
Development of Guanidine-Bisurea Bifunctional Organocatalyst Bearing Chirality at the Inner and Outer Sides of the Urea Groups, and Application to Enantioselective α-Hydroxylation of Pyranoindolizine Intermediate for Camptothecin Synthesis


Abstract
: Pyranoindolizine is a tricyclic structure found in various biologically active compounds, such as camptothecin (CPT) and its derivatives. In the case of CPTs, the chirality at the α-position in the α-hydroxyl lactone moiety of pyranoindolizine is important for the antitumor activity. This paper deals with enantioselective oxidation of the α-position in pyranoindolizine lactone, which corresponds at C20 in CPT, with cumene hydroperoxide (CHP) in the presence of newly synthesized guanidine-bisurea bifunctional organocatalysts bearing chirality on both the inner and outer sides of the urea groups.1. Introduction
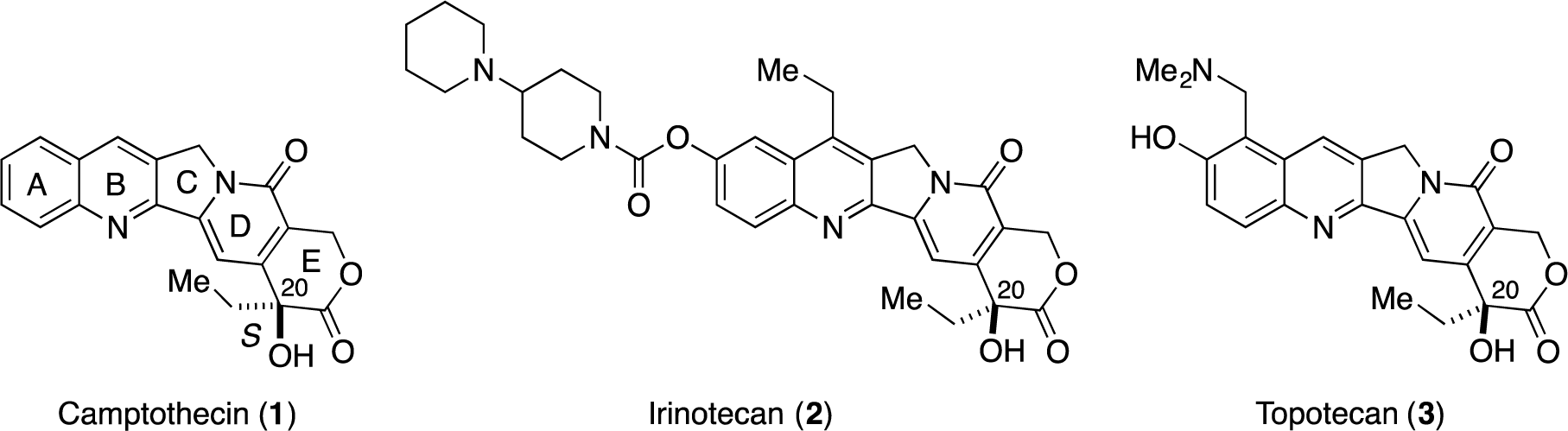
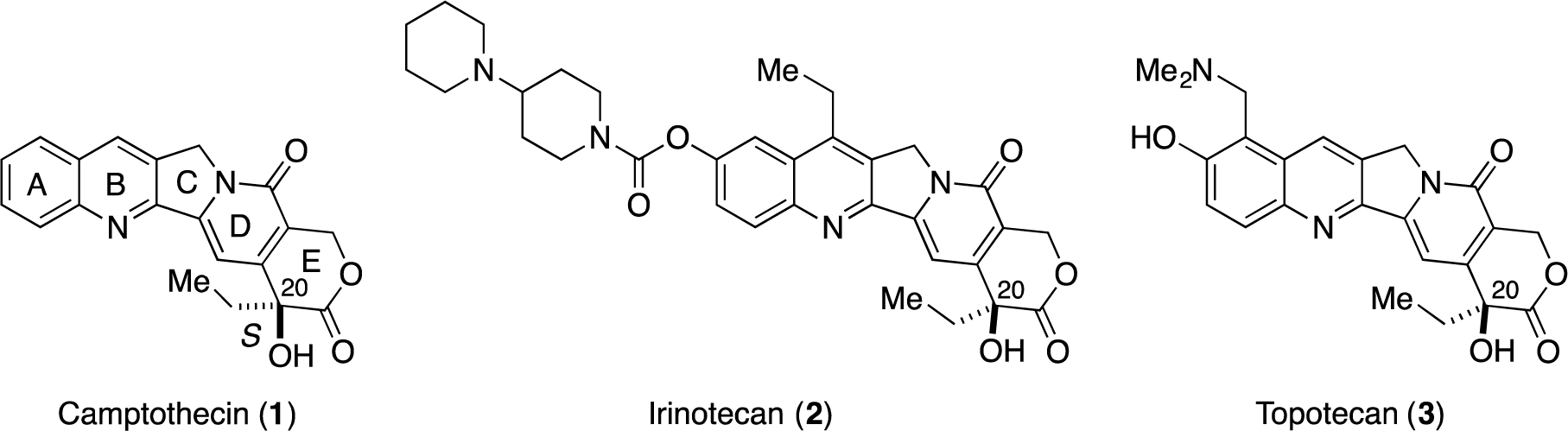
Camptothecin (CPT 1, Figure 1), which was isolated from Camptotheca acuminata by Wani and Wall in 1966 [1], exhibits potent antitumor activity by inhibiting DNA topoisomerase I (Topo I) [2–5]. To date, two camptothecin analogues, i.e., irinotecan (2) [6–9] and topotecan (3) [10–12], have been approved by the FDA to treat cancers. Structurally, CPTs contain a pyranoindolizine tricyclic structure (the C-D-E ring system) with a tert-hydroxyl group at C20, and the stereochemistry at C20 is important for their biological activity. Thus, stereoselective construction of this tert-hydoxyl group is an important issue in the synthesis of CPTs.
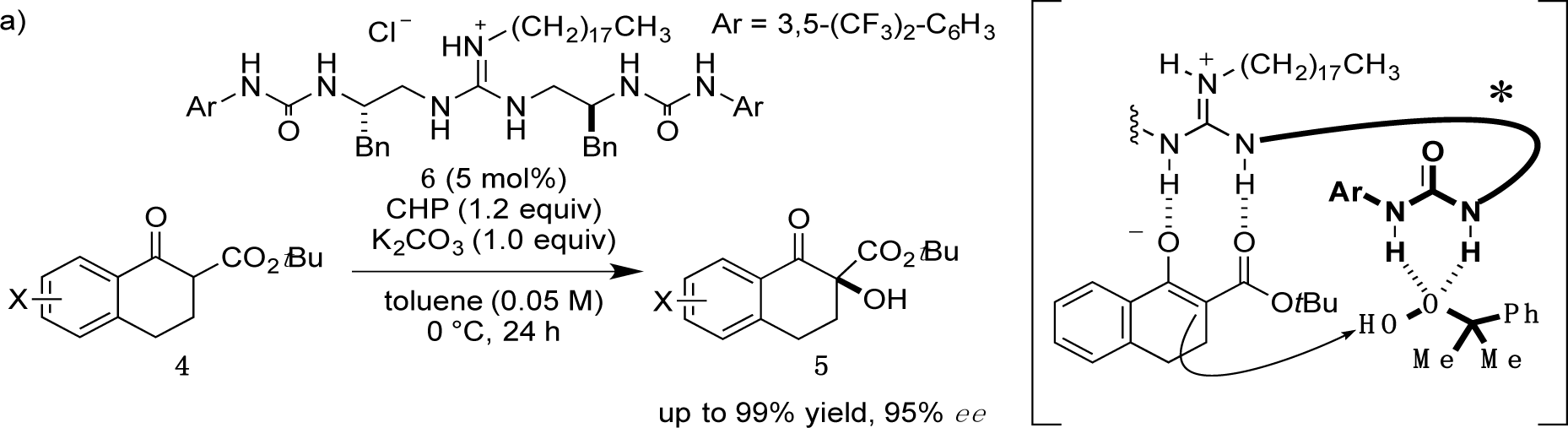
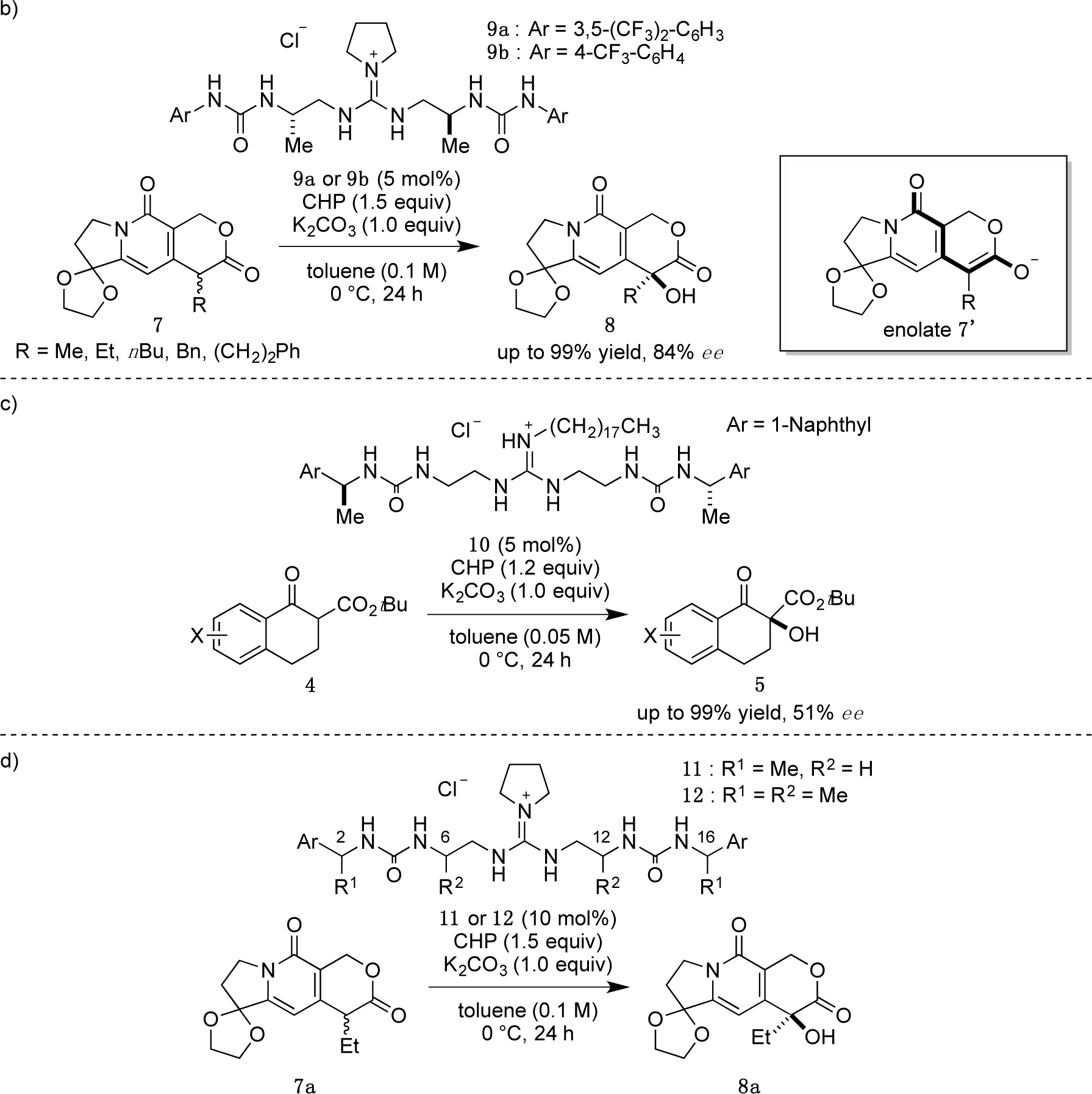
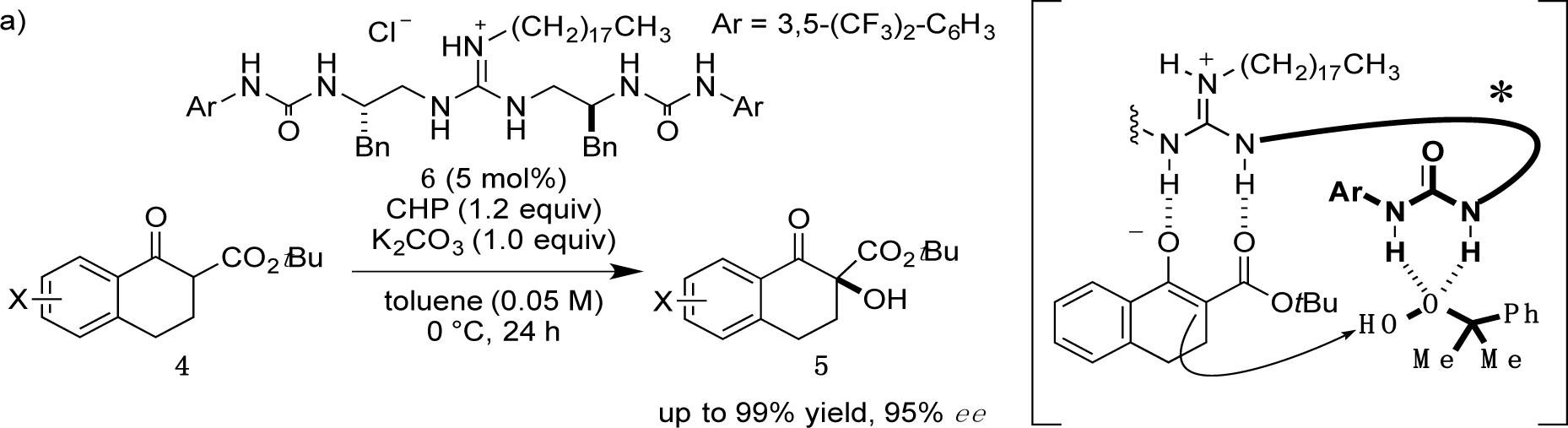
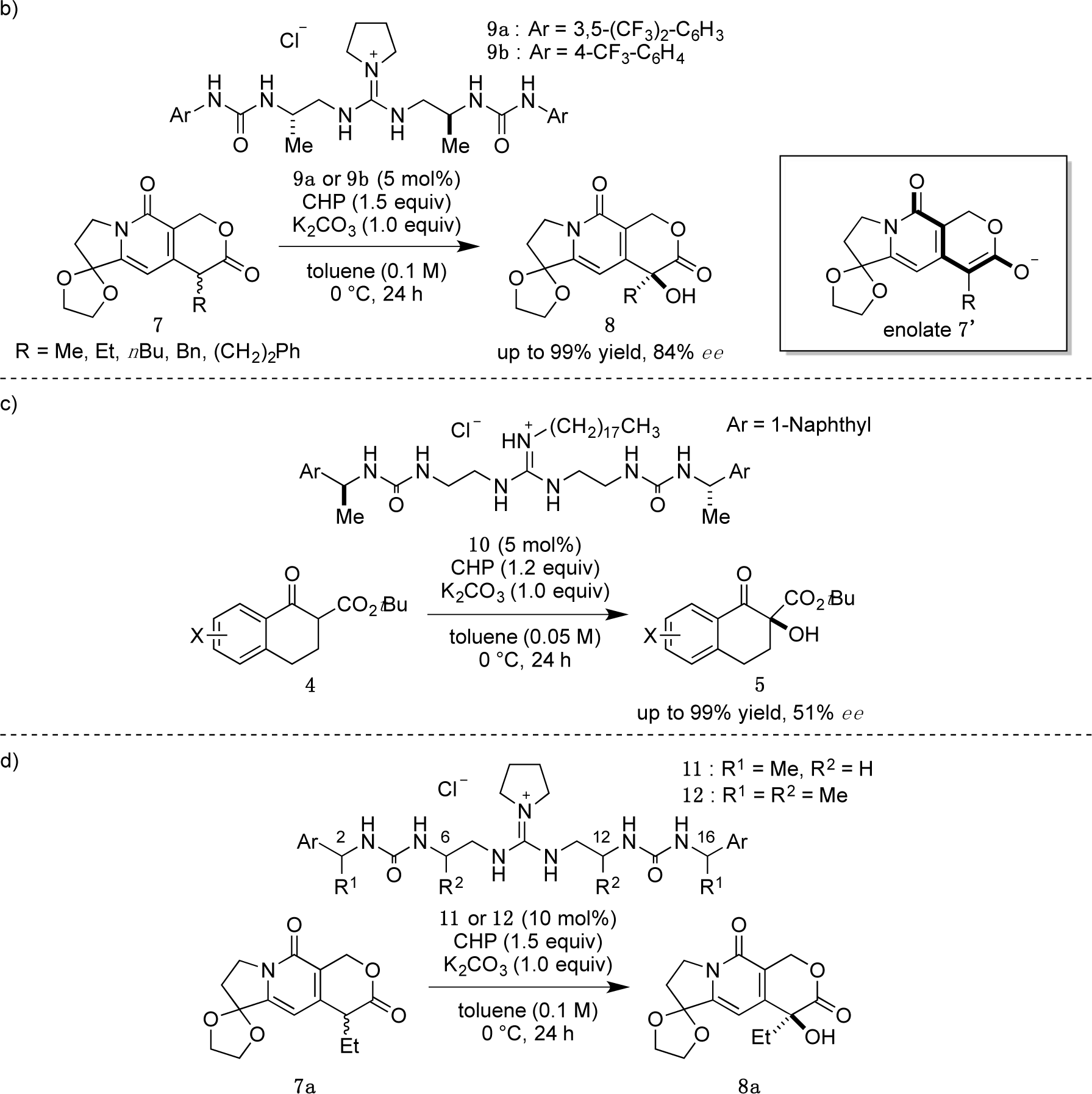
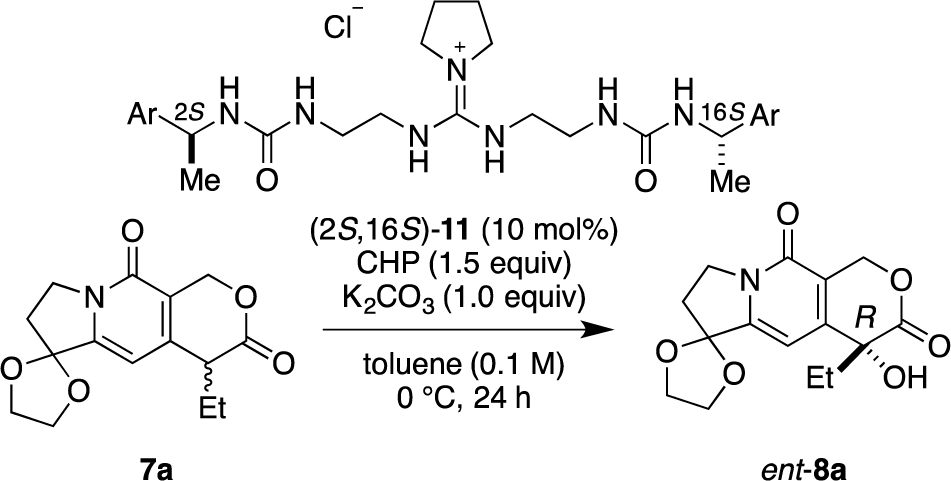
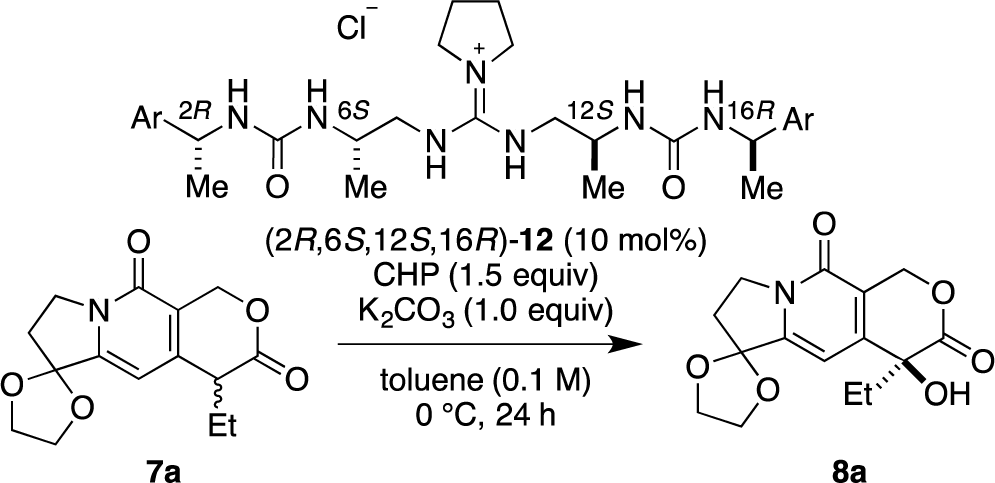
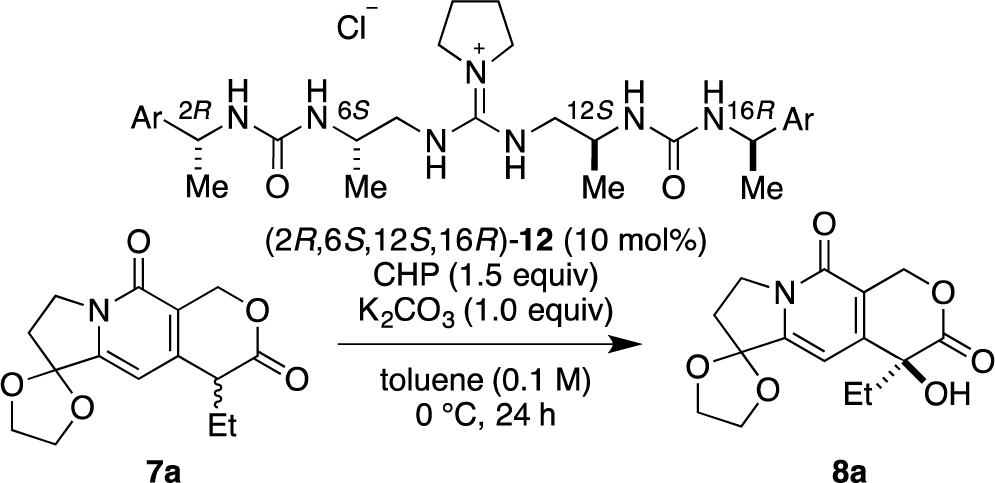
Recently, we have reported α-hydroxylation of tetralone-derived β-ketoesters 4 using cumene hydroperoxide (CHP) in the presence of guanidine-bisurea bifunctional organocatalyst 6 (Scheme 1a) [13]. In this reaction, the guanidine group and urea group were suggested to interact with the enolate of tetralone and the oxidant CHP, respectively. Based upon the proposed transition state model, we further applied this reaction to the α-hydroxylation of pyranoindolidine derivatives 7 to obtain the corresponding hydroxylated compounds 8 via the extended enolate 7′ with high enantioselectivity (Scheme 1b). The products 8 are key synthetic intermediates in the Friedländer condensation approach to CPT derivatives [14–22]. Subsequently, we developed the novel guanidine-bisurea bifunctional organocatalyst 10 (Scheme 1c) bearing a chiral center at the outer side of the urea groups [23]. Although it is conformationally more flexible than 6, we found that 10 catalyzed the α-hydroxylation of tetralone-derived β-ketoesters 4 with moderate enantioselectivity. Moreover, the catalyst 10 lacks a CF3 group in the Ar moiety, which had been believed to be mandatory for effective activation of the oxidant; nevertheless, the catalyst 10 gave the oxidative product 5 in excellent yield. Therefore, we set out to examine the effect of chirality at the outer side of urea in the newly designed catalyst 11. Herein, we described α-hydroxylation of pyranoindolidine derivative 7a in the presence of catalyst 11 bearing a chiral center at the outer side of the urea groups. We also examined the effectiveness of catalyst 12 possessing chirality at both the inner and outer sides of the urea groups (Scheme 1d).
2. Results and Discussion
Catalysts 11 and 12 were synthesized according to the procedure previously reported by our group [23]. With catalysts 11 and 12 in hand, we commenced our investigation of α-hydroxylation of the pyranoindolizine derivative 7a with catalyst (2S,16S)-11, which has chiral methyl groups with S configuration at the outer side of the urea groups, and various Ar groups. In the case of (2S,16S)-11a with phenyl as Ar, α-hydroxylation of 7a proceeded efficiently, and ent-8a was obtained in 77% yield with 50% ee in R configuration (Table 1, entry 1). With the catalyst (2S,16S)-11b, having 4-methoxyphenyl as Ar, the enantioselectivity increased to 61% ee with 88% yield (entry 2); however, when the electron-donating 4-methoxy group was replaced with the electron-withdrawing 4-nitro group, i.e., (2S,16S)-11c, the selectivity was drastically decreased to 31% ee (88% yield, entry 3). Since the nature of the Ar group in (2S,16S)-11 clearly affected the selectivity, introduction of naphthyl groups was investigated (entries 4 and 5). In the case of 1-naphthyl-substituted (2S,16S)-11d, a relatively high selectivity of 69% ee was obtained, but the 2-naphthyl-substituted catalyst of (2S,16S)-11e showed little selectivity for hydroxylated ent-8a, although very high catalytic activities (over 90% yield) were observed with both catalysts.
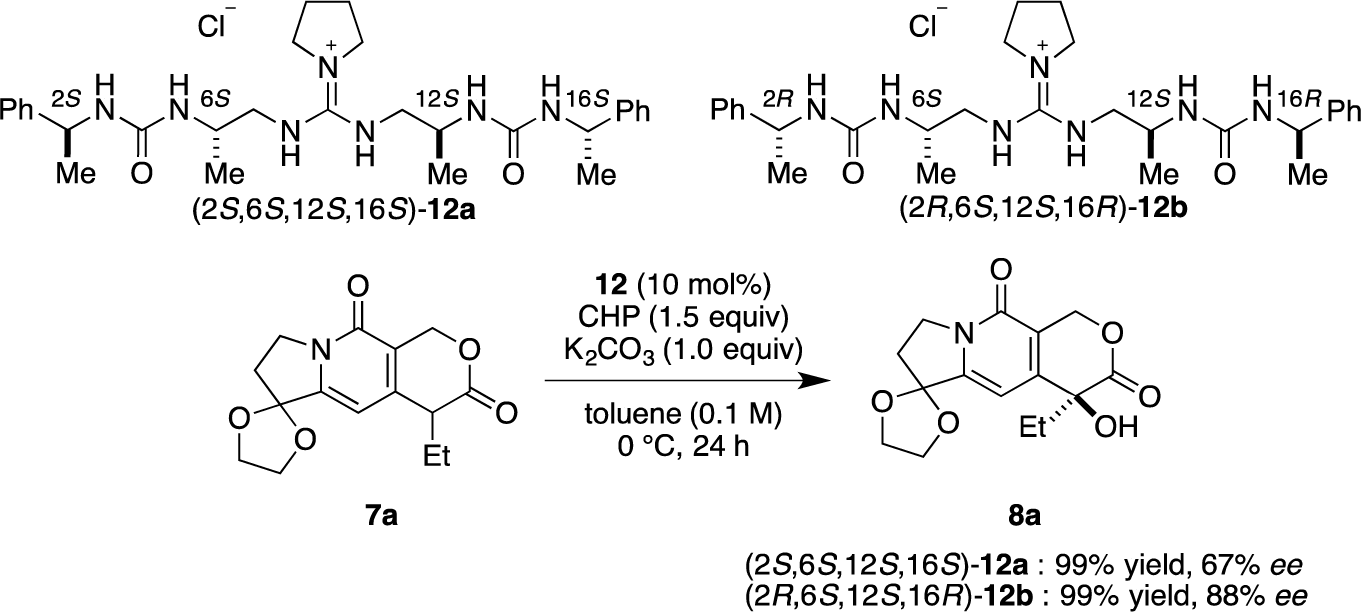
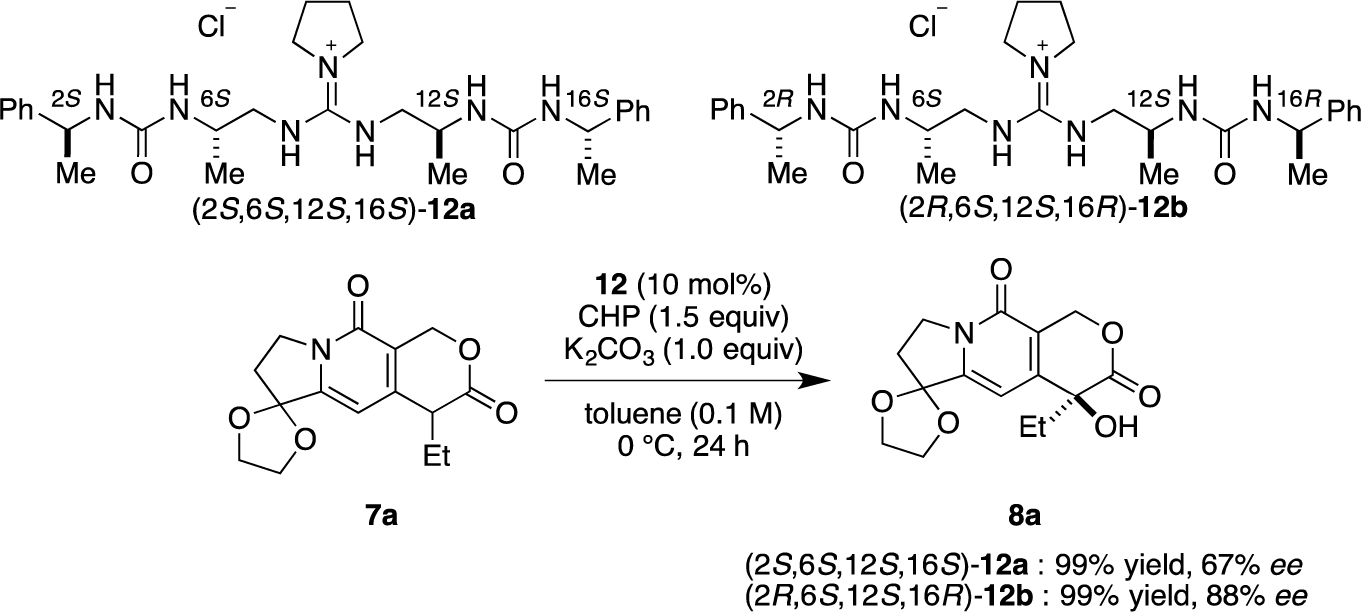
Then, we investigated the cooperative effect of inner-side chirality in the presence of outer-side chirality with (2S,6S,12S,16S)-12a and (2R,6S,12S,16R)-12b. Based on previously reported results together with the above results, inner- and outer-side chiralities with S configuration tend to induce R and S configuration of the product 8a, respectively. Therefore, (2R,6S,12S,16R)-12b should be a good matched pair for cooperation, and was expected to give the hydroxylated product 8a more selectively as compared with 12a.
Indeed, (2S,6S,12S,16S)-12a gave the α-hydroxylation product with moderate enantioselectivity (67% ee), while (2R,6S,12S,16R)-12b gave 88% ee with 99% yield (Scheme 2).
Then, we further investigated the effect of the aromatic group in (2R,6S,12S,16R)-12b on the selectivity (Table 2). The phenyl group in (2R,6S,12S,16R)-12b was varied to a 4-methoxyphenyl group (2R,6S,12S,16R)-12c, 1-naphthyl group (2R,6S,12S,16R)-12d, and 2-naphthyl group (2R,6S,12S,16R)-12e. High enantioselectivity (89% ee) was obtained in all cases (Table 2, entries 1–3).
3. Experimental Section
3.1. General Remarks
Flash chromatography was performed using silica gel 60 (spherical, particle size 0.040–0.100 mm. Kanto Co., Tokyo, Japan) or NH silica gel (spherical, particle size 0.06 mm, Fuji Silysia Chemical, Tokyo, Japan). Optical rotations were measured on a JASCO P-2200 polarimeter (JASCO, Tokyo, Japan). 1H and 13C NMR spectra were recorded on AL300 (JEOL), ECX400 (JEOL), or AVANCE400 (Bruker) instruments (JEOL, Tokyo, Japan). Chemical shifts in [d4]MeOH was reported in the scale relative to [d4]MeOH (δ = 3.30 ppm) for 1H NMR spectroscopy. For 13C NMR spectra, chemical shift was reported in the scale relative to [d4]MeOH (δ = 49.0 ppm) (internal reference). Mass spectra were recorded on a JMS-T100LC (JEOL) spectrometer (JEOL, Tokyo, Japan). HPLC analysis on a chiral stationary phase was performed on JASCO 800-series instruments (JASCO, Tokyo, Japan). A Daicel Chiralpak IA column was used with hexane/ethanol as the eluent.
3.2. Typical Procedure for a-Hydroxylation of Pyranoindolizine Derivative
A test tube equipped with a magnetic stirring bar was charged with catalyst 11 (0.01 mmol), pyranoindolizine derivative 7a (0.1 mmol), K2CO3 (0.1 mmol), and toluene (1.0 mL) at room temperature. The mixture was cooled to 0 °C and stirred for 10 min. Then, cumene hydroperoxide (0.15 mmol) was added, and stirring was continued at 0 °C for 24 h. The reaction was quenched by addition of 10% Na2S2O3 solution (2 mL) and acetic acid (0.2 mL), and the mixture was vigorously stirred at room temperature for 1 h. The organic layer was separated and the aqueous layer was extracted three times with chloroform. The combined organic solution was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate 1:1 to chloroform/methanol 100:1) to give the product 8a. Enantiomeric excess and absolute configuration were determined by HPLC analysis of the product on a chiral column (DAICEL Chiralpak IA) with n-hexane/ethanol.
3.3. Characterizations of Guanidine-Bisurea Bifunctional Organocatalysts 11 and 12
3.3.1. Catalyst (2S,16S)-11a
[α]25D = −25.6 (c = 1.00, CHCl3); 1H NMR (400 MHz, CD3OD) δ 7.34–7.30 (m, 8H), 7.26–7.21 (m, 2H), 4.82 (q, J = 7.0 Hz, 2H), 3.37–3.19 (m, 12H), 1.88–1.79 (m, 4H), 1.41 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CD3OD) δ 161.6, 158.4, 147.4, 130.4, 128.7, 127.7, 51.8, 50.9, 47.3, 41.7, 27.1, 24.6; HRMS (ESI, M-Cl) calcd for C27H40N7O2 494.3243, found 494.3293.
3.3.2. Catalyst (2S,16S)-11b
[α]25D = −28.9 (c = 0.66, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.20 (d, J = 8.6 Hz, 4H), 6.85 (d, J = 8.6 Hz, 4H), 4.74 (q, J = 7.2 Hz, 2H), 3.75 (s, 6H), 3.27–3.18 (m, 12H), 1.85–1.81 (m, 4H), 1.36 (d, J = 7.2 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.71, 160.02, 157.61, 138.52, 127.98, 114.81, 55.71, 50.37, 46.45, 40.32, 32.5, 26.25, 23.73; HRMS (ESI, M-Cl) calcd for C29H44N7O4 554.3454, found 554.3411.
3.3.3. Catalyst (2S,16S)-11c
[α]25D = −21.1 (c = 0.49, CHCl3); 1H NMR (300 MHz, CD3OD) δ 8.17 (d, J = 8.6 Hz, 4H), 7.53 (d, J = 8.6 Hz, 4H), 4.9 (q, J = 7.2 Hz, 2H), 3.27–3.17 (m, 12H), 1.89–1.82 (m, 4H), 1.42 (d, J = 7.2 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.52, 157.54, 154.64, 148.09, 128.00, 124.61, 50.77, 46.33, 40.38, 26.2, 23.19; HRMS (ESI, M-Cl) calcd for C27H38N6O6 584.2945, found 584.2930.
3.3.4. Catalyst (2S,16S)-11d
[α]25D = 56.4 (c = 0.50, CHCl3); 1H NMR (300 MHz, CD3OD) δ 8.13–8.10 (m, 2H), 7.88–7.83 (m, 2H), 7.74 (d, J = 7.5 Hz, 2H), 7.52–7.39 (m, 8H), 5.63 (q, J = 6.8 Hz, 2H), 3.27–3.11 (m, 12H), 1.62–1.57 (m, 4H), 1.51 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.49, 157.18, 142.12, 135.22, 131.76, 129.80, 128.47, 127.10, 126.59, 126.52, 124.01, 123.05, 79.48, 46.82, 46.37, 40.16, 25.90, 22.99; HRMS (ESI, M-Cl) calcd for C35H44N7O2 594.3556, found 594.3589.
3.3.5. Catalyst (2S,16S)-11e
[α]25D = −34.6 (c = 0.62, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.80–7.71 (m, 8H), 7.46–7.40 (m, 6H), 4.93 (q, J = 6.8 Hz, 2H), 3.24–3.01 (m, 12H), 1.52–1.40 (m, 4H), 1.45 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.79, 157.31, 144.05, 134.80, 133.99, 129.25, 128.78, 128.61, 127.17, 126.68, 125.53, 124.99, 51.17, 49.76, 46.50, 40.23, 25.87, 23.55; HRMS (ESI, M-Cl) calcd for C35H44N7O2 594.3556, found 594. 3589.
3.3.6. Catalyst (2S,6S,12S,16S)-12a
[α]25D = −29.6 (c = 0.96, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.34–7.18 (m, 10H), 4.8 (q, J = 6.8 Hz, 2H), 3.91–3.80 (m, 2H), 3.41–3.39 (m, 4H), 3.24 (dd, J = 13.4, 8.6 Hz, 4H), 2.00–1.98 (m, 4H), 1.4 (d, J = 6.8 Hz, 6H), 1.17 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.14, 157.62, 146.32, 129.49, 127.89, 126.81, 51.52, 50.80, 50.16, 47.07, 32.49, 26.36, 23.72; HRMS (ESI, M-Cl) calcd for C29H44N7O2 522.3556, found 522.3561.
3.3.7. Catalyst (2R,6S,12S,16R)-12b
[α]25D = 26.0 (c = 0.68, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.32–7.17 (m, 10H), 7.49 (q, J = 6.8 Hz, 2H), 3.90–3.70 (m, 4H), 3.22–2.92 (m, 8H), 1.63–1.59 (m, 4H), 1.38 (d, J = 6.8 Hz, 6H), 1.11 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.12, 157.27, 146.70, 129.53, 127.89, 126.78, 51.82, 50.90, 49.75, 46.82, 26.07, 23.98, 18.76; HRMS (ESI, M-Cl) calcd for C29H44N7O2 522.3556, found 522.3582.
3.3.8. Catalyst (2R,6S,12S,16R)-12c
[α]25D = 28.4 (c = 0.59, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.19 (d, J = 8.6 Hz, 4H), 6.85 (d, J = 8.6 Hz, 4H), 4.74 (q, J = 6.8 Hz, 2H), 3.24–2.95 (m, 4H), 1.67–1.65 (m, 2H), 1.36 (d, J = 6.8 Hz, 6H), 1.12 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.13, 160.02, 157.36, 138.63, 127.98, 114.87, 55.72, 51.87, 50.35, 49.85, 46.88, 26.13, 23.93, 18.79; HRMS (ESI, M-Cl) calcd for C31H48N7O4 582.3767, found 582.3767.
3.3.9. Catalyst (2R,6S,12S,16R)-12d
[α]25D = −63.1 (c = 0.54, CHCl3); 1H NMR (300 MHz, CD3OD) δ 8.12 (d, J = 7.2 Hz, 2H), 7.87–7.84 (m, 2H), 7.7 (d, J = 7.9 Hz, 2H), 7.53–7.37 (m, 8H), 5.65 (q, J = 6.8 Hz, 2H), 3.87–3.68 (m, 2H), 3.15–2.84 (m, 8H), 1.5 (d, J = 6.8 Hz, 6H), 1.03 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.08, 157.01, 142.47, 135.26, 131.71, 129.71, 129.85, 128.44, 127.14, 126.66, 126.58, 123.97, 123.00, 79.49, 51.88, 46.74, 25.62, 23.21, 18.65; HRMS (ESI, M-Cl) calcd for C37H48N7O4 622.3869, found 622.3851.
3.3.10. Catalyst (2R,6S,12S,16R)-12e
[α]25D = −40.0 (c = 0.67, CHCl3); 1H NMR (300 MHz, CD3OD) δ 7.79–7.61 (m, 8H), 7.46–7.37 (m, 6H), 4.9 (q, J = 6.8 Hz, 2H), 3.90–3.74 (m, 2H), 3.51–2.74 (m, 8H), 1.44 (d, J = 7.2 Hz, 6H), 1.08 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CD3OD) δ 160.20, 156.87, 144.18, 134.74, 133.92, 129.28, 128.74, 128.60, 127.16, 126.67, 125.50, 124.95, 51.95, 51.12, 49.38, 46.75, 25.50, 23.76, 18.72; HRMS (ESI, M-Cl) calcd for C37H48N7O4 622.3869, found 622.3876.
4. Conclusions
New guanidine-bisurea bifunctional organocatalysts 11 bearing chirality at the outer side of the urea groups were effective for enantioselective α-hydroxylation of pyranoindolizine derivative 7a. Catalysts bearing an aryl group with an electron-donating substituent or a 1-naphthyl group provided selectivity of 61%–69% ee. Based on these results, we synthesized catalysts 12 bearing chirality at both the outer and inner sides of the urea groups. They showed even higher enantioselectivity. Thus, chirality on the inner side of the urea groups showed a cooperative effect with outer-side chirality in the catalysts for improving stereoselectivity in the α-hydroxylation of pyranoindolizine.
Supplementary Materials
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (No. 23105013) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author Contributions
Minami Odagi and Tatsuya Watanabe performed the synthetic works; Kazuo Nagasawa contributed to the whole studies in this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J. Am. Chem. Soc 1966, 88, 3888–3890. [Google Scholar]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem 1985, 260, 14873–14878. [Google Scholar]
- Hsiang, Y.H.; Liu, L.F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res 1988, 48, 1722–1726. [Google Scholar]
- Hsiang, H.; Liu, L.F.; Wall, M.E.; Wani, M.C.; Nicholas, A.W.; Manikumar, G.; Kirschenbaum, S.; Silber, R.; Potmesil, M. DNA topoisomerase I-mediated DNA cleavage and cytotoxicity of camptothecin analogues. Cancer Res 1989, 49, 4385–4389. [Google Scholar]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar]
- Negoro, S.; Fukuoka, M.; Masuda, N.; Takada, M.; Kusunoki, Y.; Matsui, K.; Takifuji, N.; Kudoh, S.; Niitani, H.; Taguchi, T. Phase I study of weekly intravenous infusions of CPT-11, a new derivative of camptothecin, in the treatment of advanced non-small-cell lung cancer. J. Natl. Cancer Inst 1991, 83, 1164–1168. [Google Scholar]
- Kawato, Y.; Aonuma, M.; Hirata, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res 1991, 51, 4187–4191. [Google Scholar]
- Sawada, S.; Okajima, S.; Aiyama, R.; Nokata, K.; Furuta, T.; Yokokura, T.; Sugino, E.; Yamaguchi, K.; Miyasaka, T. Synthesis and antitumor activity of 20(S)-camptothecin derivatives: Carbamate-linked, water-soluble derivaties of 7-ethyl-10-hydroxycamptothecin. Chem. Pharm. Bull 1991, 39, 1446–1454. [Google Scholar]
- Wiseman, L.R.; Markham, A. Irinotecan. Drugs 1996, 52, 606–623. [Google Scholar]
- Kingsbury, W.D.; Boehm, J.C.; Jakas, D.R.; Holden, K.G.; Hecht, S.M.; Gallagher, G.; Caranfa, M.J.; McCabe, F.L.; Faucette, L.F.; Johnson, R.K.; et al. Synthesis of water-soluble (aminoalkyl) camptothecin analogs: Inhibition of topoisomerase I and antitumor activity. J. Med. Chem 1991, 34, 98–107. [Google Scholar]
- Ormrod, D.; Spencer, C.M. Topotecan. Drugs 1999, 58, 533–551. [Google Scholar]
- Riemsma, R.; Simons, J.P.; Bashir, Z.; Gooch, C.L.; Kleijnen, J. Systematic review of topotecan (Hycamtin) in relapsed small cell lung cancer. BMC Cancer 2010, 10. [Google Scholar] [CrossRef]
- Odagi, M.; Furukori, K.; Watanabe, T.; Nagasawa, K. Asymmetric α-Hydroxylation of tetralone-derived β-Ketoesters by using a guanidine-urea bifunctional organocatalyst in the presence of cumene hydroperoxide. Chem. Eur. J 2013, 19, 16740–16745. [Google Scholar]
- Wani, M.C.; Nicholas, A.W.; Wall, M.E. Plant antitumor agents. 28. Resolution of a key tricyclic synthon, 5′(RS)-1,5-dioxo-5′-hydroxy-2′H,5′H,6′H-6′-oxopyrano[3′,4′-f].DELTA.6,8-tetrahydroindolizine: Total synthesis and antitumor activity of 20(S)- and 20(R)-camptothecin. J. Med. Chem 1987, 30, 2317–2319. [Google Scholar]
- Ejima, A.; Terasawa, H.; Sugimori, M.; Tagawa, H. Asymmetric synthesis of (S)-camptothecin. Tetrahedron Lett 1989, 30, 2639–2640. [Google Scholar]
- Ejima, A.; Terasawa, H.; Sugimori, M.; Tagawa, H. Antitumour agents. Part 2. Asymmetric synthesis of (S)-camptothecin. J. Chem. Soc. Perkin Trans. 1 1990, 27–31. [Google Scholar]
- Jew, S.; Ok, K.; Kim, H.; Kim, M.G.; Kim, J.M.; Hah, J.M.; Cho, Y. Enantioselective synthesis of 20(S)-camptothecin using sharpless catalytic asymmetric dihydroxylation. Tetrahedron Asymmetry 1995, 6, 1245–1248. [Google Scholar]
- Henegar, K.E.; Ashford, S.W.; Baughman, T.A.; Sih, J.C.; Gu, R.L. Practical asymmetric synthesis of (S)-4-Ethyl-7,8-dihydro-4-hydroxy-1H-pyrano[3,4-f]indolizine-3,6,10(4H)-trione, a key intermediate for the synthesis of irinotecan and other camptothecin analogs. J. Org. Chem 1997, 62, 6588–6597. [Google Scholar]
- Imura, A.; Itoh, M.; Miyadera, A. Enantioselective synthesis of 20(S)-camptothecin using an enzyme-catalyzed resolution. Tetrahedron Asymmetry 1998, 9, 2285–2291. [Google Scholar]
- Imura, A.; Itoh, M.; Miyadera, A. Enantioselective synthesis of a key intermediate of 20(S)-camptothecin via and enzyme-catalyzed resolution. Chem. Pharm. Bull 1998, 46, 1878–1880. [Google Scholar]
- Watanabe, T.; Tsuboi, Y.; Nomura, S. Practical synthesis of (20S)-10-(3-Aminopropyloxy)-7-ethylcamptothecin, a water-soluble analogue of camptothecin. Chem. Asian J 2013, 8, 630–638. [Google Scholar]
- Watanabe, T.; Odagi, M.; Furukori, K.; Nagasawa, K. Asymmetric α-hydroxylation of a lactone with vinylogous pyridone by using a guanidine-urea bifunctional organocatalyst: Catalytic enantioselective synthesis of a key intermediate for (20S)-camptothecin analogues. Chem. Eur. J 2014, 20, 591–597. [Google Scholar]
- Odagi, M.; Takayama, K.; Furukori, K.; Watanabe, T.; Nagasawa, K. Development of novel guanidine-bisurea bifunctional organocatalysts and their application to asymmetric α-hydroxylation of tetralone-derived β-keto esters. Aust. J. Chem 2014, 67, 1017–1020. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | (2S,16S)-11 | ent-8a | ||
| Ar | yield (%)b | ee (%)c | ||
| 1 | (2S,16S)-11a | C6H5 | 77 | 50 |
| 2 | (2S,16S)-11b | 4-MeO-C6H4 | 88 | 61 |
| 3 | (2S,16S)-11c | 4-NO2-C6H4 | 88 | 31 |
| 4 | (2S,16S)-11d | 1-Naphthyl | 92 | 69 |
| 5 | (2S,16S)-11e | 2-Naphthyl | 91 | 51 |
Notes:aReaction conditions: 7a (0.1 mmol), CHP (0.15 mmol) and K2CO3 (0.1 mmol) in the presence of (2S,16S)-11 (10 mol%) in toluene (1.0 mL) at 0 °C for 24 h;bIsolated yield;cDetermined by HPLC analysis using a chiral stationary phase.
 | ||||
|---|---|---|---|---|
| Entry | (2R,6S,12S,16R)-12 | 8a | ||
| Ar | yield (%)b | ee (%)c | ||
| 1 | (2R,6S,12S,16R)-12c | 4-MeO-C6H4 | 99 | 89 |
| 2 | (2R,6S,12S,16R)-12d | 1-Naphthyl | 95 | 89 |
| 3 | (2R,6S,12S,16R)-12e | 2-Naphthyl | 75 | 89 |
Notes:aReaction conditions: 7a (0.1 mmol), CHP (0.15 mmol) and K2CO3 (0.1 mmol) in the presence of (2R,6S,12S,16R)-12 (10 mol%) in toluene (1.0 mL) at 0 °C for 24 h;bIsolated yield;cDetermined by HPLC analysis using a chiral stationary phase.
© 2015 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odagi, M.; Watanabe, T.; Nagasawa, K. Development of Guanidine-Bisurea Bifunctional Organocatalyst Bearing Chirality at the Inner and Outer Sides of the Urea Groups, and Application to Enantioselective α-Hydroxylation of Pyranoindolizine Intermediate for Camptothecin Synthesis. Symmetry 2015, 7, 43-52. https://doi.org/10.3390/sym7010043
Odagi M, Watanabe T, Nagasawa K. Development of Guanidine-Bisurea Bifunctional Organocatalyst Bearing Chirality at the Inner and Outer Sides of the Urea Groups, and Application to Enantioselective α-Hydroxylation of Pyranoindolizine Intermediate for Camptothecin Synthesis. Symmetry. 2015; 7(1):43-52. https://doi.org/10.3390/sym7010043
Chicago/Turabian StyleOdagi, Minami, Tatsuya Watanabe, and Kazuo Nagasawa. 2015. "Development of Guanidine-Bisurea Bifunctional Organocatalyst Bearing Chirality at the Inner and Outer Sides of the Urea Groups, and Application to Enantioselective α-Hydroxylation of Pyranoindolizine Intermediate for Camptothecin Synthesis" Symmetry 7, no. 1: 43-52. https://doi.org/10.3390/sym7010043
APA StyleOdagi, M., Watanabe, T., & Nagasawa, K. (2015). Development of Guanidine-Bisurea Bifunctional Organocatalyst Bearing Chirality at the Inner and Outer Sides of the Urea Groups, and Application to Enantioselective α-Hydroxylation of Pyranoindolizine Intermediate for Camptothecin Synthesis. Symmetry, 7(1), 43-52. https://doi.org/10.3390/sym7010043