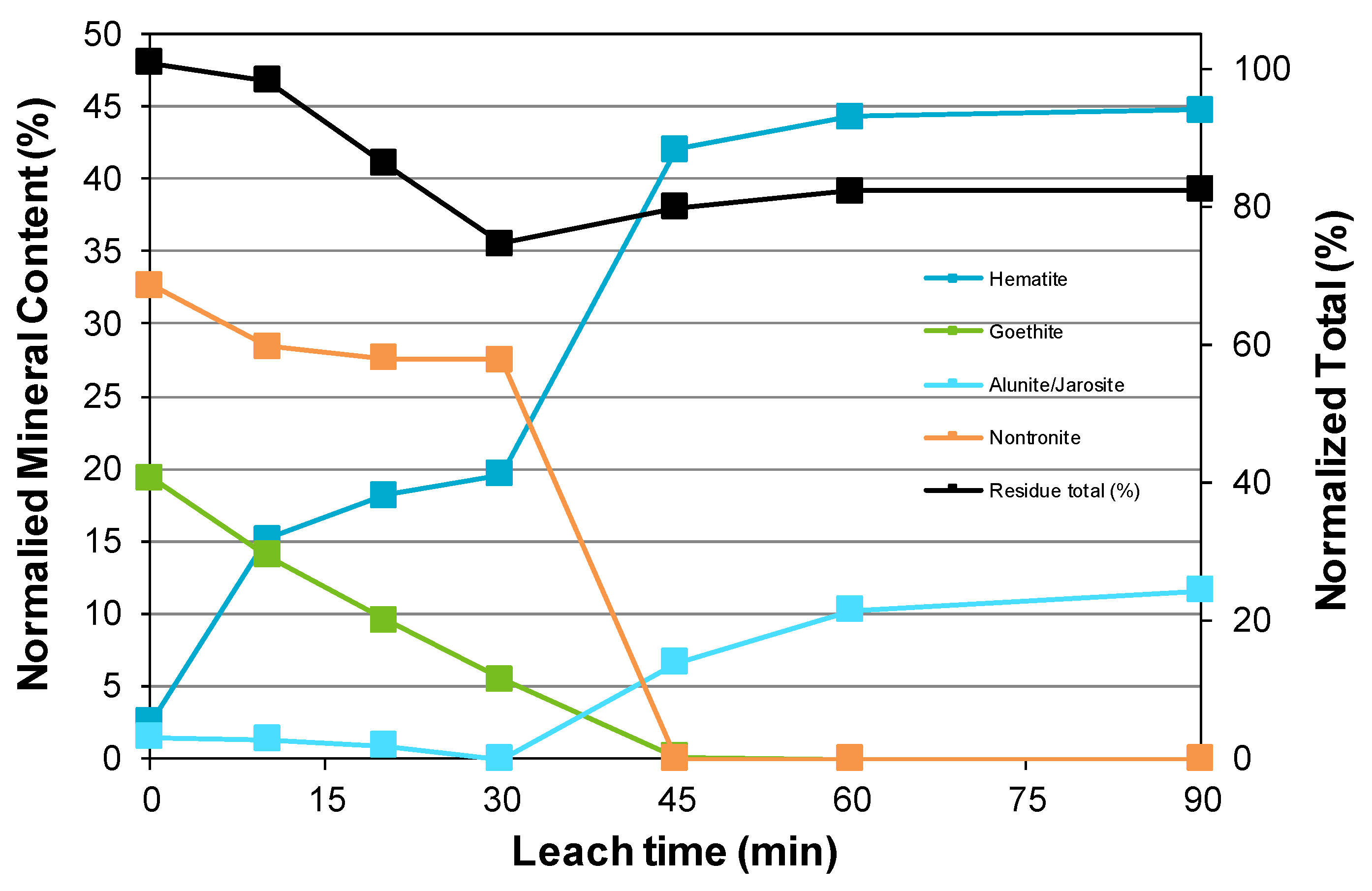
3.5. Long Terms Storage and Oxidation of Residue Samples
3.6. Comparison with Previous Studies
4. Conclusions
- High iron sulfide content feeds are highly suitable for co-processing with oxidic nickel-containing materials. Not only do these enable in situ sulfuric acid generation but the nickel and cobalt contents of the relevant minerals, typically pyrrhotite and/or pyrite. can be accessed;
- The low-grade nickel concentrate employed in this study had enough acid generating capacity to leach the nickel laterite ore without the need to provide supplementary acid;
- The mass ratio of nickel concentrate to nickel laterite can be tailored to ensure high base metal extractions and final free acidity, though the potential for the precipitation of divalent metal sulfates such as kieserite and nickel kieserite also needs to be minimized;
- Examination of the mineralogy of leach residue samples indicated that the oxidation sequence for the nickel and iron sulfide minerals is the same as that found when the nickel concentrate alone is leached; and
- For the tests conducted in this study, the iron hydrolysis products consisted mainly of hematite and an aluminum-rich sodium alunite/jarosite phase that hosts ~5% of the hydrolyzed iron in the leach residue.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gabb, J. HPAL: Upping the Pressure. Available online: https://www.gigametals.com/site/assets/files/4861/2018-03-19-hpal.pdf (accessed on 20 August 2019).
- McDonald, R.; Rodriguez, M.; Li, J.; Robinson, D.; Jackson, M.; Hosken, T. The co-processing of nickel sulphide and laterite materials using low oxygen pressures. In Proceedings of the Pressure Hydrometallurgy 2012, 42nd Annual Hydrometallurgy Meeting, Niagara Falls, ON, Canada, 30 September–3 October 2012; Collins, M.J., Filippou, D., Harlamovs, J., Peek, E., Eds.; MetSoc on behalf of the Canadian Institute of Mining, Metallurgy and Petroleum: Montreal, QC, Canada, 2012; pp. 211–225. [Google Scholar]
- Quinn, J.; Turner, J.; van der Muelen, D. The Combined Pressure Acid Leach (CPAL) process, batch and piloting testwork. In Recent Advances in Mineral Processing Plant Design; Malhotra, D., Taylor, P.R., Spiller, E., LeVier, M., Eds.; Society for Mining, Metallurgy and Exploration: Littleton, CO, USA, 2009; pp. 140–154. [Google Scholar]
- Seidel, D.C.; Fitzhugh, E.F., Jr. A hydrothermal process for oxidized nickel ores. Miner. Eng. 1968, 20, 80–86. [Google Scholar]
- Kichline, F.O. Process of Extracting Nickel and Cobalt from Ferruginous Ores. U.S. Patent 1,590,525, 29 June 1926. [Google Scholar]
- O’Neill, C.E. Acid Leaching of Lateritic Ores. U.S. Patent 3,773,891, 20 November 1973. [Google Scholar]
- Ferron, C.J.; Fleming, C.A. Co-treatment of limonitic laterites and sulphur-bearing materials as an alternative to the HPAL process. In International Nickel Laterite Symposium 2004; Imrie, W.P., Lane, D.M., Eds.; The Minerals, Metals and Materials Society: Warrendale, PA, USA, 2004; pp. 245–261. [Google Scholar]
- Opratko, V.; Bell, J.A.E.; Ferrajuolo, R.V. Acid Leaching of Laterite Ore. U.S. Patent 3,809,549, 7 May 1974. [Google Scholar]
- Salinovich, T.; Blandy, C.W.D. Mineral Processing. Australian Patent 199530448 B2, 21 March 1996. [Google Scholar]
- Rodriguez, M. Method for Recovery of Nickel from Ores. Australian Patent 2008100563 C4, 17 July 2008. [Google Scholar]
- Rodriguez, M. Method for Leaching Nickel. Australian Patent 2008101213 A4, 29 January 2009. [Google Scholar]
- O’Callaghan, J. A Hydrometallurgical Method for Leaching Base Metals. World Patent 2010/031137 A1, 25 March 2010. [Google Scholar]
- Liu, H.; Duarte, A.; Meihack, W. Consecutive or Simultaneous Leaching of Nickel and Cobalt Containing Ores. U.S. Patent 7,871,584 B2, 18 January 2011. [Google Scholar]
- Meshram, P.; Abhilash; Pandey, B.D. Advanced review on extraction of nickel from primary and secondary sources. Miner. Proc. Extr. Metall. Rev. 2019, 40, 157–193. [Google Scholar] [CrossRef]
- Oxley, A.; Smith, M.E.; Caceres, O. Why heap leach nickel laterites? Miner. Eng. 2016, 88, 53–60. [Google Scholar] [CrossRef]
- Rodriguez, M. Rheological Method for the Hydrometallurgical Recovery of Base Metals from Ores. Australian Patent 2009257204 B2, 17 December 2009. [Google Scholar]
- McDonald, R.G.; Li, J.; Austin, P.J. High temperature pressure oxidation of a low-grade nickel sulfide concentrate with control of the residue composition. Minerals 2020, 10, 249. [Google Scholar] [CrossRef]
- Wang, X.; Hart, R.D.; Li, J.; McDonald, R.G.; van Riessen, A. Quantitative analysis of turbostratically disordered nontronite with a supercell model calibrated by the PONKCS method. J. Appl. Crystallogr. 2012, 45, 1295–1302. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Hart, R.D.; Van Riessen, A.; McDonald, R. Quantitative X-ray diffraction phase analysis of poorly ordered nontronite clay in nickel laterites. J. Appl. Crystallogr. 2011, 44, 902–910. [Google Scholar] [CrossRef]
- Gerth, J. Unit-cell dimensions of pure and trace metal-associated goethites. Geochim. Cosmochim. Acta 1990, 54, 363–371. [Google Scholar] [CrossRef]
- Schwertmann, U.; Carlson, L. Aluminum influence on iron oxides: XVII. Unit-cell parameters and aluminum substitution of natural goethites. Soil Sci. Soc. Am. J. 1994, 58, 256–261. [Google Scholar] [CrossRef]
- Bertone, J.F.; Cizeron, J.; Wahi, R.K.; Bosworth, J.K.; Colvin, V.L. Hydrothermal synthesis of quartz nanocrystals. Nano Lett. 2003, 3, 655–659. [Google Scholar] [CrossRef]
- Flörke, O.W.; Graetsch, H.; Jones, J.B. Hydrothermal deposition of cristobalite. Neu. Jb. Mineral., Mon. 1990, 2, 81–95. [Google Scholar]
- Zhu, Y.; Yanagisawa, K.; Onda, A.; Kajiyoshi, K. The preparation of nano-crystallized cristobalite under hydrothermal conditions. J. Mater. Sci. 2005, 40, 3829–3831. [Google Scholar] [CrossRef]
- Bettermann, P.; Liebau, F. The transformation of amorphous silica to crystalline silica under hydrothermal conditions. Contrib. Mineral. Petr. 1975, 53, 25–36. [Google Scholar] [CrossRef]
- Carr, R.M.; Fyfe, W.S. Some observations on the crystallization of amorphous silica. Am. Mineral. 1958, 43, 908–916. [Google Scholar]
- Corwin, J.F.; Herzog, A.H.; Owen, G.E.; Yalman, R.G.; Swinnerton, A.C. The mechanism of the hydrothermal transformation of silica glass to quartz under isothermal conditions. J. Am. Chem. Soc. 1953, 75, 3933–3934. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Zhu, Y.; Onda, A.; Kajiyoshi, K. Hydrothermal synthesis of mono-dispersed quartz powders. J. Mater. Sci. 2004, 39, 2931–2934. [Google Scholar] [CrossRef]
- Campbell, A.S.; Fyfe, W.S. Hydroxyl ion catalysis of the hydrothermal crystallization of amorphous silica; a possible high temperature pH indicator. Am. Mineral. 1960, 45, 464–468. [Google Scholar]
- Alekseyev, V.A.; Medvedeva, L.S.; Kochnova, L.N.; Tytyunnik, O.A. Mechanisms of silica precipitation from hydrothermal solutions: The effects of solution evaporations and quartz seed crystals. Geochem. Int. 2010, 48, 178–182. [Google Scholar] [CrossRef]
- Görlich, E. The structure of SiO2—Current views. Ceram. Int. 1982, 8, 3–16. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Jambor, J.L. Jarosites and their application in hydrometallurgy. Rev. Mineral. Geochem. 2000, 40, 405–452. [Google Scholar] [CrossRef]
- Figueiredo, M.O.; Pereira da Silva, T. The positive environmental contribution of jarosite by retaining lead in acid mine drainage areas. Int. J. Environ. Res. Public Health 2011, 8, 1575–1582. [Google Scholar] [CrossRef]
- Makhmudov, S.A.; Kashkai, C.M. Unit cell parameters of the alunite-group minerals and their structural analogs. Tr. Azerb. Otd. Vses. Mineral. (b) O-va 1981, No. 2, 68–72. (In Russian) [Google Scholar]
- Whittington, B.I. Characterization of scales obtained during continuous nickel laterite pilot-plant leaching. Metall. Mater. Trans. B 2000, 31, 1175–1186. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Brophy, G.P.; Sheridan, M.F. Sulfate studies IV: The jarosite-natrojarosite-hydronium jarosite solid solution series. Am. Mineral. 1965, 50, 1595–1607. [Google Scholar]
- Kubisz, J. Studies on synthetic alkali-hydronium jarosites. I. Synthesis of jarosite and natrojarosite. Mineral. Polonica 1970, 1, 47–59. [Google Scholar]
- Ripmeester, J.A.; Ratcliffe, C.I.; Dutrizac, J.E.; Jambor, J.L. Hydronium ion in the alunite-jarosite group. Can. Mineral. 1986, 24, 435–447. [Google Scholar]
- Alpers, C.N.; Nordstrom, D.; Ball, J.W. Solubility of jarosite solid solutions precipitated from acid mine waters, Iron Mountain, California, U.S.A. Sci. Géol. Bull. 1989, 42, 281–298. [Google Scholar]
- Drouet, C.; Navrotsky, A. Synthesis, characterization, and thermochemistry of K-Na-H3O jarosites. Geochim. Cosmochim. Acta 2003, 67, 2063–2076. [Google Scholar] [CrossRef]
- Drouet, C.; Pass, K.L.; Baron, D.; Draucker, S.; Navrotsky, A. Thermochemistry of jarosite-alunite and natrojarosite-natroalunite solid solutions. Geochim. Cosmochim. Acta 2004, 68, 2197–2205. [Google Scholar] [CrossRef]
- Basciano, L.C.; Peterson, R.C. Jarosite–hydronium jarosite solid-solution series with full iron site occupancy: Mineralogy and crystal chemistry. Am. Mineral. 2007, 92, 1464–1473. [Google Scholar] [CrossRef]
- Basciano, L.C.; Peterson, R.C. The crystal structure of ammoniojarosite, (NH4)Fe3(SO4)2(OH)6 and the crystal chemistry of the ammoniojarosite–hydronium jarosite solid-solution series. Mineral. Mag. 2007, 71, 427–441. [Google Scholar] [CrossRef]
- Basciano, L.C.; Peterson, R.C. Crystal chemistry of the natrojarosite-jarosite and natrojarosite-hydronium jarosite solid-solution series: A synthetic study with full Fe site occupancy. Am. Mineral. 2008, 93, 853–862. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Mason, R.; Schmidt, P. Synthetic alunites of the potassium-oxonium solid solution series and some other members of the group: Synthesis, thermal and X-ray characterization. Eur. J. Mineral. 2003, 15, 913–924. [Google Scholar] [CrossRef]
- Jones, F. Crystallization of jarosite with variable Al3+ content: The transition to alunite. Minerals 2017, 7, 90. [Google Scholar] [CrossRef]
- Majzlan, J.; Stevens, R.; Boerio-Goates, J.; Woodfield, B.F.; Navrotsky, A.; Burns, P.C.; Crawford, M.K.; Amos, T.G. Thermodynamic properties, low-temperature heat-capacity anomalies, and single-crystal X-ray refinement of hydronium jarosite,(H3O)Fe3(SO4)2(OH)6. Phys. Chem. Miner. 2004, 31, 518–531. [Google Scholar] [CrossRef]
- Becker, U.; Gasharova, B. AFM observations and simulations of jarosite growth at the molecular scale: Probing the basis for the incorporation of foreign ions into jarosite as a storage mineral. Phys. Chem. Miner. 2001, 28, 545–556. [Google Scholar] [CrossRef]
- Lager, G.A.; Swayze, G.A.; Loong, C.K.; Rotella, F.J.; Richardson, J.W., Jr.; Stoffregen, R.E. Neutron spectroscopic study of synthetic alunite and oxonium-substituted alunite. Can. Mineral. 2001, 39, 1131–1138. [Google Scholar] [CrossRef][Green Version]
- Nielsen, U.G.; Majzlan, J.; Phillips, B.; Ziliox, M.; Grey, C.P. Characterization of defects and the local structure in natural and synthetic alunite (K,Na,H3O)Al3(SO4)2(OH)6 by multi-nuclear solid-state NMR spectroscopy. Am. Mineral. 2007, 92, 587–597. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Kaiman, S. Synthesis and properties of jarosite-type compounds. Can. Mineral. 1976, 14, 151–158. [Google Scholar]
- Whittington, B.I.; McDonald, R.G.; Johnson, J.A.; Muir, D.M. Pressure acid leaching of arid-region nickel laterite ore: Part I: Effect of water quality. Hydrometallurgy 2003, 70, 31–46. [Google Scholar] [CrossRef]
- Brown, M. Murrin Murrin operations—Past, present and future. In Proceedings of the ALTA (2nd) Nickel-Cobalt-Copper Conference, Perth, Australia, 23–25 May 2011; ALTA Metallurgical Services: Castlemaine, Australia; pp. 435–499. [Google Scholar]
- Willis, B. Atmospheric tank leach flowsheet development for the Çaldağ Nickel Project in Western Turkey. In Proceedings of the ALTA (5th) Nickel-Cobalt-Copper Conference, Perth, Australia, 26–28 May 2014; ALTA Metallurgical Services: Melbourne, Australia; pp. 66–78. [Google Scholar]
- Rubisov, D.H.; Krowinkel, J.M.; Papangelakis, V.G. Sulphuric acid pressure leaching of laterites—Universal kinetics of nickel dissolution for limonites and limonitic/saprolitic blends. Hydrometallurgy 2000, 58, 1–11. [Google Scholar] [CrossRef]
- Madhuchhanda, M.; Devi, N.B.; Rao, K.S.; Rath, P.C.; Paramguru, R.K. Galvanic interaction between sulfide minerals and pyrolusite. J. Solid State Electr. 2000, 4, 189–198. [Google Scholar] [CrossRef]
- Elias, M.; Donaldson, M.J.; Giorgetta, N.E. Geology, mineralogy, and chemistry of lateritic nickel-cobalt deposits near Kalgoorlie, Western Australia. Econ. Geol. 1981, 76, 1775–1783. [Google Scholar] [CrossRef]
- Tindall, G.P.; Muir, D.M. Transformation of iron oxide in nickel laterite processing. In: Iron Control and Disposal. In Proceedings of the Second International Symposium on Iron Control in Hydrometallurgy, Ottawa, QC, Canada, 20–23 October 1996; Dutrizac, J.E., Harris, G.B., Eds.; Canadian Institute of Mining, Metallurgy and Petroleum: Montréal, QC, Canada, 1996; pp. 249–262. [Google Scholar]
- Queneau, P.B.; Doane, R.E.; Cooperrider, M.W.; Berggren, M.H.; Rey, P. Control of autoclave scaling during acid pressure leaching of nickeliferous laterite ore. Metall. Trans. B 1984, 15, 433–440. [Google Scholar] [CrossRef]
- Scarlett, N.V.; Madsen, I.C.; Whittington, B.I. Time-resolved diffraction studies into the pressure acid leaching of nickel laterite ores: A comparison of laboratory and synchrotron X-ray experiments. J. Appl. Crystallogr. 2008, 41, 572–583. [Google Scholar] [CrossRef]
- Talla, D.; Wildner, M. Investigation of the kieserite–szomolnokite solid-solution series, (Mg,Fe)SO4 H2O, with relevance to Mars: Crystal chemistry, FTIR, and Raman spectroscopy under ambient and martian temperature conditions. Am. Mineral. 2019, 104, 1732–1749. [Google Scholar] [CrossRef]
- Kobylin, P.M.; Sippola, H.; Taskinen, P.A. Thermodynamic model for acidic Fe(II) sulphate from solubility data. Calphad 2012, 38, 185–193. [Google Scholar] [CrossRef]
- Marshall, W.L.; Slusher, R. Aqueous systems at high temperature. XV. Solubility and hydrolytic instability of magnesium sulfate in sulfuric acid-water and deuterosulfuric acid-deuterium oxide solutions, 200° to 350°C. J. Chem. Eng. Data 1965, 10, 353–358. [Google Scholar] [CrossRef]
- Marshall, W.L.; Gill, J.S.; Slusher, R. Aqueous systems at high temperature—VI Investigations on the system NiO-SO3-H2O and its D2O analogue from 10−4 to 3 m SO3, 150–450 °C. J. Inorg. Nucl. Chem. 1962, 24, 889–897. [Google Scholar] [CrossRef]
- Liu, H.; Papangelakis, V.G. Chemical modeling of high temperature aqueous processes. Hydrometallurgy 2005, 79, 48–61. [Google Scholar] [CrossRef]
- Shibayama, K.; Yokogawa, T.; Sato, H.; Enomoto, M.; Nakai, O.; Ito, T.; Mizuno, F.; Hattori, Y. Taganito HPAL Plant Project. Min. Eng. 2016, 88, 61–65. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Q.; Hu, H.; Ding, Z.; Yin, Z. Characteristics of scales formed from pressure leaching of Yuanjiang laterite. Hydrometallurgy 2011, 109, 131–139. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feed | Ni | Co | Cu | Mg | Mn | Fe | Al | Cr | Ca | Na | Si | S |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentrate | 8.11 | 0.138 | 1.28 | 0.651 | 0.037 | 45.0 | 0.318 | 0.175 | 0.421 | 0.082 | 2.48 | 30.3 |
| Laterite | 1.90 | 0.131 | 0.011 | 3.32 | 0.456 | 24.6 | 2.47 | 0.857 | 0.170 | 0.201 | 16.8 | 0.02 |
| Bulong Laterite Blend | % (w/w) | Nickel Concentrate | % (w/w) |
|---|---|---|---|
| Nontronite | 55 | Pyrrhotite 4C | 39 |
| Goethite | 26 | Pyrite | 13 |
| Spinel * | 7 | Pentlandite | 8 |
| Maghemite | 2 | Violarite | 9 |
| Magnesite | 1 | Chalcopyrite | 3 |
| Quartz | 3 | Quartz | 4 |
| Clinochlore | 3 | Clinochlore | 3 |
| Actinolite | 1 | Actinolite | 2 |
| Lizardite | 0.7 | Talc | 4 |
| Talc | 0.3 | Hematite | 4 |
| Hydronium jarosite | 5 | ||
| Nickel hexahydrite | 1.8 |
| Study | BlendSolids (% w/w) | |||||||||
| Ni | Co | Fe | Mn | Al | Cr | Mg | Ca | Si | S | |
| Current | 4.07 | 0.133 | 31.8 | 0.310 | 1.72 | 0.618 | 2.39 | 0.257 | 11.8 | 10.6 |
| Quinn et al. (B) | 1.24 | 0.055 | 12.2 | nr | 4.74 | nr | 5.46 | nr | nr | 3.20 * |
| Quinn et al. (P) | 1.41 | 0.020 | 15.4 | 0.072 | 3.72 | nr | 4.54 | 1.03 | nr | 2.52 * |
| Process Liquor in (mg/L) | FA (g/L) | |||||||||
| Ni | Co | Fe | Fe(II) | Al | Cr | Mg | Ca | Na | ||
| Current | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | 148 | 51 | 1380 | Nil |
| Quinn et al. (B) | 4350 | 326 | 41,500 | 1790 | 8550 | 834 | 21,300 | 371 | nr | 15.3 |
| Quinn et al. (P) | 3820 | 314 | 37,500 | 1540 | 7990 | nr | 15,900 | nr | 21,100 | 24.6 |
| Process Liquor out (mg/L) | FA (g/L) | |||||||||
| Ni | Co | Fe | Fe(II) | Al | Cr | Mg | Ca | Na | ||
| Current | 17,100 | 637 | 4820 | 1340 | 435 | 101 | 11,200 | 635 | 664 | 54.9 |
| Quinn et al. (B) | 13,000 | 669 | 5620 | nr | 2510 | nr | nr | nr | nr | 56.2 |
| Quinn et al. (P) | 8700 | 394 | 6710 | 2280 | 968 | nr | nr | nr | 880 | 46.7 |
| Extraction (%) | ||||||||||
| Ni | Co | Fe | Mn | Al | Cr | Mg | Ca | |||
| Current | 98.0 | 97.8 | 3.3 | 96.0 | 5.4 | 3.4 | 89.3 | 61.8 | ||
| Quinn et al. (B) | 97.5 | 96.4 | nr | nr | nr | nr | nr | nr | ||
| Quinn et al. (P) | 91.9 | 78.8 | nr | nr | nr | nr | nr | nr | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonald, R.G.; Li, J. The High Temperature Co-Processing of Nickel Sulfide and Nickel Laterite Sources. Minerals 2020, 10, 351. https://doi.org/10.3390/min10040351
McDonald RG, Li J. The High Temperature Co-Processing of Nickel Sulfide and Nickel Laterite Sources. Minerals. 2020; 10(4):351. https://doi.org/10.3390/min10040351
Chicago/Turabian StyleMcDonald, Robbie G., and Jian Li. 2020. "The High Temperature Co-Processing of Nickel Sulfide and Nickel Laterite Sources" Minerals 10, no. 4: 351. https://doi.org/10.3390/min10040351
APA StyleMcDonald, R. G., & Li, J. (2020). The High Temperature Co-Processing of Nickel Sulfide and Nickel Laterite Sources. Minerals, 10(4), 351. https://doi.org/10.3390/min10040351