Dissolution, Stability and Solubility of Tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] at 25–45 °C and pH 2–12
by
 , , ,
, , ,
Zongqiang Zhu
1,2 ,
,
Jun Zhang
1,
Yinian Zhu
1,*,
Jie Liu
1,
Shen Tang
2,
Lihao Zhang
2,* and
Yaru Wang
1 1
College of Environmental Science and Engineering, Guilin University of Technology, Guilin 541004, China
2
Collaborative Innovation Center for Water Pollution Control and Water Safety in Karst Area, Guilin University of Technology, Guilin 541004, China
*
Authors to whom correspondence should be addressed.
Minerals 2020, 10(10), 921; https://doi.org/10.3390/min10100921
Submission received: 30 September 2020
/
Revised: 9 October 2020
/
Accepted: 16 October 2020
/
Published: 19 October 2020
(This article belongs to the Special Issue Elemental and Isotopic Approaches to Characterize Sources and Processes Controlling the Budgets of Toxic Metals in the Environment)
Abstract
:Tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] was synthesized and characterized to investigate its possible immobilization for arsenic in acidic and alkali environments by a long-term dissolution of 330 d. The synthetic tooeleite was platy crystallites of ~1μm across, giving the lattice parameters of a = 6.4758 Å, b = 19.3737 Å and c = 8.9170 Å. For the tooeleite dissolution, the dissolved arsenic concentration showed the lowest value of 427.3~435.8 mg/L As at initial pH 12 (final pH 5.54). The constituents were dissolved preferentially in the sequence of SO42− > AsO33− > Fe3+ in the aqueous medium at initial pH 2–12. The dissolved iron, arsenite and sulfate existed mainly as FeSO4+/Fe3+, H3AsO30 and SO42− at initial pH 2, and in the form of Fe(OH)30/Fe(OH)2+, H3AsO30 and SO42− at initial pH 12, respectively. The tooeleite dissolution was characterized by the preferential releases of SO42− anions from solid surface into aqueous medium, which was fundamentally controlled by the Fe-O/OH bond breakages and the outer OH− group layers. From the data of the dissolution at 25 °C and initial pH 2 for 270–330 d, the ion-activity product [logˍIAP], which equaled the solubility product [Ksp] at the dissolution equilibrium, and the Gibbs free energy of formation [ΔGfo] were estimated as −200.28 ± 0.01 and −5180.54 ± 0.07 kJ/mol for the synthetic tooeleite, respectively.
1. Introduction
As arsenic is found widely in Earth’s crust and is one of the chemicals of greatest health concern, inorganic arsenic compounds were classified by the International Agency for Research on Cancer in Group 1 (carcinogenic to humans) [1]. The extremely toxic arsenic is common in wastes from the mining-metallurgical industry for non-ferrous and precious metals. It can be released into the environment and, finally, threaten human beings [1,2]. The elimination of the most toxic inorganic As(III) is more difficult owing to its higher solubility and mobility than As(V) [3,4]. It is still a practical challenge to eliminate trivalent arsenic effectively from contaminated waters with very high arsenic concentration and low pH [5].
As-bearing minerals are important in the dissolution–precipitation equilibria and geochemical cycling of arsenic [6,7]. It is difficult to assess the arsenic contamination scale exactly [8]. The weathering of the mining tailings of metallic sulfide ores can lead to the formations of acid mine drainages (AMDs), containing very high contents of iron, sulfate and toxic metal(loid)s including As, Pb, Zn, Cd, Co, Cu, Hg, Mo, Ni, Ru, Sb, Se, Sn, Te, Bi, etc. [9,10,11]. The oxidation of ferrous ions and the progressive neutralization of AMDs can commonly result in the precipitation of iron oxy-hydroxides and sulfates within the sequence ferrihydrite, goethite, lepidocrocite, jarosite and schwertmannite [12,13,14,15]. Arsenic can be removed by adsorption onto different mineral surfaces or by the structural substitution into various minerals, consequently restraining its mobility [14,15,16]. Therefore, the arsenic substitution into mineral crystal structure can offer an alternative remediation strategy for arsenic-polluted wastewaters [17,18,19], e.g., calcium arsenates [20], scorodite [21], reinerite and cafarsite [22], nealite [23], alunite group minerals [14,24,25], etc.
The precipitation of these minerals could be strengthened by the bacteria metabolism, e.g., Acidithiobacillus ferrooxidans could enhance the oxidation of ferrous ions into ferric ions by dissolved oxygen and result in the precipitation of tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] [10,13,14,15], a sole mineral of ferric arsenite sulfate that had drawn the attention of many scholars [26,27] and was proposed as a possible compound to fix trivalent arsenic due to its stable occurrence in mine areas [28,29,30]. Tooeleite is an As(III) mineral, even though for a long time it was first considered as a novel arsenate mineral from the US Mine at Gold Hill in Tooele County, Utah, USA, where tooeleite naturally occurred with arseno-pyrite, pyrite, scorodite and jarosite [31,32]. In fact, tooeleite was an arsenite mineral and could be simply synthesized from the aqueous solution of the pure constituents at temperature >90 °C [28]. The X-ray diffraction (XRD) data of the mineral indicated the orthorhombic structure which was accepted as No. 44-1468 by the International Centre for Diffraction Data (ICDD) [32]. In the crystal structure of tooeleite, the FeO6-octahedra are linked with the AsO3-pyramids by both corner- and edge-linkages, forming positively charged As-Fe (oxy-)hydroxide layers, and the isolated SO4-tetrahedra reside in the interlayer space [31]. The high acidity/iron/sulfate concentration with proper Fe/As ratio played a very important role in the precipitation of well-crystalline tooeleite [28]. Tooeleite has also been found in the acid hydrometallurgical wastes in SãoBento gold mine, Brazil [33]. Tooeleite was recognized as the unique arsenite-sulfate in the extremely As(III)-rich AMD at the Carnoulès mine, Gard, France [15] and its precipitation decreased considerably the As(III) concentration in the Amous river downstream [34]. In the seriously polluted AMD, As(III) reached 80–280 mg/L in the acidic spring discharge of the waste-piles [34]. Characterization of the stromatolite and sediments showed the uncommon precipitation of As(III)-rich minerals, particularly the nano-crystalline tooeleite and amorphous Fe(III)-As(III)/As(V) (oxy-)hydroxides, which gave a better understanding of the natural process and might contribute to planning effective arsenic-elimination procedures [31]. The low pH environment was helpful in the tooeleite formation [35]. The tooeleite stability and crystallinity could be affected by the coexisting arsenate and sulfate ions [27]. Tooeleite precipitation could be an alternate technique to eliminate As(III) directly from high-As acid wastewaters [35]. The precipitation experiment indicated that the As(III) elimination efficiency could reach up to 99% at the As(III) concentrations greater than 0.75 g/L with the Fe/As mole ratio of 0.8~2.0 and the initial pH of 1.8~4.5 [5]. The leaching test of tooeleite with 0.01 M Na-acetate buffer of pH 4.93 exhibited that the leached arsenic concentration reached only about 9 mg/L [7]. However, a previous batch experiment showed also that tooeleite coprecipitated at pH 2~3.5 and would quickly transform to an amorphous ferric arsenite at pH > 4, which showed a higher As solubility in the leaching test [30]. In the dissolution experiment of tooeleite for 30 days, the concentration of arsenic released from solid into solution decreased in the first two days, which was possibly related to the re-immobilization of arsenite onto surface, and then slowly increased up to 100 mg/L [35]. The leached arsenic concentrations were >100 mg/L [36] or 350~650 mg/L [27], which indicated that the mineral tooeleite seemed an unlikely candidate for arsenite fixation at contaminated sites [10,37]. Nevertheless, the incongruent dissolution of tooeleite would lead to the precipitation of poorly crystallized iron hydroxides that could restrain the arsenite release again into the neighboring environment under alkaline conditions. Thus, it is required to evaluate the long-term fixation ability of tooeleite in the further work [27].
The solubilities of arsenic-containing minerals are significant in geological and environmental chemistry [16]. However, very few researches about tooeleite have been reported in literature, and henceforth the essential geochemical and mineralogical information is still lacking [29]. Moreover, there are many controversies in the literature about the dissolution process, solubility and stability of tooeleite, further research is needed on the co-incorporation and release of arsenic in/from the mineral and its long-term stability [25,27].
This work aimed to synthesize crystalline tooeleite by a simple hydrothermal method. Different instruments are then applied to inspect the structure and morphology of the obtained tooeleite. Its dissolution mechanism, solubility and long-term stability at diverse solution pHs and temperatures are examined, and simultaneously its suitability as storage materials for arsenic is also discussed.
2. Materials and Methods
2.1. Synthesis
The synthesis of tooeleite was completed in the same manner with slight modifications after the pre-experimental result as that used for measuring the heat capacity of tooeleite by relaxation calorimetry [37]. Firstly, 248.65 mL of ultrapure water were mixed with 1.35 mL 98% H2SO4 to prepare a sulfuric acid solution in a polyethylene bottle, into which 5.00 g of As2O3 was then added. The resulting slurry was heated in a 90 °C waterbath with a constant stirring at 600 rpm for 4h and then air-cooled. The undissolved solids were removed by filtration. After that, 10.50 g of Fe2(SO4)3·9H2O were added, and the resulting slurry was mixed and heated in a 90 °C water bath with a constant stirring at 600 rpm for 1 h. After air-cooling to room temperature, the mixed solution was adjusted to pH = 3.00 with 10 mol/L NaOH solution and then was heated in a 90 °C water bath with a constant stirring at 600 rpm for another 1 h. Finally, the suspension solution was air-cooled and separated using vacuum filtration. The obtained precipitates were rinsed 3 times using 50 mL ultrapure water and oven-dried at 110 °C for 24 h. The color of the tooeleite precipitate obtained in our work was yellow, like that observed for the mineral tooeleite in literature [32].
2.2. Characterization
The bulk elemental composition of the synthetic tooeleite was determined by digesting 50 mg of the prepared tooeleite in 20 mL of 6 M HCl solution, which was then diluted to 50 mL with ultrapure water. The iron, arsenic and sulfur concentrations were analyzed using an inductively coupled plasma-optical emission spectrometer (ICP-OES, Optima 7000DV, Perkin-Elmer Ltd., Waltham, MA, USA) or an atomic absorption spectrometer (AAS, PinAAcle 900T, Perkin-Elmer Ltd., Waltham, MA, USA) when the concentrations of iron or arsenic were less than the ICP-OES detection limits. The contents of the crystal water were then calculated based on the thermogravimetric analysis (TGA), which was completed using a thermal analyzer (TA, STA 409, Netzsch-Gerätebau GmbH, Selb, Germany) from room temperature to 1130 °C at 10 °C/min in a 20 mL/min N2 atmosphere. The synthesis for tooeleite was repeated ten times to obtain enough products for the following tests, and all of them were characterized by an X-ray diffractometer (XRD, X’Pert PRO, PANalytical B.V., Almelo, the Netherlands) with Cu-Kα radiation of 1.540598 Å at 40 kV/40 mA and recognized by comparing with the reference for tooeleite (00-044-1468) from the International Centre for Diffraction Data (ICDD) to check the reproducibility in the synthesis procedure. The functional groups and the morphology of the tooeleite were observed using a Fourier transform infrared spectrophotometer (FT-IR, Nicolet Nexus 470, Thermo Fisher Scientific Inc., Waltham, MA, USA) over the 400~4000 cm−1 range and a field emission scanning electron microscope (FE-SEM, S-4800, Hitachi Ltd., Tokyo, Japan), respectively.
2.3. Dissolution Experiments
In each dissolution test, five grams of the synthetic tooeleite were weighed into a 100 mL polythene bottle, in which 100 mL of HNO3 or NaOH solution of different pHs (pH 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12) were then added. The capped bottle was placed in a thermostatic water bath (25 °C) and agitated by a magnetic stirring bar. The pH drifted freely and was recorded periodically. The experiments studying the effect of the temperature (25 °C, 35 °C or 45 °C) were conducted at initial pH 2. From each flask, 5 mL of the solution were collected periodically at 23 intervals form 1 h to 7920 h (330 d), filtered into a volumetric flask and instantly stabilized with 0.2% HNO3 to 25 mL. After each sampling, 5 mL of HNO3 or NaOH solution that had been adjusted to the pH measured at sampling were replenished into the bottle to hold a relatively constant solid/solution ratio. The variation of the solution components due to this sampling-replenishing was amended using the mass balance. The total iron, arsenic and sulfur concentrations were determined by the ICP-OES or AAS instrument. After 330 d dissolution, the residual solid of tooeleite was taken out from the bottle and characterized using XRD, FT-IR and FE-SEM as previously described to examine the possible variation of the mineral properties during dissolution. The dissolution experiment at different temperatures was made in triplicate (25 °C, initial pH = 2.00) or duplicate (35 °C and 45 °C, initial pH = 2.00) to check the repeatability.
2.4. Thermodynamic Calculation
The elemental speciation calculation for the tooeleite dissolution was conducted using the PHREEQC program (Version 3.6.2, U.S. Geological Survey, Denver, CO, USA) [38]. The aqueous activities of Fe3+, AsO33−, SO42− and OH− were first computed and then the ion-activity product [logˍIAP] was determined according to its definition, which was equal to the solubility product [logˍKsp] for the synthetic tooeleite at the dissolution equilibrium. The minteq.v4.dat database was recompiled based on the MINTEQA2 database (Version 3.0, U.S. Environmental Protection Agency, Athens, GA, USA) [39], including the thermodynamic data of all aqueous species and solid phases for the speciation simulation, in which the Debye–Hückel equation was chosen automatically because the aqueous ionic strength in the present work was <0.07889 mol/L.
3. Results and Discussion
3.1. Chemical Analysis
The proportions of the major constituents were calculated from the ICP-OES and TGA analysis and described as: Fe2O3 44.99%, As2O3 38.57%, SO3 7.52% and H2O 8.92%, which correspond to the chemical formula of tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] [28], and also agreed well with the values of Fe2O3 44.3%, As2O3 36.2% and SO3 9.7%, giving a total of 90.2 wt.% [31], and Fe2O3 46.0%, As2O3 35.1% and SO3 9.2%, total 85.5 wt% [32].
3.2. XRD
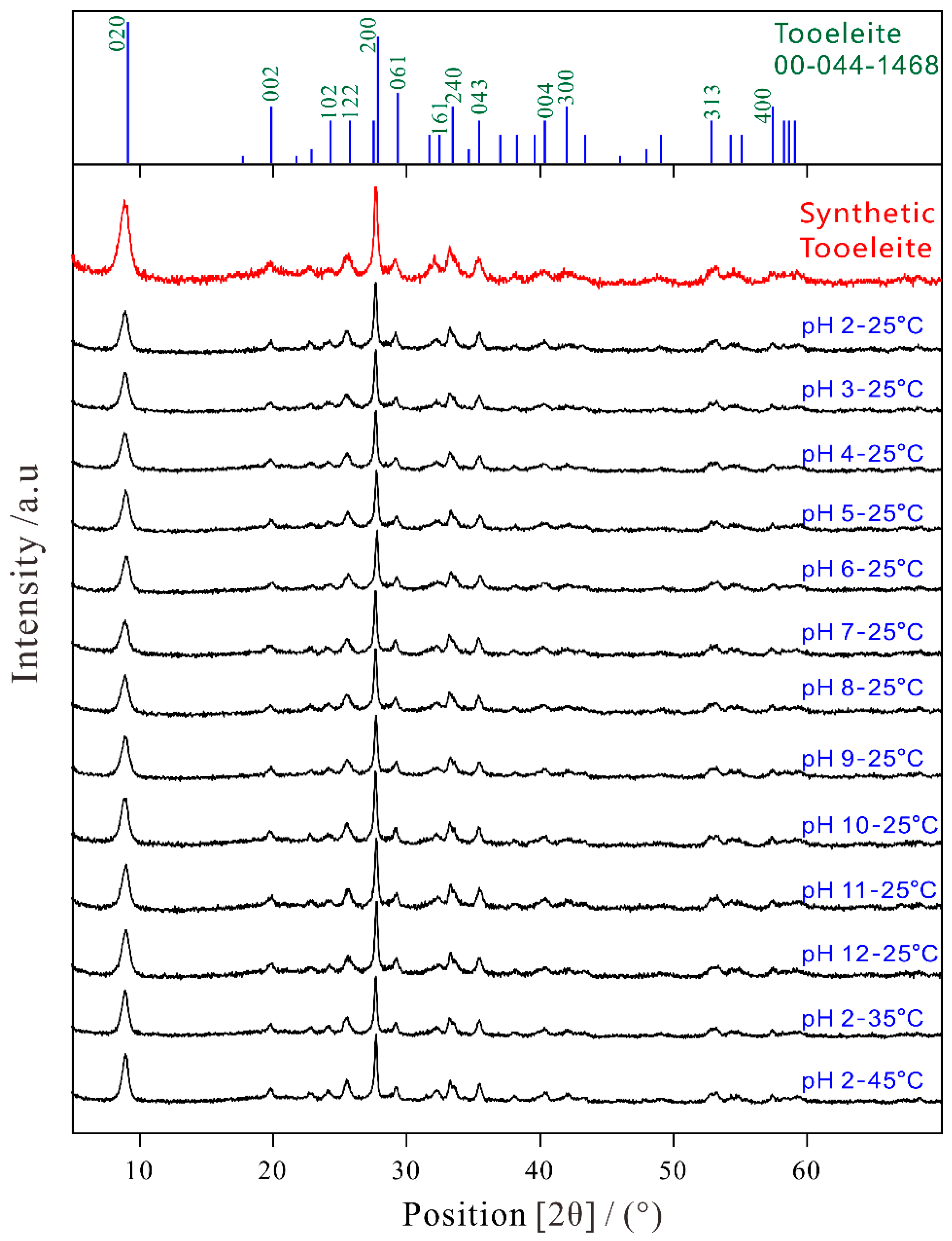
The XRD spectra of the synthetic tooeleite before and after 330 d dissolution are illustrated in Figure 1. The structural characterization was made by Rietveld refinements using the MDI Jade and the PANalytical HighScore Plus programs. The single crystal data of tooeleite (ICDD Powder Diffraction File as No. 00-044-1468) were applied as initial structural models [28]. The most intense peaks of all samples were principally in the identical positions with the similar intensities and matched very well the peaks for tooeleite [28] and the structural analysis showed that only the phase tooeleite was in the solid product with the space group Pbc57 and the orthorhombic structure. The most intense peaks (dobs, Iobs, hkl) in the powder XRD spectrum of the synthetic tooeleite were 9.69(76)020, 4.48(13)002, 3.46(18)122, 3.21(100)200, 3.06(21)061, 2.79(26)161, 2.69(35)240, 2.53(23)043 and 1.72(16)313, giving the cell parameters of a = 6.4758 Å, b = 19.3737 Å and c = 8.9170 Å, which were close to a = 6.4160 Å, b = 19.4500 Å and c = 8.9410 Å for tooeleite [28]. The XRD spectra of tooeleite, collected after 330 days of dissolution, did not show any evidence of other mineral phases even if it could not be excluded that new phases with low abundance and/or poor crystallinity presented eventually. Similar results were also reported in the literature for the hydroxyapatite dissolution in simple aqueous solutions [40].
3.3. FT-IR
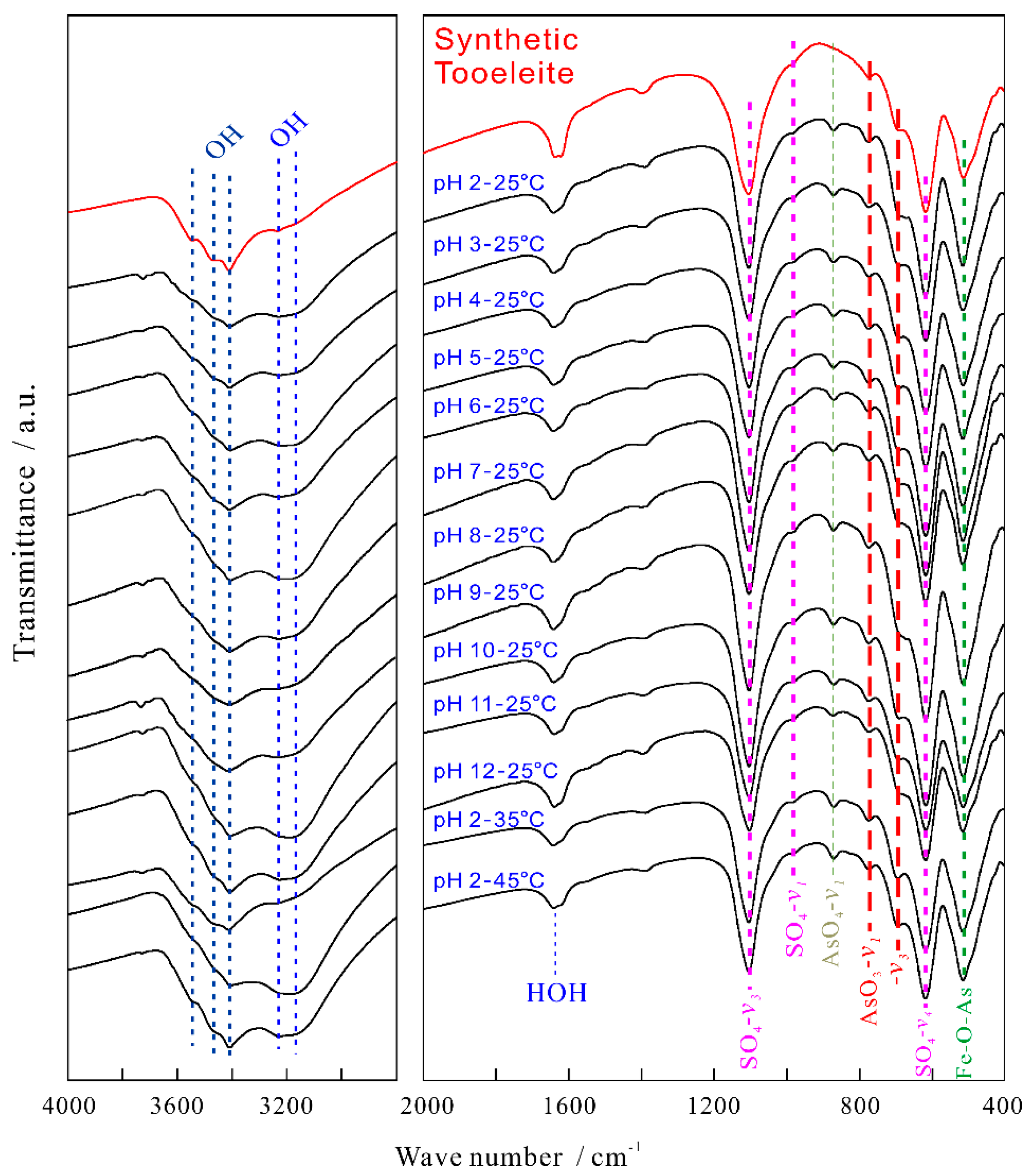
The FT-IR spectra were recorded for the synthetic tooeleite before and after dissolution for 330 d (Figure 2) and interpreted based on the literature data [5,22,41]. No obvious variation was detected in the FT-IR spectra after dissolution. All FT-IR spectra of the synthetic tooeleite presented the bending or stretching vibrations of AsO33−, SO42− and OH− as reported previously [42], but the split stretching vibration of the FeO6 octahedra [41] was not observed in this work.
The 2800~3700 cm−1 spectra of the OH stretching were characterized by the bands at 3549, 3466, 3406~3415, 3225~3242 and 3184~3199 cm−1. The bands at 3184~3242 cm−1 and 3406~3549 cm−1 were ascribed to the OH stretching vibrations of the adsorbed H2O and the crystal water in the tooeleite structure, respectively, which confirmed the presence of strong H-bonds in the crystal structure. The strong bands at 1637~1643 cm−1 were easily assigned to the bending of the sorbed H2O, i.e., the framework deformation vibration of H2O [5].
The structural SO42− of the tooeleite crystal exhibited three fundamental vibrations: the v1 symmetric stretching vibration (982~985 cm−1), the v3 antisymmetric stretching vibration (1101~1105 cm−1) and the v4 anti-symmetric bending vibration (615 cm−1) (Figure 2). The degenerate v3 mode of SO42− was not split into three bands as same as in the previous researches [26].
AsO33− had a planar triangular shape and its fundamental vibrations in water had been studied by many researchers [27,43], which yet showed some differences in the related band positions. The characteristic bands at 752 and 680 cm−1 were ascribed to the v1 and v3 vibrations of AsO33−, respectively [43]. The bands of the v1 and v3 vibrations were found at 690 and 672 cm−1 [44]. The v1 and v3 vibration bands of AsO33− were found to exist at 653 and 631 cm−1 [45]. The v1 and v3 vibrations of AsO33− were recorded at 772 and 696 cm−1 for the synthetic tooeleite, respectively [27]. In this work, the arsenite IR spectra of tooeleite in 687~773 cm−1 were observed as illustrated in Figure 2. The moderate bands at 771~773 cm−1 and the weaker strong bands at 687~682 cm−1 were ascribed to the antisymmetric stretching vibration (v1) and the As-O stretching vibration (v3) of AsO33−, respectively. The bands at 511~513 cm−1 were assigned to the Fe-O-As vibration [5,46].
The very low intense bands at 870–872 cm−1 were detected for the tooeleite after dissolution, which were ascribed to the v1 symmetric stretching vibration of AsO43− and thought as a spectroscopic evidence for the oxidization of AsO33− to AsO43−, which was yet not confirmed by the XPS and XRD analysis [26].
3.4. FE-SEM
The morphologies of the synthetic tooeleite that was recognized by XRD were investigated using FE-SEM (Figure 3). The pure tooeleite consisted of platy crystallites of ~1 μm across, which aggregated together and exhibited a reticulated flower structure, which was in agreement with some previous research [6,7,27,28,35,37]. No significant morphological variation of the synthetic tooeleite was observed after dissolution at initial pH 2~12 and 25~45 °C for 330 d (Figure 3).
3.5. Evolution of the Aqueous Solutions and Dissolution Mechanism
The dissolution of the synthetic tooeleite is normally described by the following reaction (Equation (1)).
Fe6(AsO3)4(SO4)(OH)4·4H2O = 6Fe3+ + 4AsO33− + SO42− + 4OH− + 4H2O
Theoretically, 1.00 mol of the synthetic tooeleite could liberate 4.00 mol OH− after Equation (1). Consequently, the dissolution in strong acidic solution could cause a solution pH rising, indicating the H+ consumption, or the dissolution in strong alkali solution could cause a solution pH reduction, indicating the OH+ consumption, and the aqueous complexations could control the speciation reactions of all released constituents (Equations (2)–(4)).
H+ + SO42− = HSO4−
nH+ + AsO33− = HnAsO3(3−n)− n = 1~3
mFe3+ + nOH− = Fem(OH)n(3m−n)+ m = 1, n = 1~4; m = 2, n = 2; m = 3, n = 4
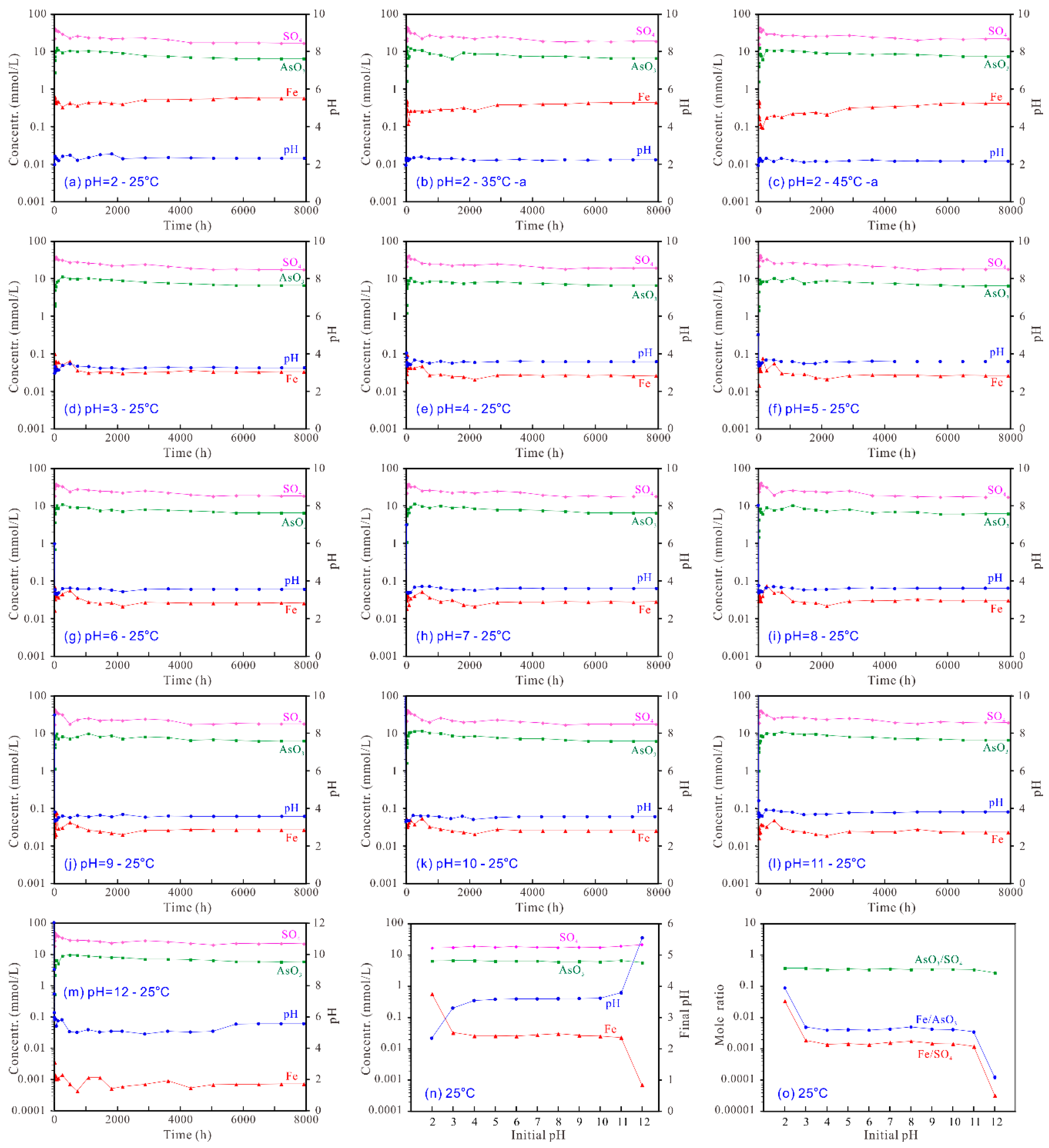
For the dissolution of tooeleite at 25 °C and initial pH 2.00~3.00 (Figure 4a,d), the aqueous pH rose up to 2.44~3.36 in the early 1 h of dissolution and then fluctuated and gradually decreased to 2.33~3.30 after 6480 h dissolution. For the dissolution at initial pH 4.00~12.00, the solution pH decreased gradually in the early 4300 h and then reached the steady state. The final solution pHs at the end of the experiment increased from 3.54 to 5.54 with the increasing initial pH from 4.00 to 12.00 (Figure 4n). The final solution pHs for the dissolution at initial pH 2.00 decreased with the increasing temperature, i.e., from 2.31~2.33 at 25 °C, 2.27~2.28 at 35 °C to 2.21~2.23 at 45 °C (Figure 4 a–c).
The dissolved Fe3+ concentration increased rapidly up to 0.6285 mmol/L in the early 1 h and then decreased to 0.3277 mmol/L from 1 h to 240 h; after that increased gradually to a steady state of 0.5747~0.5770 mmol/L after 6480 h of dissolution at initial 25 °C and pH 2.00 (Figure 4a). For the dissolution at 25 °C and initial pH 12.00, the dissolved Fe3+ concentration increased rapidly up to 0.003449 mmol/L in the early 1 h and then fluctuated with a general decreasing trend to a steady state of 0.000702~0.000718 mmol/L after 6480 h (Figure 4m). The final dissolved Fe3+ concentration decreased with the increasing initial pH from 2.00 to 12.00 with two sharp decreases from 2.00 to 3.00 and from 11.00 to 12.00 (Figure 4n). The final dissolved Fe3+ concentration for the dissolution at initial pH 2.00 decreased with the increasing temperature, i.e., from 0.5747~0.5770 mmol/L at 25 °C, 0.4324~0.4339 mmol/L at 35 °C to 0.4186~0.4234 mmol/L at 45 °C (Figure 4a–c).
The dissolved AsO33− concentration increased quickly up to the peak values of 9.37~13.36 mmol/L in the early 72~480 h and then slightly decreased to a steady state of 5.78~6.70 mmol/L after 6480 h of dissolution (Figure 4a–m). For the tooeleite dissolution, the dissolved arsenic concentrations at the end of the experiment showed the lowest value of 5.70~5.82 mmol/L, i.e., 427.3~435.8 mg/L As, at initial pH 12 (final pH 5.54) (Figure 4n). The final dissolved AsO33− concentration for the dissolution at initial pH 2.00 increased with the increasing temperature, i.e., from 6.43~6.48 mmol/L at 25 °C, 6.56~6.66 mmol/L at 35 °C to 7.50~7.53 mmol/L at 45 °C (Figure 4a–c).
The dissolved SO42− concentration increased rapidly up to 37.61~46.83 mmol/L in the early 6~72 h and then decreased gradually to a steady state of 16.82~21.90 mmol/L after 6480 h of dissolution (Figure 4a–m). The final dissolved SO42− concentration increased slightly from 16.82~16.93 mmol/L to 21.37~21.90 mmol/L with the increasing initial pH from 2.00 to 12.00 (Figure 4n). The final dissolved SO42− concentration for the dissolution at initial pH 2.00 increased with the increasing temperature, i.e., from 16.82~16.93 mmol/L at 25 °C, 18.54~19.05 mmol/L at 35 °C to 21.48~21.80 mmol/L at 45 °C (Figure 4a–c).
For the tooeleite dissolution at 25 °C and initial pH 2.00, the aqueous Fe/AsO3 mole ratio reached 0.227921 in the early 1 h and decreased with time gradually to 0.035462 in 240 h and then increased gradually to a steady state; the solution Fe/SO4 mole ratio reached 0.028147 in the early 1 h and decreased gradually to 0.011208 in 240 h and then increased slowly to a steady state; the solution AsO3/SO4 mole ratio reached 0.457605 in the early 480 h and then decreased progressively with slight fluctuation to a steady state. After 6480 h, the aqueous pH and the total dissolved Fe3+, AsO33− and SO42− concentrations attained a steady state with the Fe/AsO3 mole ratios of 0.088631~0.089192, the Fe/SO4 mole ratios of 0.033950~0.034309 and the AsO3/SO4 mole ratios of 0.382033~0.384663, which were obviously lower than the stoichiometric Fe/AsO3, Fe/SO4 and AsO3/SO4 mole ratios of 1.50, 6.00 and 4.00 for the synthetic tooeleite, respectively (Figure 4o; Figure S1—Supplementary Material).
For the tooeleite dissolution at 25 °C and initial pH 12.00, the aqueous Fe/AsO3 mole ratio reached 0.006608 in the early 1 h and then decreased slowly with a mild fluctuation to a steady state; the solution Fe/SO4 mole ratio reached 0.000190 in the early 1 h and then decreased slowly with a mild fluctuation to a steady state; the solution AsO3/SO4 mole ratio reached 0.336264 in the early 480 h and then decreased slowly with a mild fluctuation to a steady state. After 6480 h, the aqueous pH and the detached Fe3+, AsO33− and SO42− concentrations attained a steady state with the Fe/AsO3 mole ratios of 0.000121~0.000126, the Fe/SO4 mole ratios of 0.000033 and the AsO3/SO4 mole ratios of 0.260367~0.272172, which were obviously smaller than the stoichiometric Fe/AsO3, Fe/SO4 and AsO3/SO4 mole ratios of 1.50, 6.00 and 4.00 for the synthetic tooeleite, respectively (Figure 4o; Figure S1—Supplementary Material).
It is concluded that all constituents were preferentially dissolved from solid into solution in the sequence of SO42− > AsO33− > Fe3+ in dissolution, indicating an incongruent dissolution of the synthetic tooeleite and/or formation of Fe-rich residuals. The total dissolved Fe3+, AsO33− and SO42− concentrations decreased with the increasing initial pH, which indicated that tooeleite was more soluble in the strong acidic solution. The decrease of the Fe/AsO3, Fe/SO4 and AsO3/SO4 mole ratios with the increasing initial pH suggested that the Fe3+ and AsO33− ions were more easily released at lower pH in comparison to SO42− ions.
For the tooeleite dissolution at initial pH 2.00, the dissolved iron, arsenite and sulfate existed mainly as FeSO4+/Fe3+, H3AsO30 and SO42− at initial pH 2.00, respectively; all aqueous solutions were unsaturated with ferrihydrite [Fe(OH)3], maghemite [γ-Fe2O3], arsenolite [As2O3], claudetite [As2O3] and Fe2(SO4)3 from the PHREEQC simulation, i.e., all of these possible iron sulfates/oxides were thermodynamically unfavorable to precipitate. Instead, all solutions were saturated or very closely near-saturated with hematite [Fe2O3] (SI = 3.33~5.62), H-jarosite (H3O)Fe3(SO4)2(OH)6] (SI = 2.20~4.03), goethite [α-FeO(OH)] (SI = 0.46~1.61) and lepidocrocite [γ-FeO(OH)] (SI = −0.33~0.73). It was possible to form iron-rich precipitates.
For the tooeleite dissolution at initial pH 12.00, the dissolved iron, arsenite and sulfate existed mainly in the form of Fe(OH)30/Fe(OH)2+, H3AsO30 and SO42− at initial pH 12.00, respectively; all solutions were unsaturated with arsenolite [As2O3], claudetite [As2O3], Fe2(SO4)3 and H-jarosite, i.e., all of these possible iron sulfates/oxides were thermodynamically unfavorable to precipitate. Instead, all aqueous solutions were saturated or very closely near-saturated with hematite [Fe2O3] (SI = 7.65~14.88), goethite [α-FeO(OH)] (SI = 2.63~6.24), lepidocrocite [γ-FeO(OH)] (SI = 1.75~5.36), maghemite [γ-Fe2O3], ferrihydrite [Fe(OH)3] (SI = −0.07~3.54) and (SI = −0.15~7.07). It was possible to form iron-rich precipitates. Although the XRD spectra exhibited that no other minerals than tooeleite existed, it could not be confirmed here that they did not exist in smaller quantity, which was under the detection limit (Figure 1).
Two different mechanisms for the tooeleite dissolution happened at low and high pHs. At initial pH < 3, the aqueous pH increased gradually with time, suggesting a hydrion-consuming (Equation (5)); on contrary, at initial pH > 3, the aqueous pH decreased progressively with time, showing a hydroxyl-consumption (Equation (6)).
Fe6(AsO3)4(SO4)(OH)4·4H2O + 16H+ = 5Fe3+ + FeSO4+ + 4H3AsO30 + 8H2O
Fe6(AsO3)4(SO4)(OH)4·4H2O + 8H2O + OH− = 5Fe(OH)30 + Fe(OH)2+ + 4H3AsO30 + SO42−
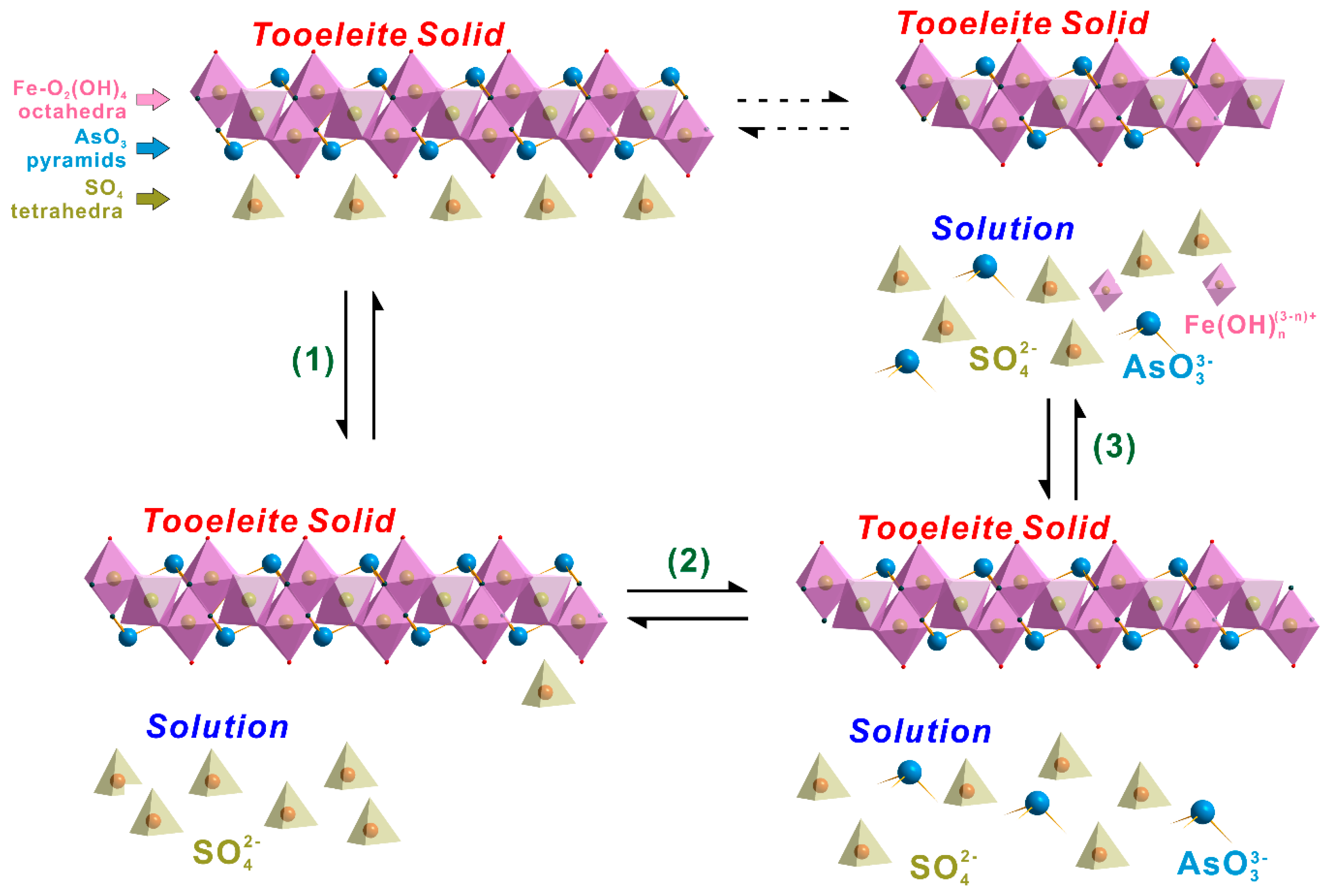
Because all constituents were preferentially released from solid into solution in the order of SO42− > AsO33− > Fe3+, the tooeleite dissolution could be expressed by the favored releasing of sulfate anions from solid surface into solution, followed by the releasing of arsenite anions and Fe3+ ions, while Fe3+ cations were favorably left behind as a residual octahedral layer of the solid (Figure 5). This was also confirmed by the SEM-EDS analysis on the tooeleite surface, which showed that the Fe/(AsO3 + SO4) and AsO3/(AsO3 + SO4) mole ratios on the tooeleite surface increased from 1.01~1.16 and 0.76~0.80 to 1.01~1.24 and 0.76~0.81 after 330 d dissolution at 25 °C and initial pH 2.00, respectively (Table S1, Supplementary Material). The dissolution was fundamentally controlled by the breakages of Fe-O/OH bonds in the lattice structure of tooeleite and restrained by the outer OH− layers, which was also found in the alunite dissolution previously [47]. Tooeleite dissolved congruently at initial pH 2 and incongruent when initial pH >3 [29].
3.6. Solubility Calculation
The aqueous activities of Fe3+, AsO33−, SO42− and OH− in the final steady state (6480 h, 7200 h and 7920 h) were first computed using the PHREEQC program with its built-in minteq.v4.dat database [38], which was supplemented with the thermodynamic data of some aqueous metal-arsenite species [19,48], and then the ion-activity product [logˍIAP] was computed according to its definition. At the dissolution equilibrium, the saturation index for the synthetic tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] [log_IAP/Ksp] will be zero, i.e., the ion activity product [IAP] equals the solubility product [Ksp] [38]. The aqueous species Fe3+, Fe(OH)2+, Fe(OH)3, Fe(OH)4−, Fe2(OH)24+, Fe3(OH)45+, FeSO4+, Fe(SO4)2− and FeH2AsO32+ were considered in the simulation for the total Fe; H3AsO3, AsO33−, HAsO32−, H2AsO3− and FeH2AsO32+ for the total arsenite; SO42−, HSO4−, FeSO4+ and Fe(SO4)2− for the total sulfate. The main speciation reactions with the equilibrium constants in the PHREEQC computing are listed in Table 1.
The dissolution reaction of tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] can be expressed by Equation (1).
Its ion-activity product [IAP] is defined by Equation (7).
where {} are the activities of the aqueous species of Fe3+, AsO33−, SO42− and OH−.
IAP = {Fe3+}6{AsO33−}4{SO42−}{OH−}4
Based on the following thermodynamic properties, ΔGfo[H2O] = −237.18 kJ/mol, ΔGfo[Fe3+] =−4.6 kJ/mol, ΔGfo[AsO33−] = −421.8 kJ/mol, ΔGfo[SO42−] = −744.6 kJ/mol and ΔGfo[OH−] = −157.3 kJ/mol [19,49,50], the Gibbs free energy of formation, ΔGfo[Fe6(AsO3)4(SO4)(OH)4·4H2O] for the synthetic tooeleite was also calculated. The standard free energy of reaction (ΔGro) at 25 °C and 0.101 MPa can be calculated from Equation (8).
For Equation (1),
Rearranging,
ΔGro = −5.708 logKsp
ΔGro = 6 ΔGfo[Fe3+] + 4 ΔGfo[AsO33−] + ΔGfo[SO42−] + 4 ΔGfo[OH−]+ 4 ΔGfo[H2O]
− ΔGfo[Fe6(AsO3)4(SO4)(OH)4·4H2O]
− ΔGfo[Fe6(AsO3)4(SO4)(OH)4·4H2O]
ΔGfo[Fe6(AsO3)4(SO4)(OH)4·4H2O]
= 6 ΔGfo[Fe3+] + 4 ΔGfo[AsO33−] + ΔGfo[SO42−] + 4 ΔGfo[OH−]+ 4 ΔGfo[H2O] − ΔGro
= 6 ΔGfo[Fe3+] + 4 ΔGfo[AsO33−] + ΔGfo[SO42−] + 4 ΔGfo[OH−]+ 4 ΔGfo[H2O] − ΔGro
Because the relatively higher solid/water ratio (5 g/100 mL) was applied in the dissolution experiment, only <8% of the solid dissolved into water, i.e., the bulk constituent of the synthetic tooeleite showed no significant change after 7920 h of dissolution. The analytical data of the dissolution at 25 °C and initial pH 2 for 270 d (6480 h), 7200 h (300 d) and 7920 h (330 d) together with the calculated thermodynamic properties for the synthetic tooeleite are given in Table 2.
The dissolution tests were carried out until the differences in the ion activity products [IAPs] determined from the last three samples were within the analytical uncertainty of ±0.13 log units. The dissolution system reached equilibrium and all solutions could be considered to be saturated with tooeleite [51]. The ion-activity product [logˍIAP], which equaled the solubility product [logˍKsp] at equilibrium, and the free energy of formation [ΔGfo] were computed to be −200.28 ± 0.01 and −5180.54 ± 0.07 kJ/mol for the synthetic tooeleite, respectively.
Very rare Ksp and ΔGfo data for tooeleite could be found in literature. The solubility product [logˍKsp] and the standard Gibbs free energy for tooeleite were determined to be 7.7 and −3605 kJ/mol through the batch experiment of 30 d without the speciation consideration of the dissolved Fe3+, AsO33− and SO42− in the calculation, respectively [29]. The result of this work was not in good agreement with the solubility product of logˍKsp ≈ 23 for tooeleite [10,31], which was estimated from the residual iron and arsenic analysis in the synthetic experiment [28]. However, it was very close to the thermodynamic properties of synthetic tooeleite that were measured by the calorimetry technique [37]. The ΔGfo and logˍKsp values were calculated to be −5396.3 ± 9.3 kJ/mol and −17.25 ± 1.80 for the reaction Equation (11), respectively [37]; and the logˍKsp was re-calculated to be −238.09 for the reaction Equation (1).
Fe6(AsO3)4(SO4)(OH)4·4H2O + 16H+ = 6Fe3+ + 4H3AsO3 + SO42− + 8H2O
Tooeleite was stable only at high arsenite and sulfate concentrations and formed under the oxidation of Fe2+ to Fe3+ and the firm conservation of the trivalent oxidation state of arsenic [37].
4. Conclusions
For the dissolution of the synthetic tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O], the dissolved arsenic concentrations exhibited a minimum of 427.3~435.8 mg/L As at 25 °C and initial pH 12.00 with the final pH 5.54. The constituents were dissolved preferentially in the sequence of SO42− > AsO33− > Fe3+ at 25 °C and initial pH 2.00~12.00. The dissolved iron, arsenite and sulfate were present mainly as FeSO4+/Fe3+, H3AsO30 and SO42− at 25 °C and initial pH 2.00, and as Fe(OH)30/Fe(OH)2+, H3AsO30 and SO42− at 25 °C and initial pH 12.00, respectively. The tooeleite dissolution was characterized by the preferential release of SO42− anions from solid surface into aqueous medium, which was controlled by the Fe-O/OH bond breakages and hindered by the outer OH− group layers.
From the data of the dissolution at 25 °C and initial pH 2.00 for 6480~7920 h, the ion-activity product [logˍIAP], which was very close to the solubility product [logˍKsp], and the Gibbs free energy of formation [ΔGfo] were computed to be −200.28 ± 0.01 and −5180.54 ± 0.07 kJ/mol for the synthetic tooeleite, respectively.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-163X/10/10/921/s1, Figure S1: Change of the mole ratios between the solution components in the tooeleite dissolution at 25 °C and initial pH 2 or 12, Table S1: SEM-EDS analysis results of tooeleite before and after dissolution.
Author Contributions
Conceptualization, Y.Z.; Data curation, Z.Z., J.Z. and Y.W.; Formal analysis, L.Z. and J.Z.; Funding acquisition, Y.Z.; Investigation, J.Z., J.L. and S.T.; Methodology, J.Z. and J.L.; Project administration, Y.Z.; Visualization, Z.Z. and Y.Z.; Writing—Original Draft, Y.Z.; Writing—Review and Editing, Z.Z., L.Z. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (41763012, 42063003 and 21707024), the Guangxi Science and Technology Planning Project (GuiKe-AD18126018) and the Provincial Natural Science Foundation of Guangxi (2018GXNSFAA050044).
Acknowledgments
The manuscript has greatly benefited from insightful comments by editors and three anonymous reviewers. The authors would like to acknowledge the Science and Education Combined with Science and Technology Innovation Base of Guangxi Key Laboratory of Environmental Pollution Control Theory and Technology for the help in solid and solution analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Guidelines for Drinking-Water Quality; fourth edition incorporating the first addendum; World Health Organization: Geneva, Switzerland, 2017; pp. 1–541. [Google Scholar]
- Davis, M.A.; Signes-Pastor, A.J.; Argos, M.; Slaughter, F.; Pendergrast, C.; Punshon, T.; Gossai, A.; Ahsan, H.; Karagas, M.R. Assessment of human dietary exposure to arsenic through rice. Sci. Total Environ. 2017, 586, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Nriagu, J.; Haack, S. Arsenic species and chemistry in groundwater of southeast Michigan. Environ. Pollut. 2002, 120, 379–390. [Google Scholar] [CrossRef]
- Paikaray, S.; Göttlicher, J.; Peiffer, S. As(III) retention kinetics, equilibrium and redox stability on biosynthesized schwertmannite and its fate and control on schwertmannite stability on acidic (pH 3.0) aqueous exposure. Chemosphere 2012, 86, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.; Yue, M.; Yang, J.; Wang, Q.; Li, Q.; Liu, H. Formation of tooeleite and the role of direct removal of As(III) from high-arsenic acid wastewater. J. Hazard. Mater. 2016, 320, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, Y.; Hu, K.; Zhang, G.; Jiang, D.; Li, Q.; Ye, B.; Chai, L.; Wang, Q.; Liu, H.; et al. Structural substitution for SO4 group in tooeleite crystal by As(V) and As(III) oxoanions and the environmental implications. Chemosphere 2018, 213, 305–313. [Google Scholar] [CrossRef]
- Chai, L.; Yue, M.; Li, Q.; Zhang, G.; Zhang, M.; Wang, Q.; Liu, H.; Liu, Q. Enhanced stability of tooeleite by hydrothermal method for the fixation of arsenite. Hydrometallurgy 2018, 175, 93–101. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Acid mine drainage remediation options: A review. Sci. Total Environ. 2005, 338, 3–14. [Google Scholar] [CrossRef]
- Olías, M.; Cánovas, C.R.; Nieto, J.M.; Sarmiento, A.M. Evaluation of the dissolved contaminant load transported by the Tinto and Odiel rivers (South West Spain). Appl. Geochem. 2006, 21, 1733–1749. [Google Scholar] [CrossRef]
- Egal, M.; Casiot, C.; Morin, G.; Parmentier, M.; Bruneel, O.; Lebrun, S.; Elbaz-Poulichet, F. Kinetic control on the formation of tooeleite, schwertmannite and jarosite by Acidithiobacillus ferrooxidans strains in an As(III)-rich acid mine water. Chem. Geol. 2009, 265, 432–441. [Google Scholar] [CrossRef]
- Iakovleva, E.; Mäkilä, E.; Salonen, J.; Sitarz, M.; Wang, S.; Sillanpää, M. Acid mine drainage (AMD) treatment: Neutralization and toxic elements removal with unmodified and modified limestone. Ecol. Eng. 2015, 81, 30–40. [Google Scholar] [CrossRef]
- Maillot, F.; Morin, G.; Juillot, F.; Bruneel, O.; Casiot, C.; Ona-Nguema, G.; Wang, Y.; Lebrun, S.; Aubry, E.; Vlaic, G.; et al. Structure and reactivity of As(III)- and As(V)-rich schwertmannites and amorphous ferric arsenate sulfate from the Carnoulès acid mine drainage, France: Comparison with biotic and abiotic model compounds and implications for As remediation. Geochim. Cosmochim. Acta 2013, 104, 310–329. [Google Scholar] [CrossRef]
- Morin, G.; Calas, G. Arsenic in soils, mine tailings, and former industrial sites. Elements 2006, 2, 97–101. [Google Scholar] [CrossRef]
- Roussel, C.; Néel, C.; Bril, H. Minerals controlling arsenic and lead solubility in an abandoned gold mine tailings. Sci. Total Environ. 2000, 263, 209–219. [Google Scholar] [CrossRef]
- Morin, G.; Juillot, F.; Casiot, C.; Bruneel, O.; Personneä, J.C.; Elbaz-Poulichet, F.; Leblanc, M.; Ildefonse, P.; Calas, G. Bacterial formation of tooeleite and mixed As(III) or As(V)-Fe(III) gels in the Carnoulès AMD, France. A XANES, XRD, and SEM study. Environ. Sci. Technol. 2003, 37, 1705–1712. [Google Scholar] [CrossRef]
- Zhu, Y.; Jiang, Z.; Zhu, Z.; Liu, H.; Zhang, L.; Lin, J. Arsenic immobilization from aqueous solution by the precipitation of the pseudo-octahedral arsenate-substituted natroalunite solid solutions. Sci. Total Environ. 2019, 669, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, M.C.F. Arsenic: An environmental problem limited by solubility. Pure Appl. Chem. 2002, 74, 1843–1850. [Google Scholar] [CrossRef]
- Gieré, R.; Sidenko, N.V.; Lazareva, E.V. The role of secondary minerals in controlling the migration of arsenic and metals from high-sulfide wastes (Berikul gold mine, Siberia). Appl. Geochem. 2003, 18, 1347–1359. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Majzlan, J.; Konigsberger, E. Thermodynamic properties for arsenic minerals and aqueous Species. Rev. Mineral. Geochem. 2014, 79, 217–255. [Google Scholar] [CrossRef]
- Nishimura, T.; Robins, R.G. A re-evaluation of the solubility and stability regions of calcium arsenites and calcium arsenates in aqueous solution at 25 °C. Min. Proc. Ext. Met. Rev. 1998, 18, 283–308. [Google Scholar] [CrossRef]
- Paktunc, D.; Bruggeman, K. Solubility of nanocrystalline scorodite and amorphous ferric arsenate: Implications for stabilization of arsenic in mine wastes. Appl. Geochem. 2010, 25, 674–683. [Google Scholar] [CrossRef]
- Frost, R.L.; Bahfenne, S. Raman spectroscopic study of the arsenite minerals leiteite ZnAs2O4, reinerite Zn3(AsO3)2 and cafarsite Ca5(Ti,Fe,Mn)7(AsO3)12·4H2O. J. Raman Spectrosc. 2010, 41, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.; Silmarilly, B. The mineral nealite Pb4Fe2+(AsO3)2Cl4·2H2O-A Raman spectroscopic study. Spectrosc. Lett. 2011, 44, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Nieto, J.M.; Capitán, M.A.; Sáez, R.; Almodóvar, G.R. Beudantite: A natural sink for As and Pb in sulphide oxidation processes. Appl. Earth Sci. 2003, 112, 293–296. [Google Scholar] [CrossRef]
- Hudson-Edwards, K.A. Uptake and release of arsenic and antimony in alunite-jarosite and beudantite group minerals. Am. Mineral. 2019, 104, 633–640. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, H.; Frost, R.L.; Dong, F. The mineral tooeleite Fe6(AsO3)4SO4(OH)4·4H2O–An infrared and Raman spectroscopic study-environmental implications for arsenic remediation. Spectrochim. Acta A 2013, 103, 272–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Deng, S.; Zhao, F.; Cheng, H.; Frost, R.L. Spectroscopic characterization and solubility investigation on the effects of As(V) on mineral structure tooeleite (Fe6(AsO3)4SO4(OH)4·H2O). Spectrochim. Acta A 2015, 134, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Robins, R.G. Confirmation that tooeleite is a ferric arsenite sulfate hydrate, and is relevant to arsenic stabilisation. Miner. Eng. 2008, 21, 246–251. [Google Scholar] [CrossRef]
- Liu, J.; Huang, X.; Chen, S.; Liu, J.; Wu, C. Mineralogical research on synthesized tooeleite. Acta Petrol. Mineral. 2012, 31, 901–906. (In Chinese) [Google Scholar]
- Opio, F.K.; Peacey, J.; Jamieson, H.E. Arsenic immobilization from industrial effluents. In One Century of the Discovery of Arsenicosis in Latin America (1914–2014): As 2014-Proceedings of the 5th International Congress on Arsenic in the Environment; CRC Press/Balkema: Boca Raton, FL, USA, 2014; pp. 766–768. [Google Scholar]
- Morin, G.; Rousse, G.; Elkaim, E. Crystal structure of tooeleite, Fe6(AsO3)4SO4(OH)4·4H2O, a new iron arsenite oxyhydroxysulfate mineral relevant to acid mine drainage. Am. Mineral. 2007, 92, 193–197. [Google Scholar] [CrossRef]
- Cesbron, F.P.; Williams, S.A. Tooeleite, a new mineral from the U.S. Mine, Tooele County, Utah. Mineral. Mag. 1992, 56, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Cesbron, M.; Gaspar, J.; Bessler, K.E.; Magela, G. Process mineralogy of bacterial oxidized gold ore in São Bento Mine (Brasil). Hydrometallurgy 2006, 83, 114–123. [Google Scholar]
- Casiot, C.; Lebrun, S.; Morin, G.; Bruneel, O.; Personné, J.-C.; Elbaz-Poulichet, F. Sorption and redox processes controlling arsenic fate and transport in a stream impacted by acid mine drainage. Sci. Total Environ. 2005, 347, 122–130. [Google Scholar] [CrossRef]
- Li, X.; Zhao, F.; Deng, S. The removal of Arsenic(III) from acid mine drainage by mineral trap of tooeleite (Fe6(AsO3)4SO4(OH)4·4H2O). In An Interdisciplinary Response to Mine Water Challenges; Sui, W., Sun, Y., Wang, C., Eds.; China University of Mining and Technology Press: Xuzhou, China, 2014; pp. 671–674. [Google Scholar]
- Raghav, M.; Shan, J.; Sáez, A.E.; Ela, W.P. Scoping candidate minerals for stabilization of arsenic-bearing solid residuals. J. Hazard. Mater. 2013, 263, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Majzlan, J.; Dachs, E.; Benisek, A.; Koch, C.B.; Bolanz, R.; Göttlicher, J.; Steininger, R. Thermodynamic properties of tooeleite, Fe63+(As3+O3)4(SO4)(OH)4·4H2O. Chemie der Erde 2016, 76, 419–428. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3, A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Techniques and Methods, Book 6, Chap. A43; U.S. Geological Survey: Denver, CO, USA, 2013; pp. 1–497.
- Allison, J.D.; Brown, D.S.; Novo-Gradac, K.J. MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.0 User’s Manual; Environmental Research Laboratory, Office of Research and Development, U.S. Environmental Protection Agency: Athens, GA, USA, 1991; pp. 1–106.
- Boudia, S.; Zuddas, P.; Fernane, F.; Fiallo, M.; Sharrock, P. Mineralogical transformation during hydroxyapatite dissolution in simple aqueous solutions. Chem. Geol. 2018, 477, 85–91. [Google Scholar]
- Frost, R.L.; Palmer, S.J.; Spratt, H.J.; Martens, W.N. The molecular structure of the mineral beudantite PbFe3(AsO4,SO4)2(OH)6-Implications for arsenic accumulation and removal. J. Mol. Struct. 2011, 1004, 88–93. [Google Scholar]
- Murphy, P.J.; Smith, A.M.L.; Hudson-Edwards, K.A.; Dubbin, W.E.; Wright, K. Raman and IR spectroscopic studies of alunite-supergroup compounds containing Al, Cr3+, Fe3+ and V3+ at the B site. Can. Mineral. 2009, 47, 663–681. [Google Scholar] [CrossRef] [Green Version]
- Loehr, T.M.; Plane, R.A. Raman spectra and structures of arsenious acid and arsenites in aqueous solution. Inorg. Chem. 1968, 7, 1708–1714. [Google Scholar]
- Tossell, J. Theoretical studies on arsenic oxide and hydroxide species in minerals and in aqueous solution. Geochim. Cosmochim. Acta 1997, 61, 1613–1623. [Google Scholar]
- Bahfenne, S.; Frost, R.L. Raman spectroscopic study of the mineral finnemanite Pb5(As3+O3)3Cl. J. Raman Spectrosc. 2010, 41, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Haseen, U. Development of polyacrylamide chromium oxide as anew sorbent for solid phase extraction of As(III) from food and environmental water samples. RSC Adv. 2015, 5, 7311–7323. [Google Scholar] [CrossRef]
- Acero, P.; Hudson-Edwards, K.A.; Gale, J.D. Influence of pH and temperature on alunite dissolution: Rates, products and insights on mechanisms from atomistic simulation. Chem. Geol. 2015, 419, 1–9. [Google Scholar] [CrossRef]
- Marini, L.; Accornero, M. Prediction of the thermodynamic properties of metal-arsenate and metal-arsenite aqueous complexes to high temperatures and pressures and some geological consequences. Environ. Geol. 2007, 52, 1343–1363. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry, Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1996; pp. 976–1004. [Google Scholar]
- Nordstrom, D.K.; Archer, D.G. Arsenic thermodynamic data and environmental geochemistry. In Arsenic in Ground Water, 1st ed.; Welch, A.H., Stollenwerk, K.G., Eds.; Springer: Boston, MA, USA, 2003; Chapter 1; pp. 1–25. [Google Scholar]
- Baron, D.; Palmer, C.D. Solid-solution aqueous-solution interactions between jarosite and its chromate analog. Geochim. Cosmochim. Acta 2002, 66, 2841–2853. [Google Scholar] [CrossRef]
Figure 1.
XRD spectra of the synthetic tooeleite before and after dissolution for 330 d.

Figure 2.
FT-IR spectra of the synthetic tooeleite before and after dissolution for 330 d.
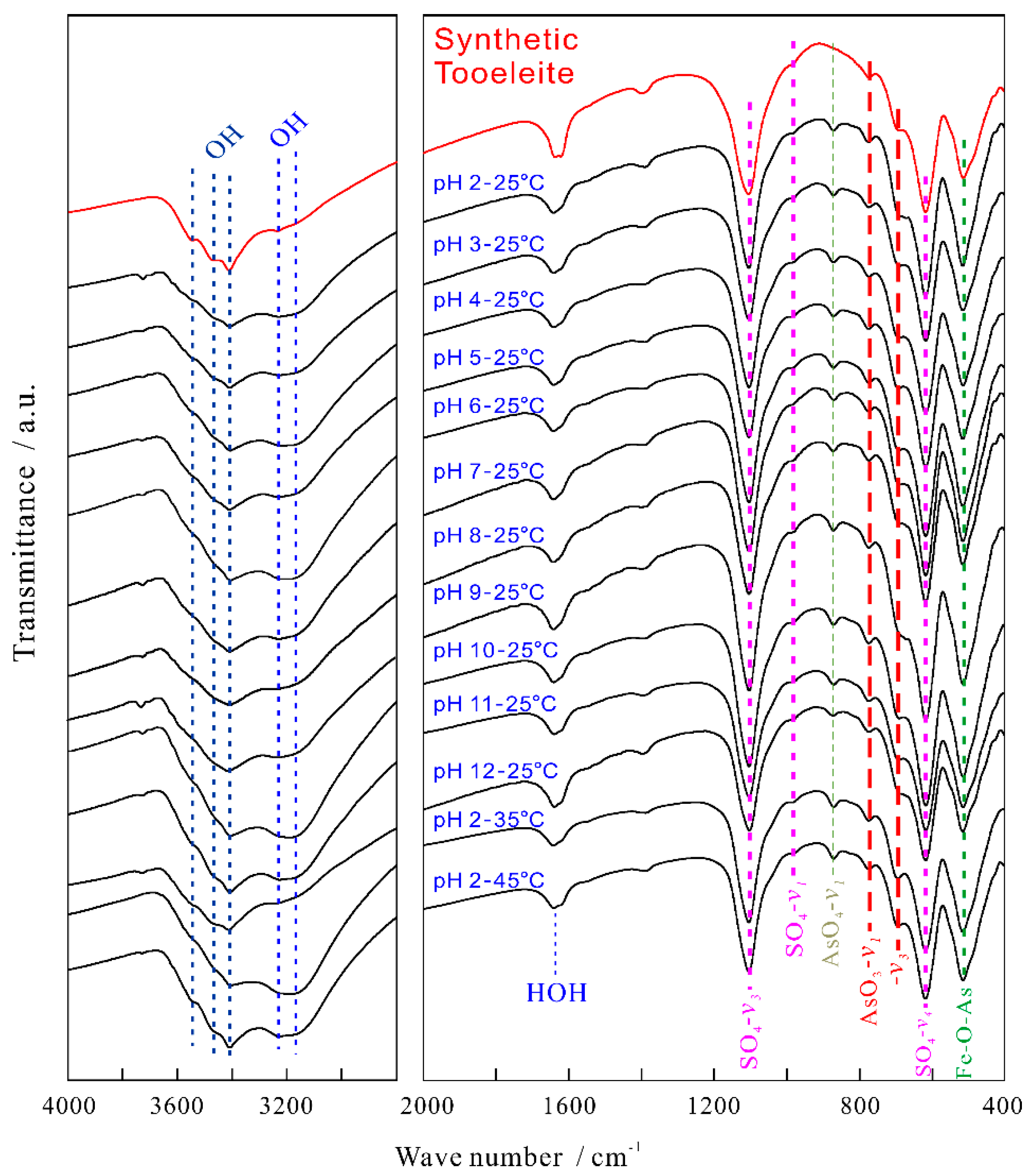
Figure 3.
FE-SEM images of the synthetic tooeleite before and after dissolution for 330 d.

Figure 4.
Aqueous evolution during the dissolution of the synthetic tooeleite at 25–45 °C and initial pH 2–12 for 330 d (a–m) and the variation of the solution components with initial pH at the end of the experiment (n,o).
Figure 4.
Aqueous evolution during the dissolution of the synthetic tooeleite at 25–45 °C and initial pH 2–12 for 330 d (a–m) and the variation of the solution components with initial pH at the end of the experiment (n,o).
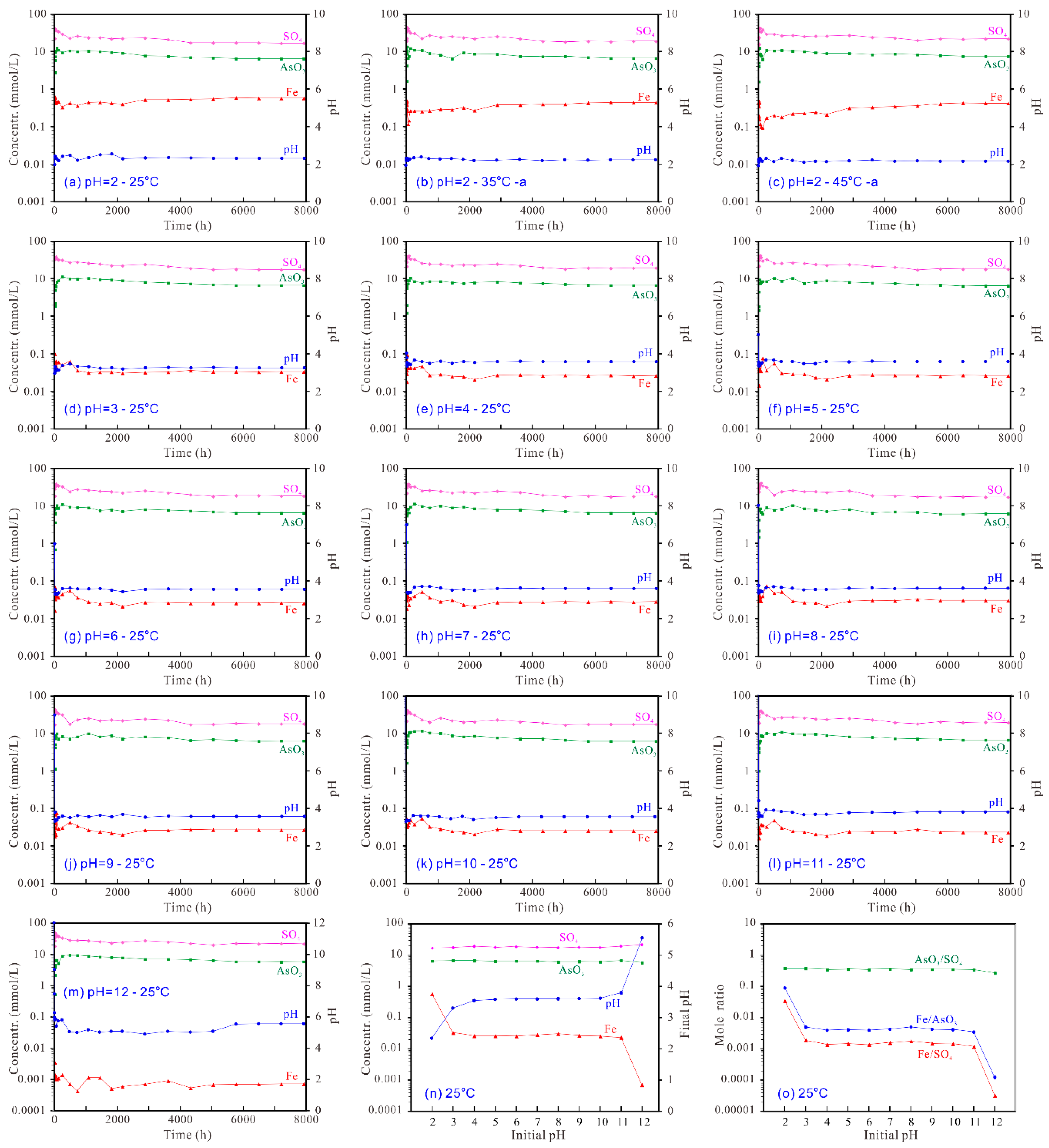
Figure 5.
Dissolution mechanism of tooeleite in aqueous solution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Main speciation reactions involved in the PHREEQC simulation.
| Speciation Reactions | log_K | Speciation Reactions | log_K |
|---|---|---|---|
| Fe3+ + H2O = FeOH2+ + H+ | −2.187 | Fe3+ + SO42− = FeSO4+ | 4.05 |
| Fe3+ + 2H2O = Fe(OH)2+ + 2H+ | −4.594 | Fe3+ + 2SO42− = Fe(SO4)2− | 5.38 |
| Fe3+ + 3H2O = Fe(OH)3 + 3H+ | −12.56 | H3AsO3 = AsO33− + 3H+ | −34.744 |
| Fe3+ + 4H2O = Fe(OH)4− + 4H+ | −21.588 | H3AsO3 = HAsO32− + 2H+ | −21.33 |
| 2Fe3+ + 2H2O = Fe2(OH)24+ + 2H+ | −2.854 | H3AsO3 = H2AsO3− + H+ | −9.29 |
| 3Fe3+ + 4H2O = Fe3(OH)45+ + 4H+ | −6.288 | H3AsO3 + H+ = H4AsO3+ | −0.305 |
| H+ + SO42− = HSO4− | 1.99 | Fe3+ + H2AsO3− = FeH2AsO32+ | 7.28 |
Table 2.
Analytical data and solubility determination of the synthetical tooeleite.
| Temp (°C) | Initial pH | Time (h) | Analytical Data (mmol/L) | log_IAP | Average log_IAP | ΔGfo [kJ/mol] | Average ΔGfo [kJ/mol] | |||
|---|---|---|---|---|---|---|---|---|---|---|
| pH | Fe | AsO3 | SO4 | |||||||
| a25 | 2.00 | 6480 | 2.33 | 0.5747 | 6.4841 | 16.9276 | −200.29 | −200.28 | −5180.58 | −5180.54 |
| 7200 | 2.33 | 0.5770 | 6.4694 | 16.8184 | −200.27 | ±0.01 | −5180.47 | ±0.07 | ||
| 7920 | 2.33 | 0.5759 | 6.4347 | 16.8434 | −200.29 | −5180.58 | ||||
| 25 | 3.00 | 6480 | 3.30 | 0.0328 | 6.6603 | 17.3767 | −194.64 | −194.61 | −5148.31 | −5148.18 |
| 7200 | 3.30 | 0.0335 | 6.6109 | 17.4889 | −194.60 | ±0.03 | −5148.09 | ±0.13 | ||
| 7920 | 3.30 | 0.0333 | 6.6710 | 17.6698 | −194.61 | −5148.14 | ||||
| 25 | 4.00 | 6480 | 3.53 | 0.0265 | 6.6580 | 19.0295 | −193.19 | −193.18 | −5140.07 | −5140.01 |
| 7200 | 3.54 | 0.0258 | 6.5549 | 19.1262 | −193.21 | ±0.04 | −5140.19 | ±0.24 | ||
| 7920 | 3.54 | 0.0263 | 6.6403 | 19.1511 | −193.14 | −5139.77 | ||||
| 25 | 5.00 | 6480 | 3.57 | 0.0265 | 6.3787 | 17.8881 | −192.94 | −192.96 | −5138.61 | −5138.75 |
| 7200 | 3.57 | 0.0259 | 6.4427 | 17.8164 | −192.98 | ±0.02 | −5138.86 | ±0.14 | ||
| 7920 | 3.57 | 0.0261 | 6.4240 | 17.7509 | −192.97 | −5138.77 | ||||
| 25 | 6.00 | 6480 | 3.58 | 0.0258 | 6.4854 | 19.0389 | −192.92 | −192.92 | −5138.50 | −5138.50 |
| 7200 | 3.58 | 0.0259 | 6.4214 | 18.4713 | −192.91 | ±0.01 | −5138.47 | ±0.03 | ||
| 7920 | 3.58 | 0.0257 | 6.4561 | 18.5586 | −192.92 | −5138.53 | ||||
| 25 | 7.00 | 6480 | 3.58 | 0.0281 | 6.4668 | 17.7135 | −192.67 | −192.68 | −5137.07 | −5137.15 |
| 7200 | 3.58 | 0.0277 | 6.4961 | 17.8289 | −192.70 | ±0.02 | −5137.28 | ±0.13 | ||
| 7920 | 3.58 | 0.0279 | 6.5388 | 17.9692 | −192.67 | −5137.11 | ||||
| 25 | 8.00 | 6480 | 3.59 | 0.0305 | 6.0450 | 17.3829 | −192.50 | −192.44 | −5136.08 | −5135.77 |
| 7200 | 3.60 | 0.0304 | 6.1104 | 17.1334 | −192.42 | ±0.06 | −5135.63 | ±0.31 | ||
| 7920 | 3.60 | 0.0304 | 6.1050 | 17.1552 | −192.41 | −5135.59 | ||||
| 25 | 9.00 | 6480 | 3.60 | 0.0270 | 6.2265 | 17.8881 | −192.70 | −192.70 | −5137.25 | −5137.26 |
| 7200 | 3.60 | 0.0269 | 6.2906 | 17.9973 | −192.69 | ±0.01 | −5137.20 | ±0.08 | ||
| 7920 | 3.60 | 0.0268 | 6.2692 | 17.9442 | −192.71 | −5137.34 | ||||
| 25 | 10.00 | 6480 | 3.61 | 0.0255 | 6.1958 | 17.5825 | −192.78 | −192.78 | −5137.73 | −5137.71 |
| 7200 | 3.61 | 0.0257 | 6.1291 | 17.4172 | −192.78 | ±0.00 | −5137.72 | ±0.02 | ||
| 7920 | 3.61 | 0.0257 | 6.1531 | 17.5201 | −192.78 | −5137.69 | ||||
| 25 | 11.00 | 6480 | 3.79 | 0.0231 | 6.6203 | 19.4006 | −191.82 | −191.85 | −5132.25 | −5132.42 |
| 7200 | 3.78 | 0.0230 | 6.6536 | 19.5971 | −191.89 | ±0.04 | −5132.60 | ±0.18 | ||
| 7920 | 3.78 | 0.0232 | 6.7017 | 19.3788 | −191.85 | −5132.41 | ||||
| 25 | 12.00 | 6480 | 5.54 | 0.0007 | 5.8167 | 21.3715 | −193.66 | −193.64 | −5142.75 | −5142.63 |
| 7200 | 5.54 | 0.0007 | 5.7033 | 21.9048 | −193.64 | ±0.02 | −5142.60 | ±0.12 | ||
| 7920 | 5.54 | 0.0007 | 5.7794 | 21.7863 | −193.63 | −5142.54 | ||||
| a25 | 2.00 | 6480 | 2.30 | 0.5690 | 6.6309 | 17.1740 | −200.75 | −200.68 | −5183.20 | −5182.79 |
| 7200 | 2.30 | 0.5775 | 6.4895 | 17.0835 | −200.74 | ±0.13 | −5183.12 | ±0.74 | ||
| 7920 | 2.31 | 0.5834 | 6.6136 | 17.2332 | −200.55 | −5182.05 | ||||
| a25 | 2.00 | 6480 | 2.31 | 0.5792 | 6.5495 | 17.5388 | −200.62 | −200.64 | −5182.47 | −5182.58 |
| 7200 | 2.30 | 0.5811 | 6.6283 | 17.3954 | −200.72 | ±0.08 | −5183.04 | ±0.46 | ||
| 7920 | 2.31 | 0.5828 | 6.5549 | 17.3268 | −200.58 | −5182.23 | ||||
| b35 | 2.00 | 6480 | 2.28 | 0.4324 | 6.5589 | 18.5399 | −199.39 | −199.39 | ||
| 7200 | 2.28 | 0.4339 | 6.6456 | 19.0451 | −199.42 | ±0.03 | ||||
| 7920 | 2.28 | 0.4337 | 6.6616 | 18.6366 | −199.37 | |||||
| b35 | 2.00 | 6480 | 2.28 | 0.4393 | 6.5495 | 19.0233 | −199.40 | −199.35 | ||
| 7200 | 2.28 | 0.4361 | 6.7324 | 18.6459 | −199.33 | ±0.05 | ||||
| 7920 | 2.28 | 0.4412 | 6.7017 | 18.7800 | −199.33 | |||||
| c45 | 2.00 | 6480 | 2.23 | 0.4234 | 7.4958 | 21.4776 | −197.80 | −197.83 | ||
| 7200 | 2.23 | 0.4201 | 7.5279 | 21.7957 | −197.85 | ±0.03 | ||||
| 7920 | 2.23 | 0.4186 | 7.5172 | 21.6678 | −197.85 | |||||
| c45 | 2.00 | 6480 | 2.21 | 0.4627 | 7.3397 | 20.7354 | −197.81 | −197.80 | ||
| 7200 | 2.21 | 0.4643 | 7.4558 | 20.8944 | −197.79 | ±0.02 | ||||
| 7920 | 2.21 | 0.4623 | 7.3984 | 20.9225 | −197.82 | |||||
a,b,c Dissolution tests in triplicate (25 °C, initial pH = 2.00) or duplicate (35 °C and 45 °C, initial pH = 2.00).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhu, Z.; Zhang, J.; Zhu, Y.; Liu, J.; Tang, S.; Zhang, L.; Wang, Y. Dissolution, Stability and Solubility of Tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] at 25–45 °C and pH 2–12. Minerals 2020, 10, 921. https://doi.org/10.3390/min10100921
AMA Style
Zhu Z, Zhang J, Zhu Y, Liu J, Tang S, Zhang L, Wang Y. Dissolution, Stability and Solubility of Tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] at 25–45 °C and pH 2–12. Minerals. 2020; 10(10):921. https://doi.org/10.3390/min10100921
Chicago/Turabian StyleZhu, Zongqiang, Jun Zhang, Yinian Zhu, Jie Liu, Shen Tang, Lihao Zhang, and Yaru Wang. 2020. "Dissolution, Stability and Solubility of Tooeleite [Fe6(AsO3)4(SO4)(OH)4·4H2O] at 25–45 °C and pH 2–12" Minerals 10, no. 10: 921. https://doi.org/10.3390/min10100921
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.