Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene


Abstract
:1. Introduction
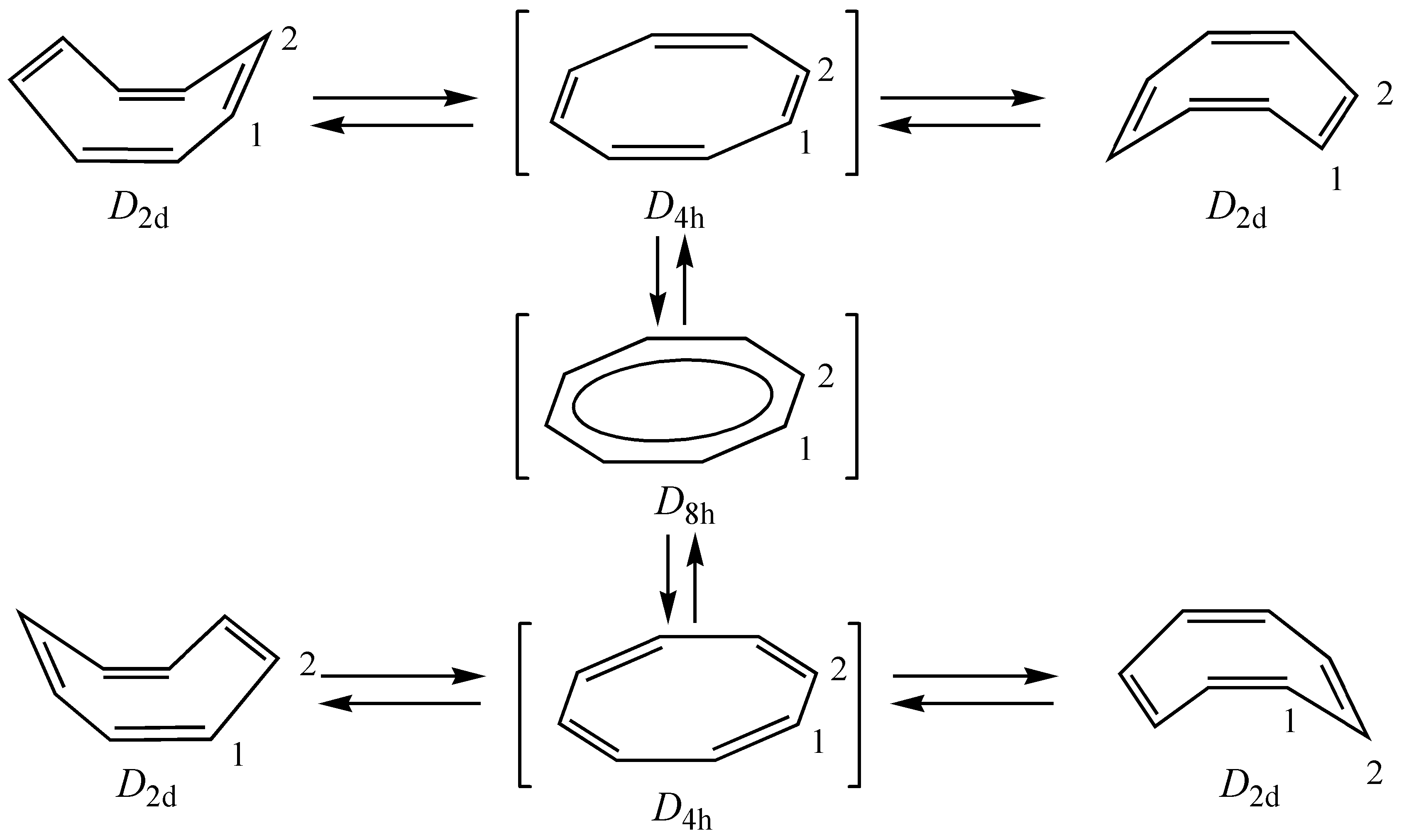
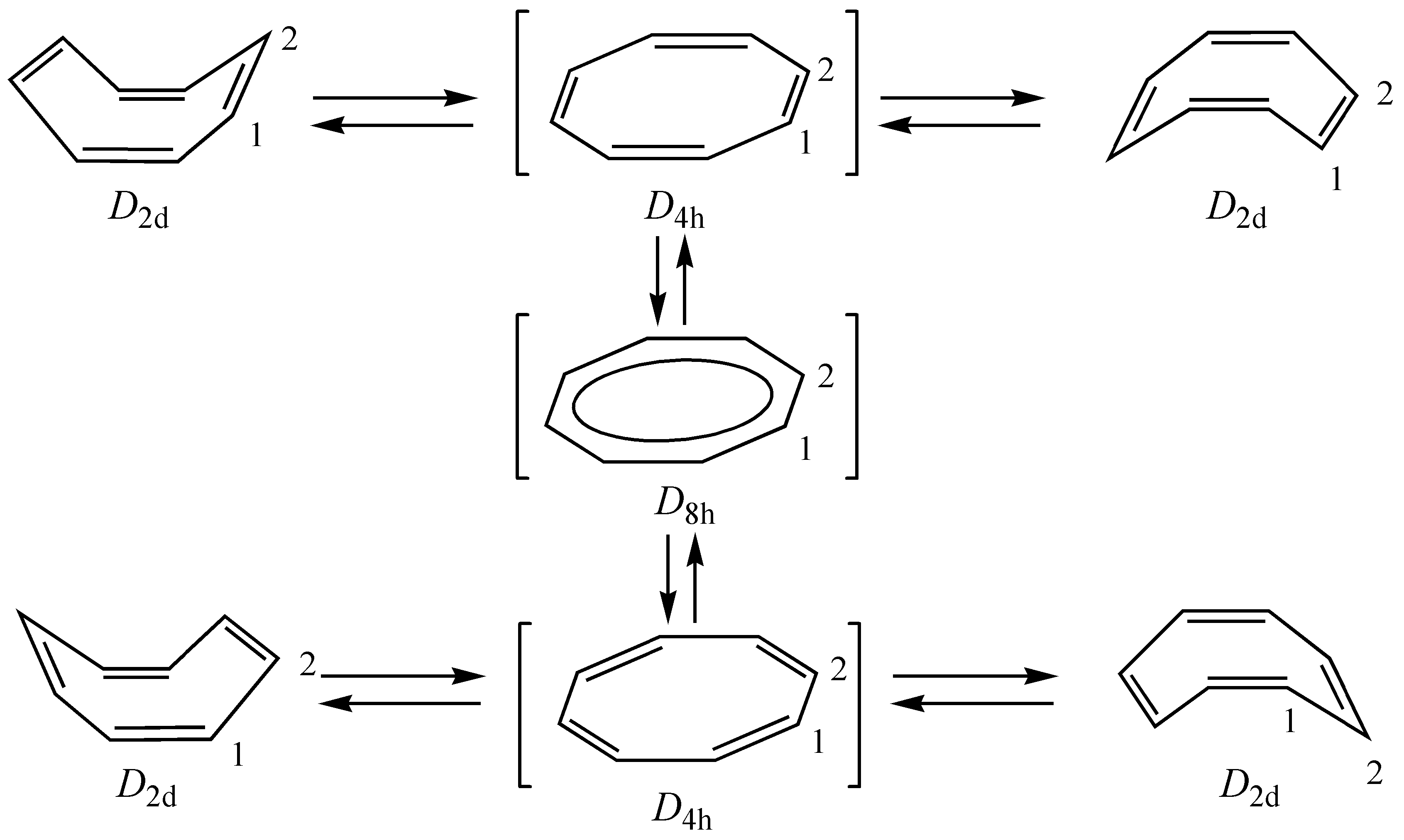
2. Unsubstituted COT
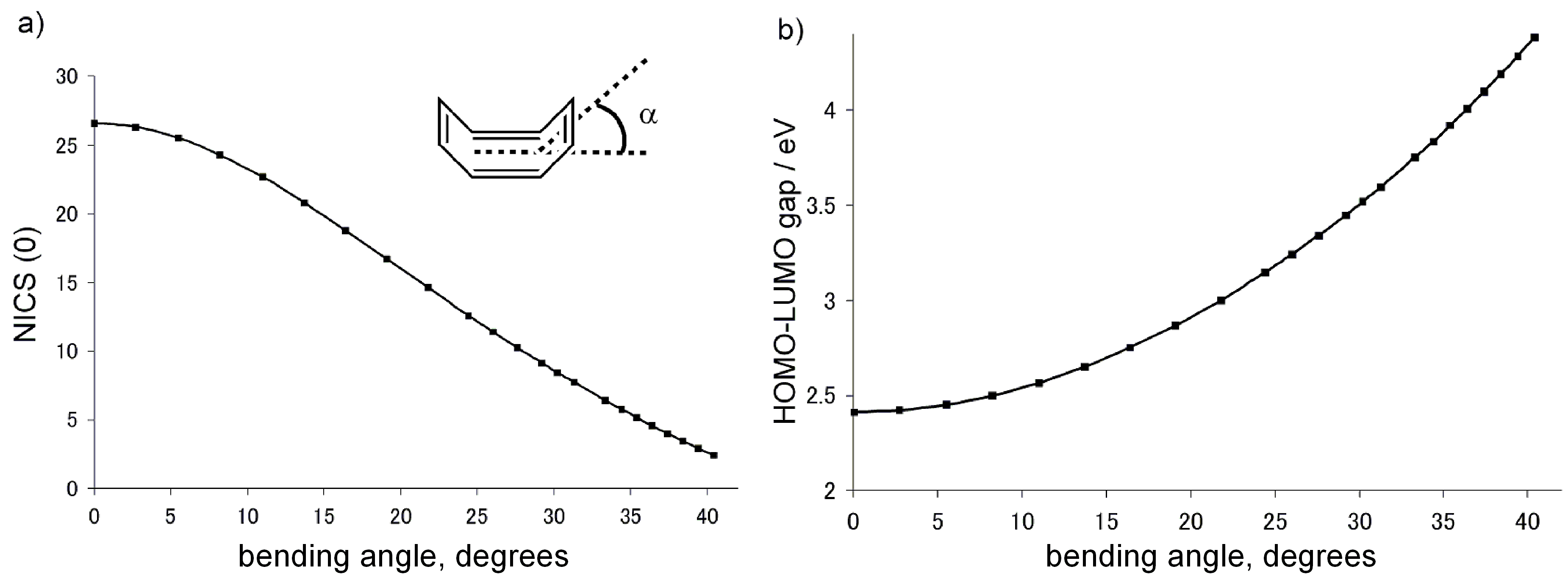
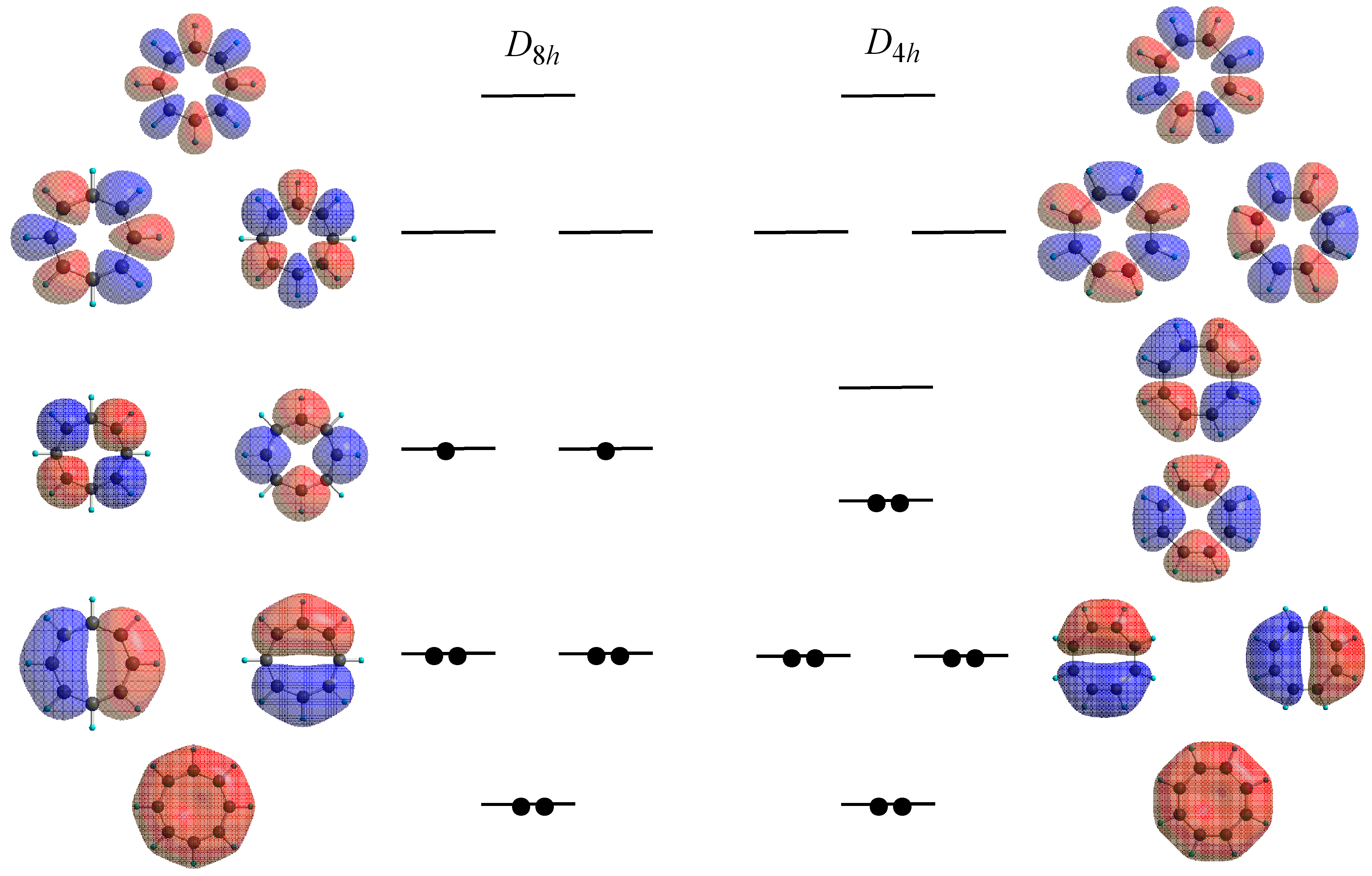
2.1. Computational Studies Based on NICS
2.2. Computational Studies Based on CTOCD-DZ
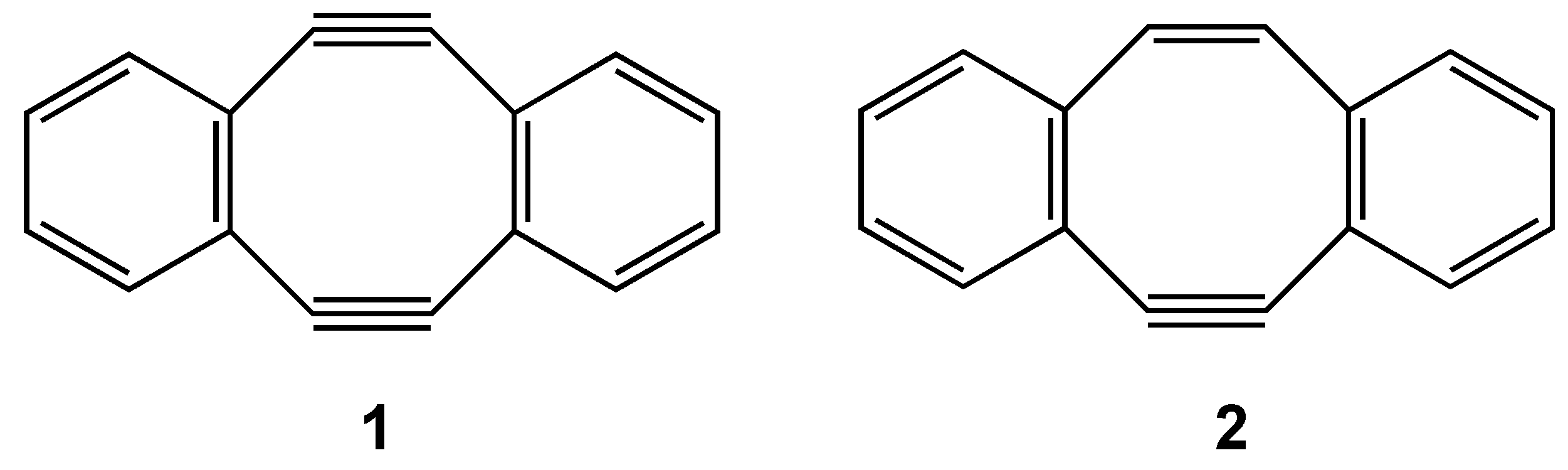
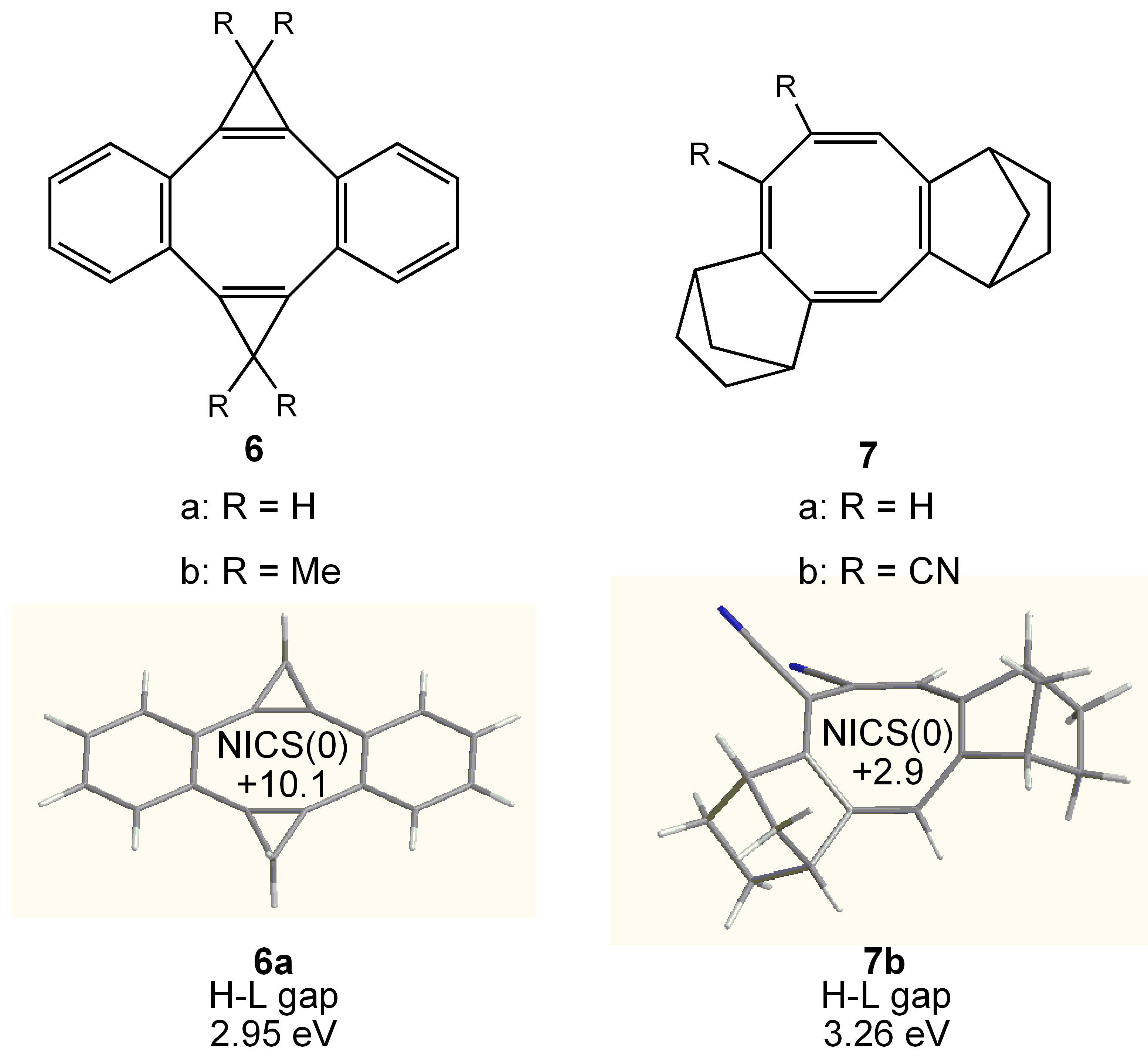
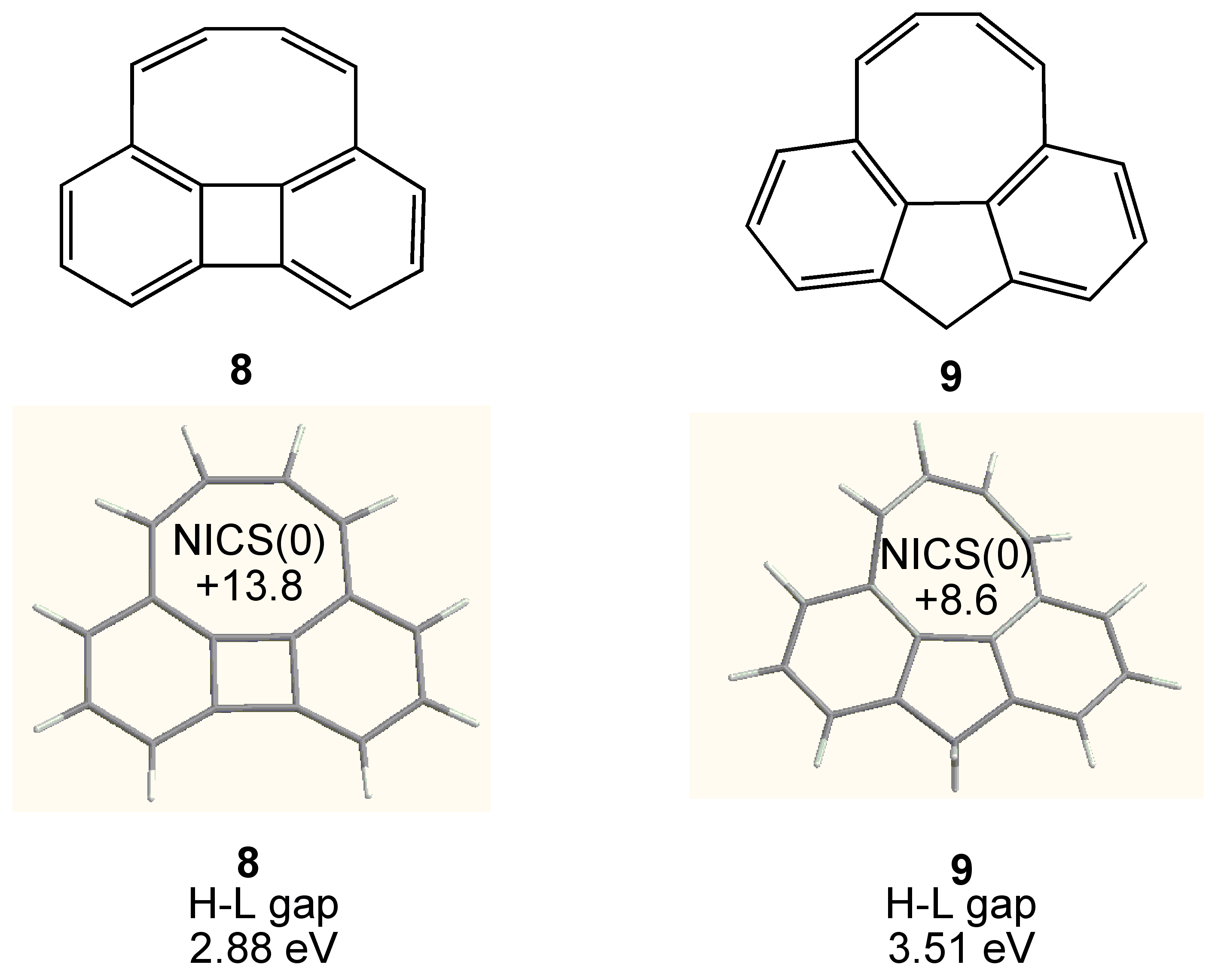
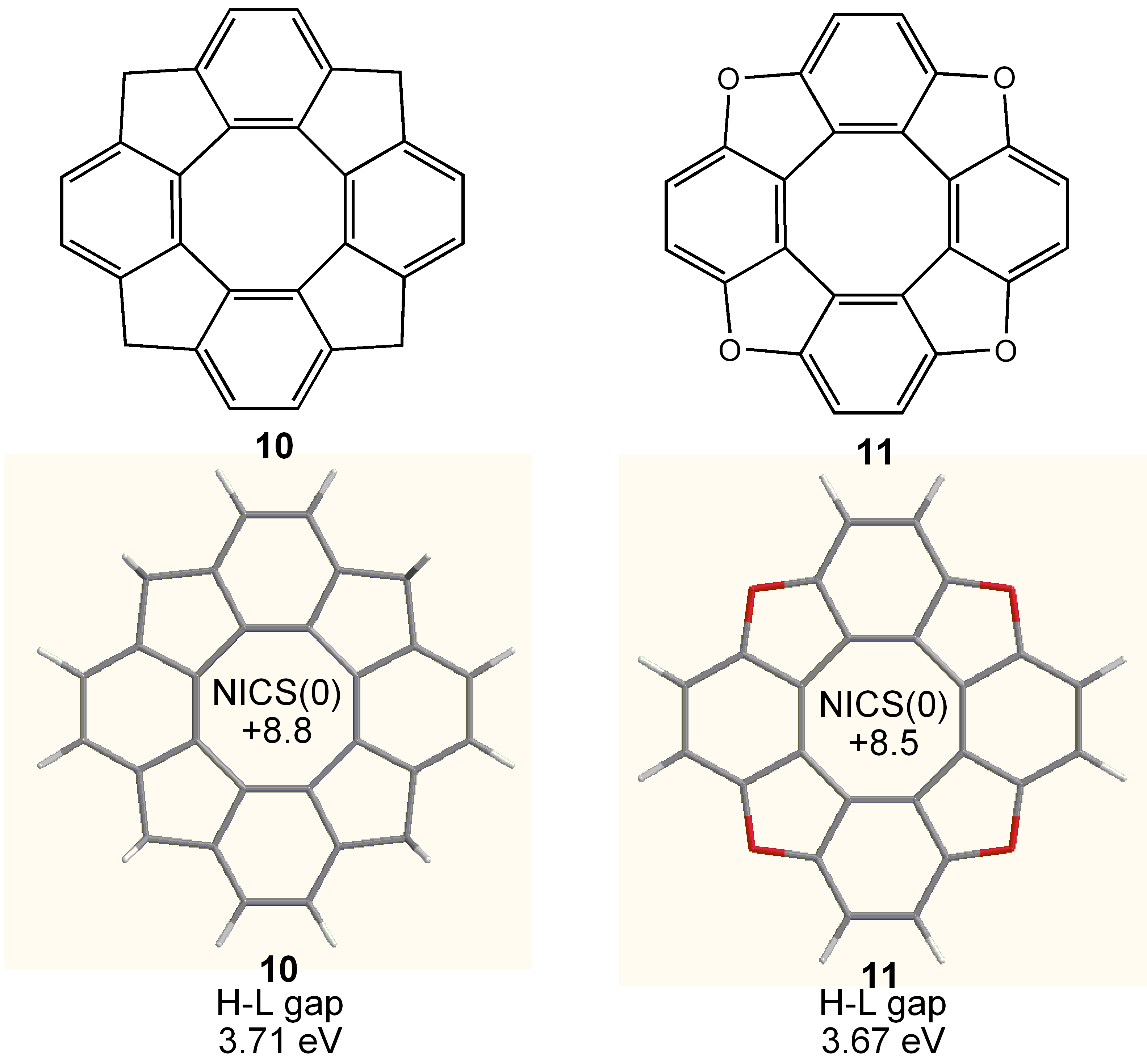
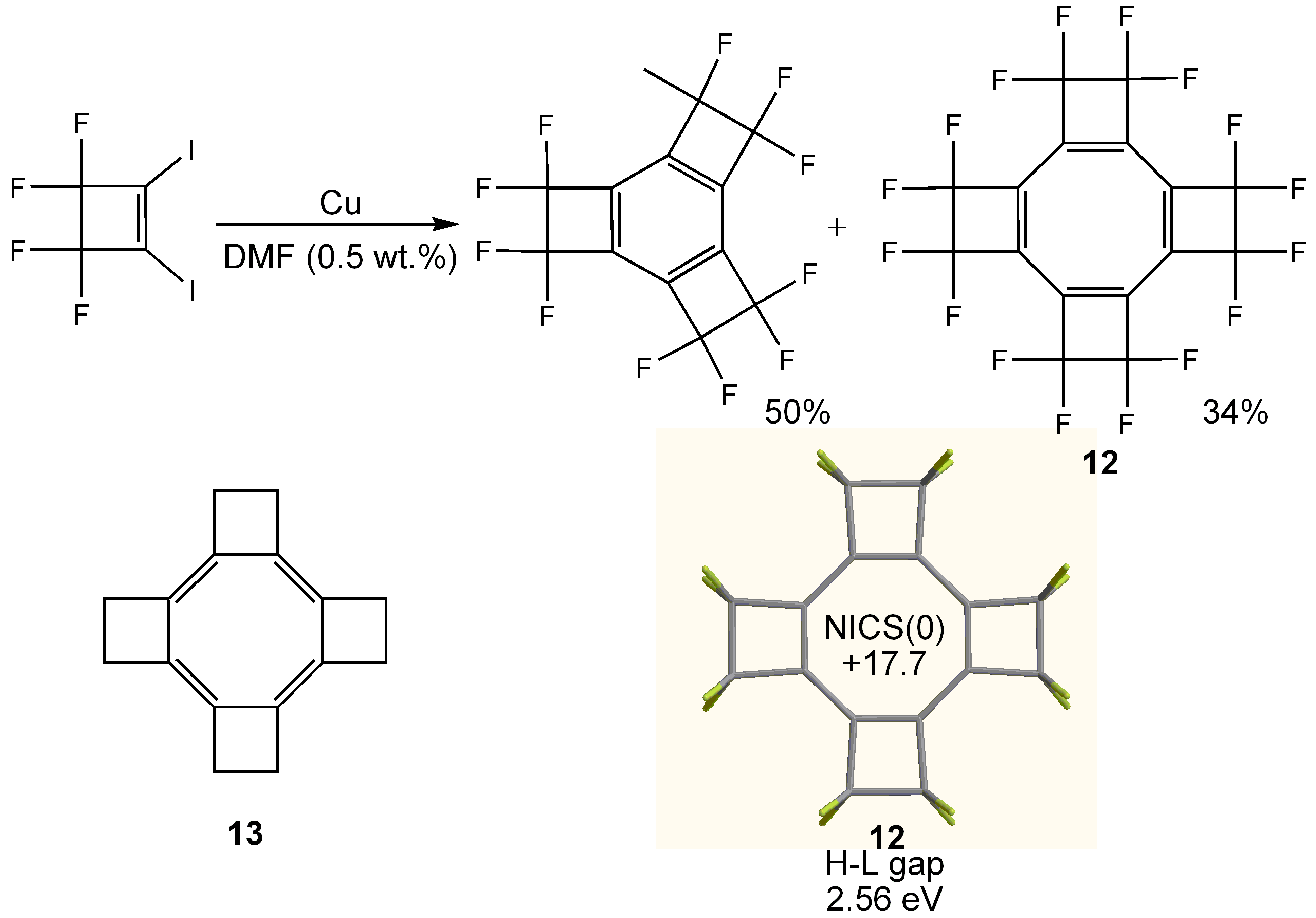
3. COTs Planarized by Annelation
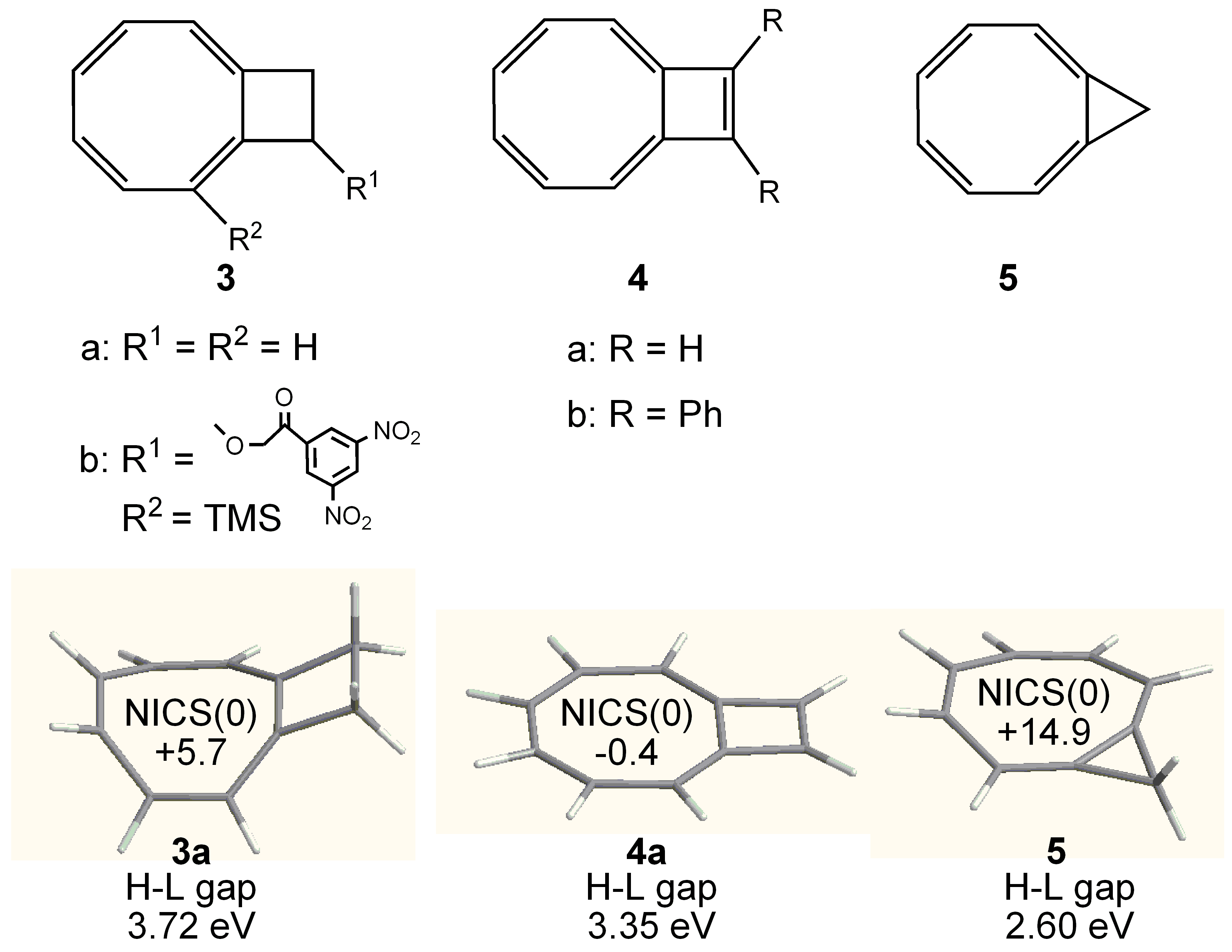
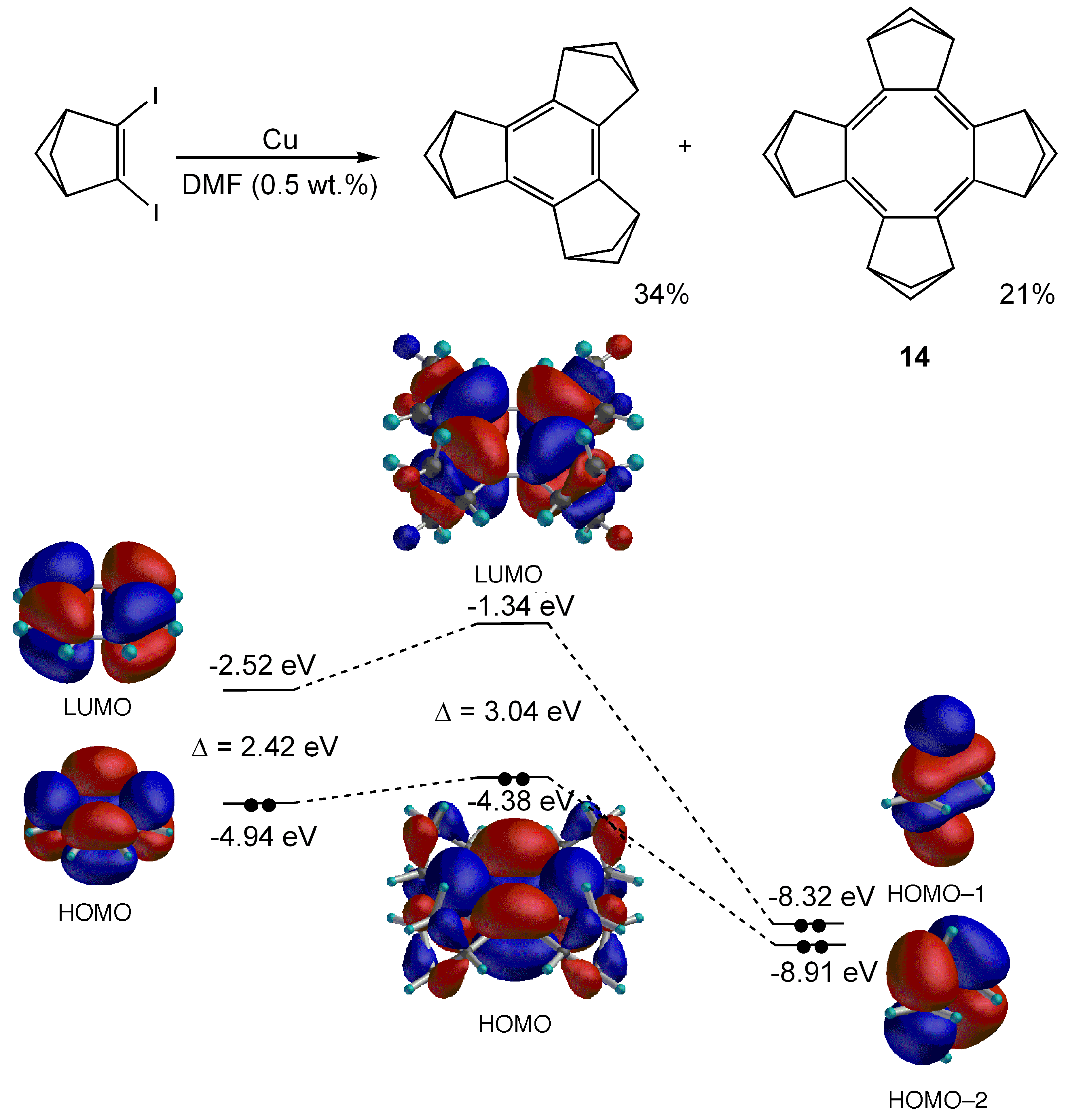
3.1. Attempted syntheses
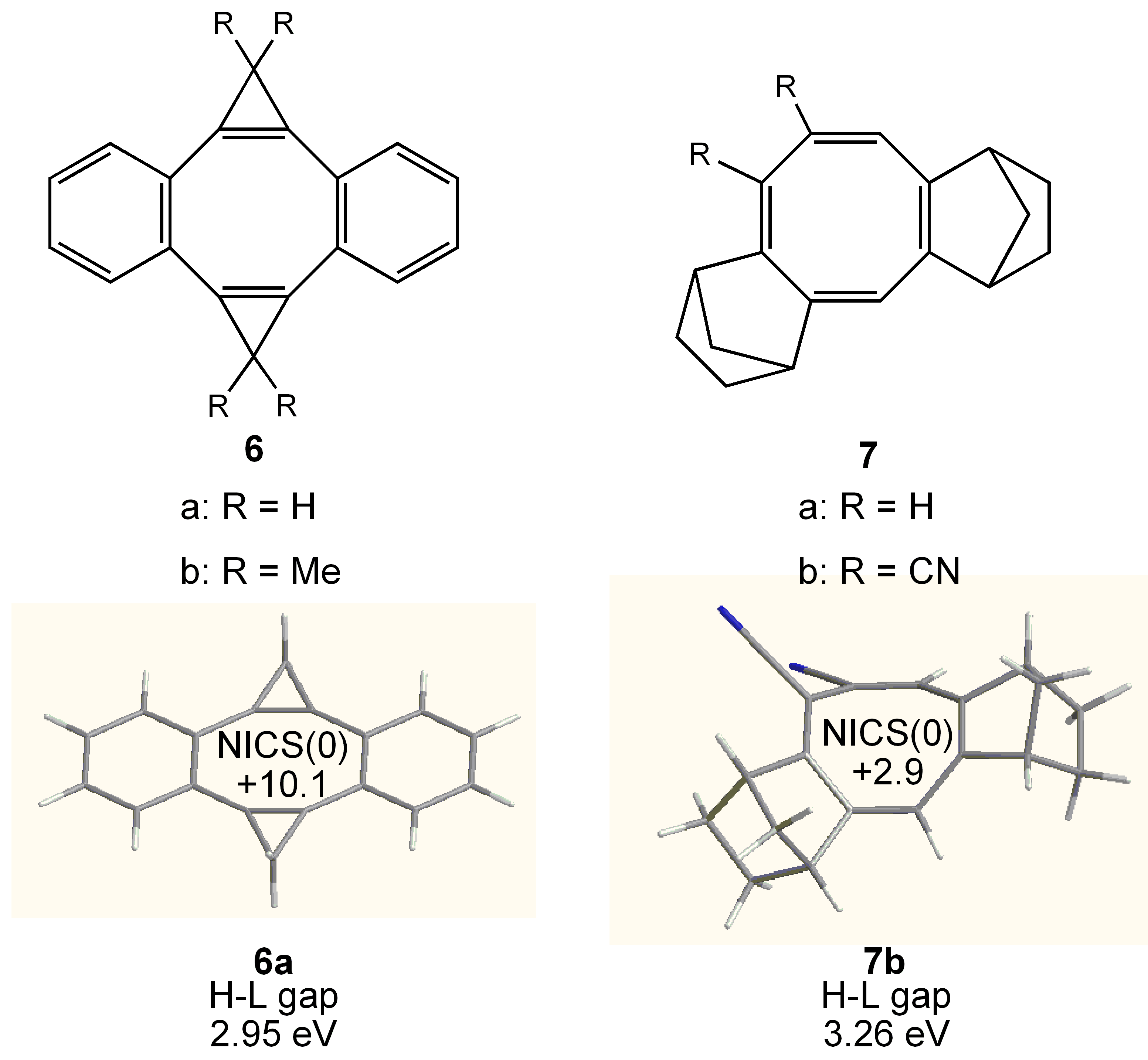
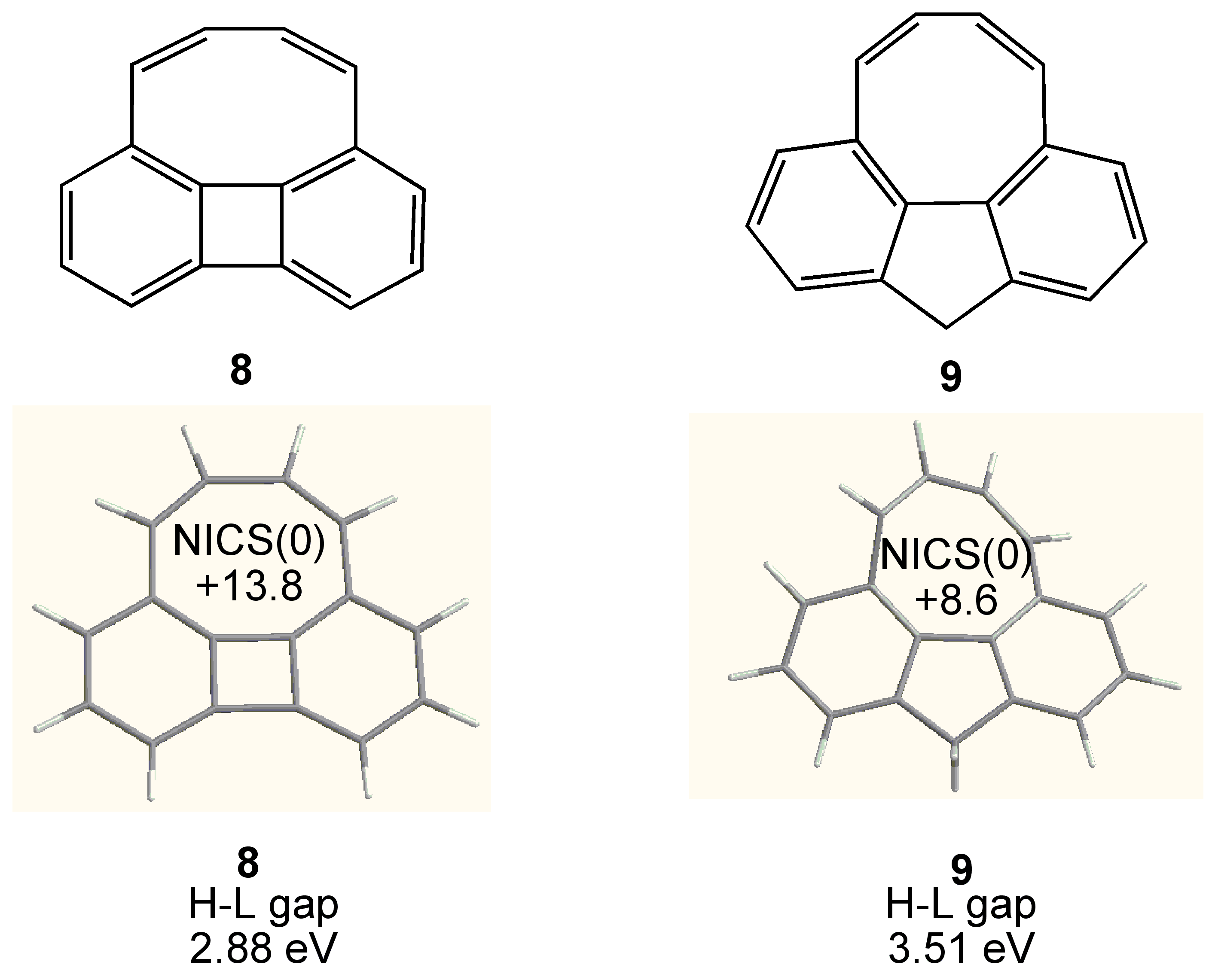
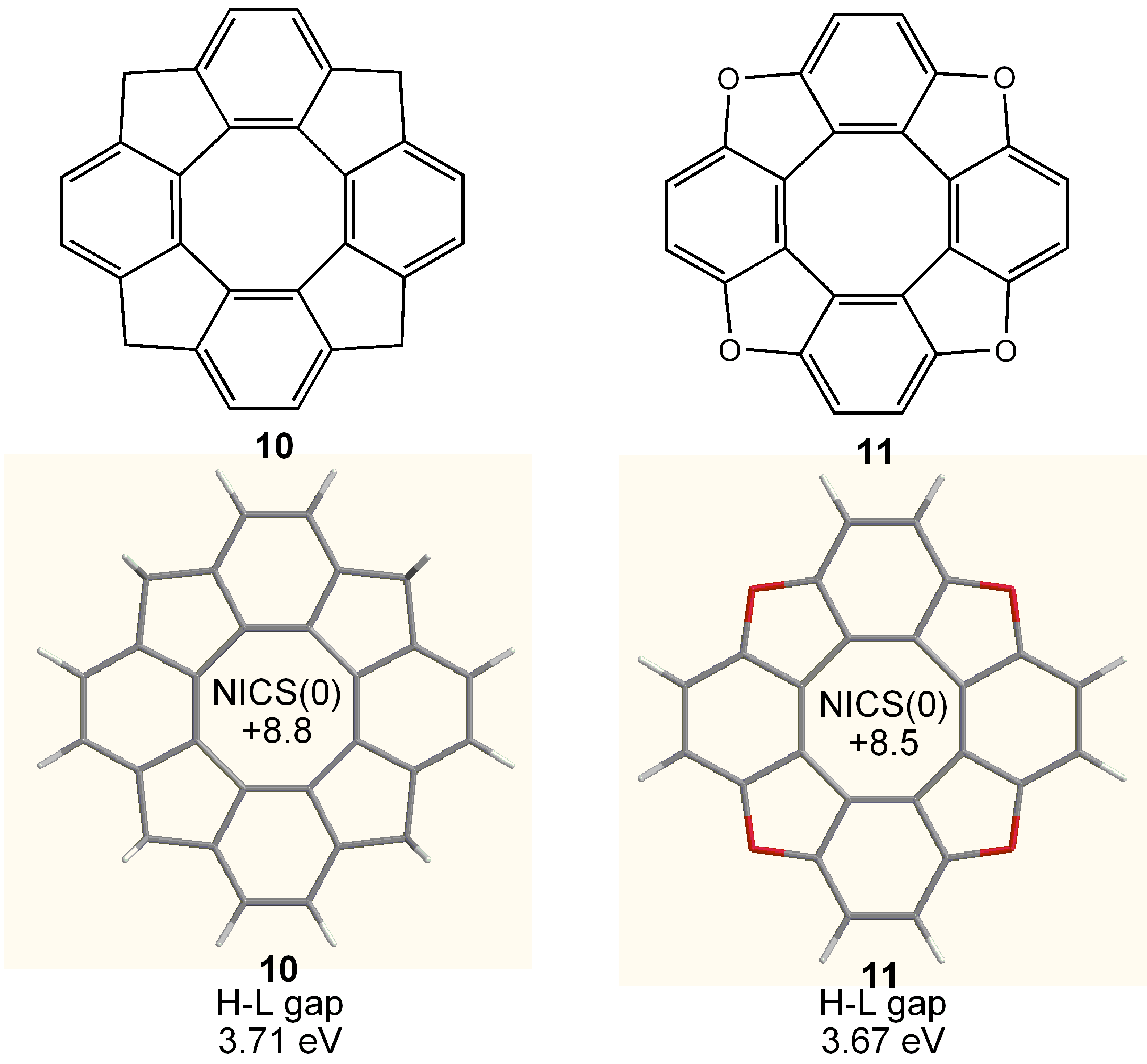
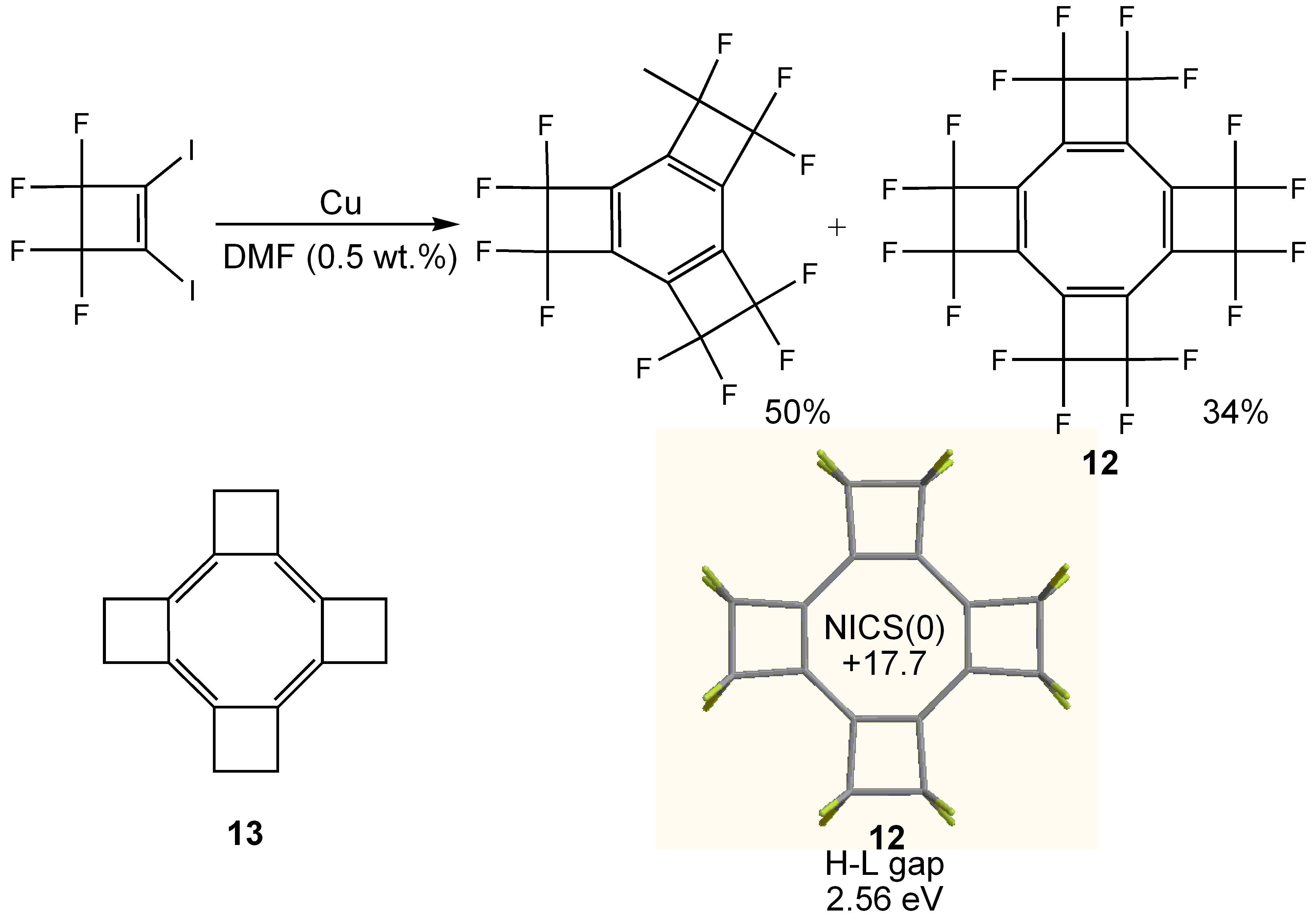
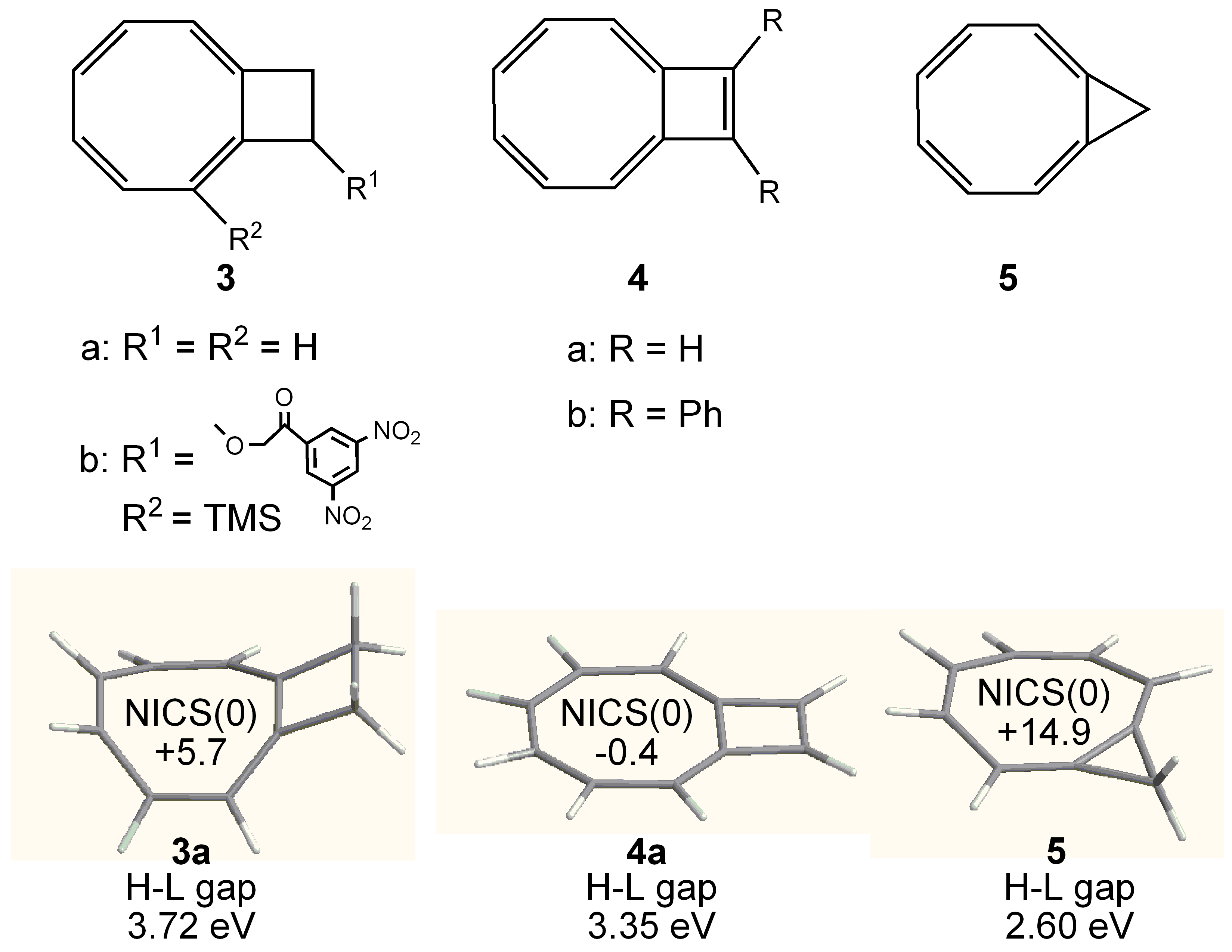
3.2. Planar COT Annelated with Four Cyclobutene Rings
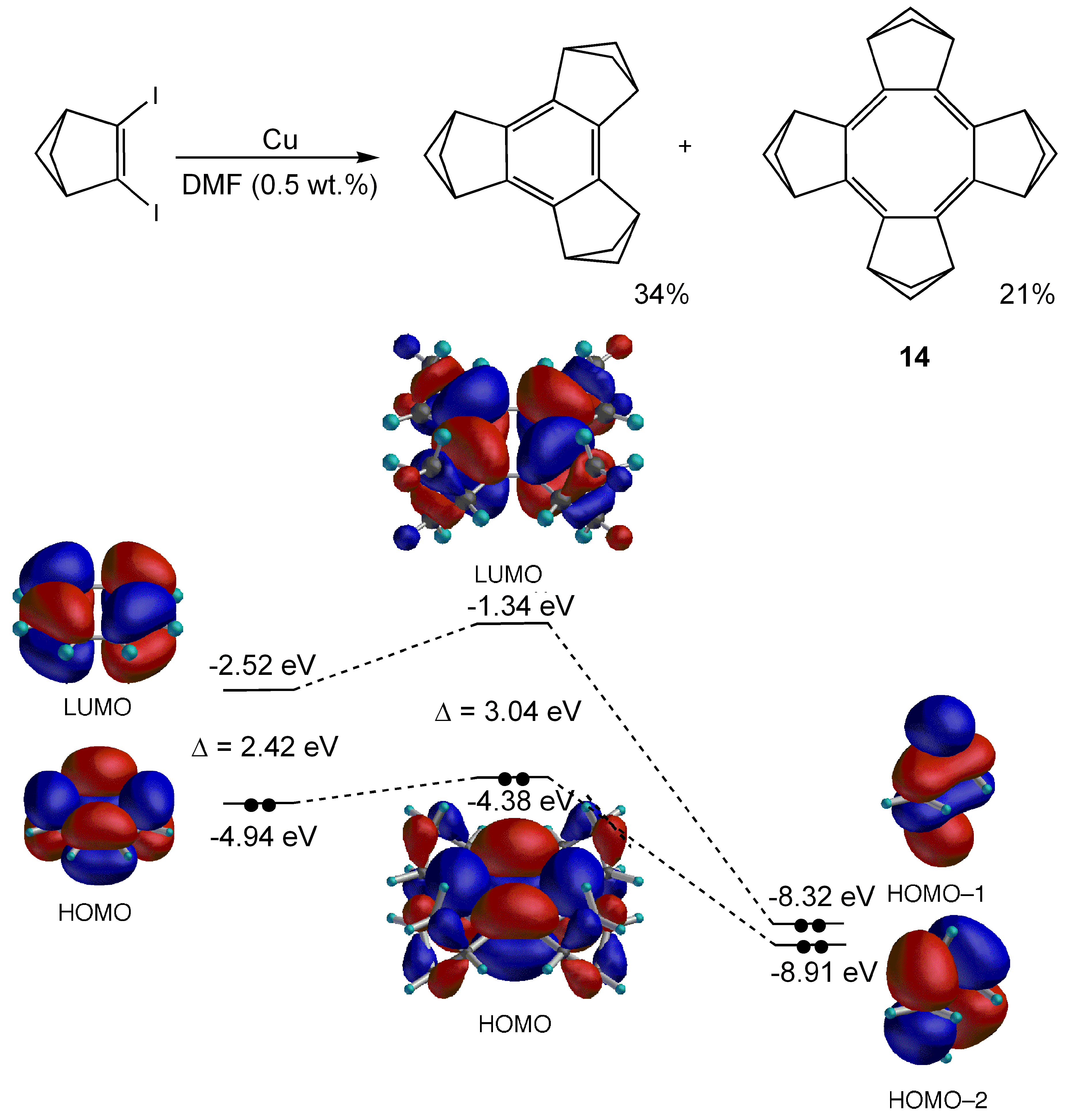
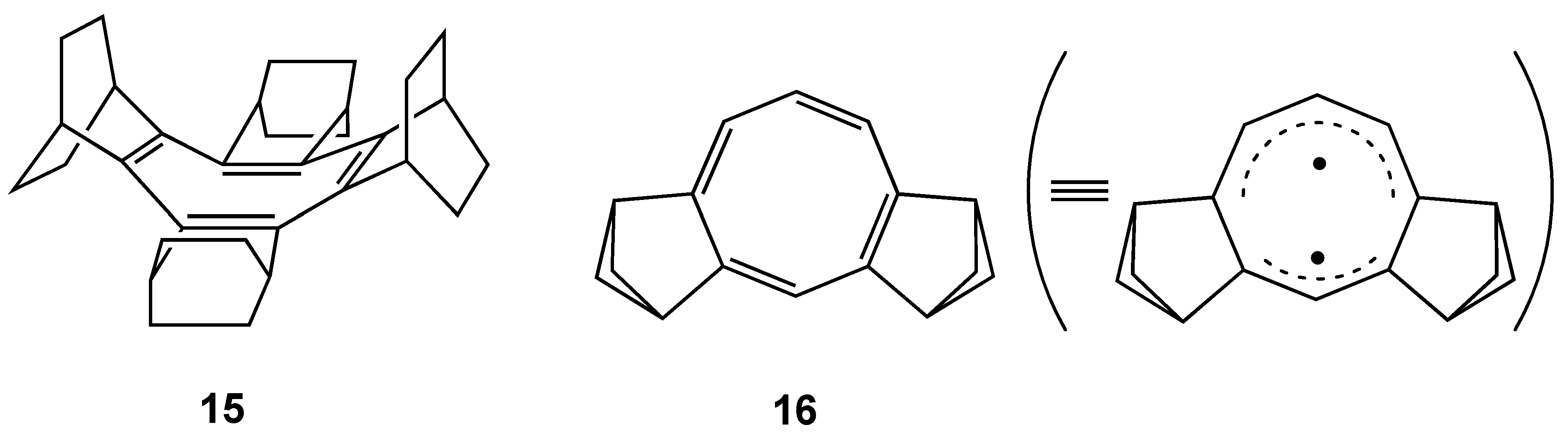
3.3. Planar COT Annelated with Plural Bicyclo[2.1.1]hexene Rings
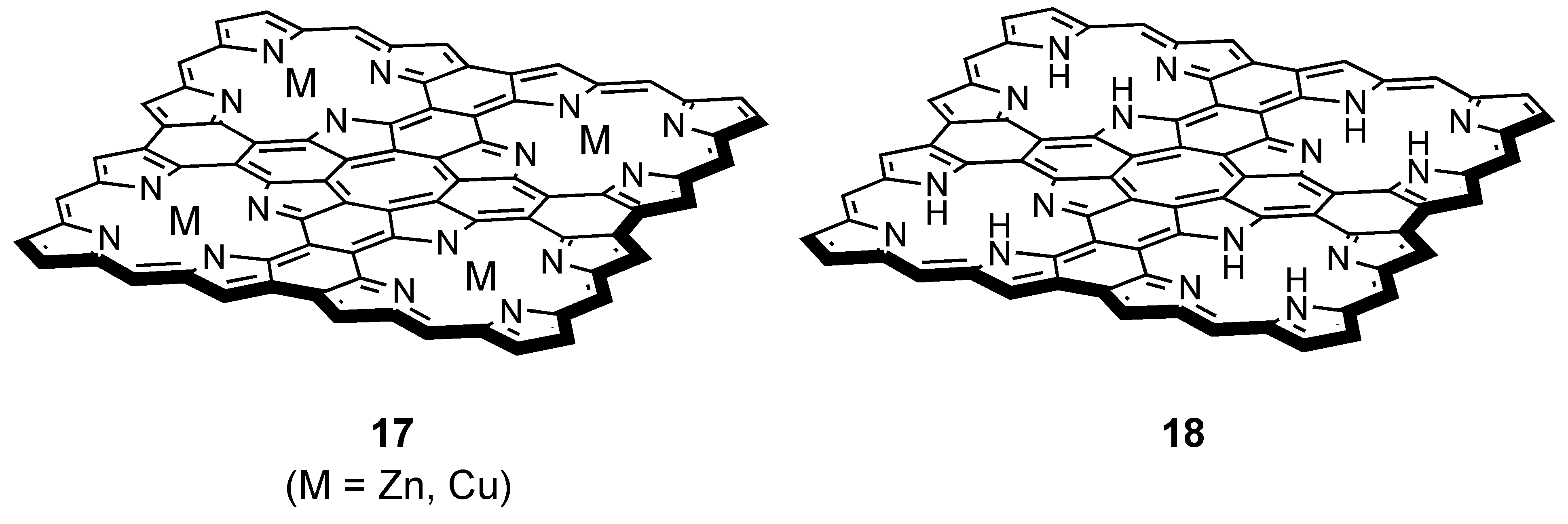
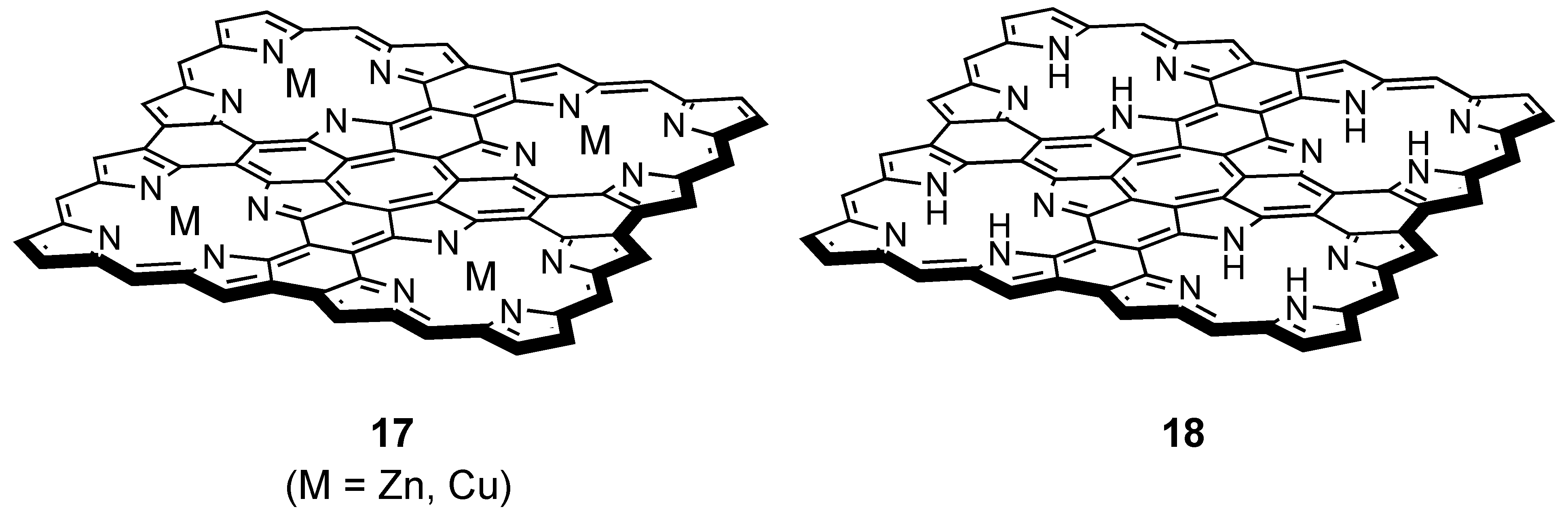
3.4. Planar COT Annelated with Porphyrin Rings
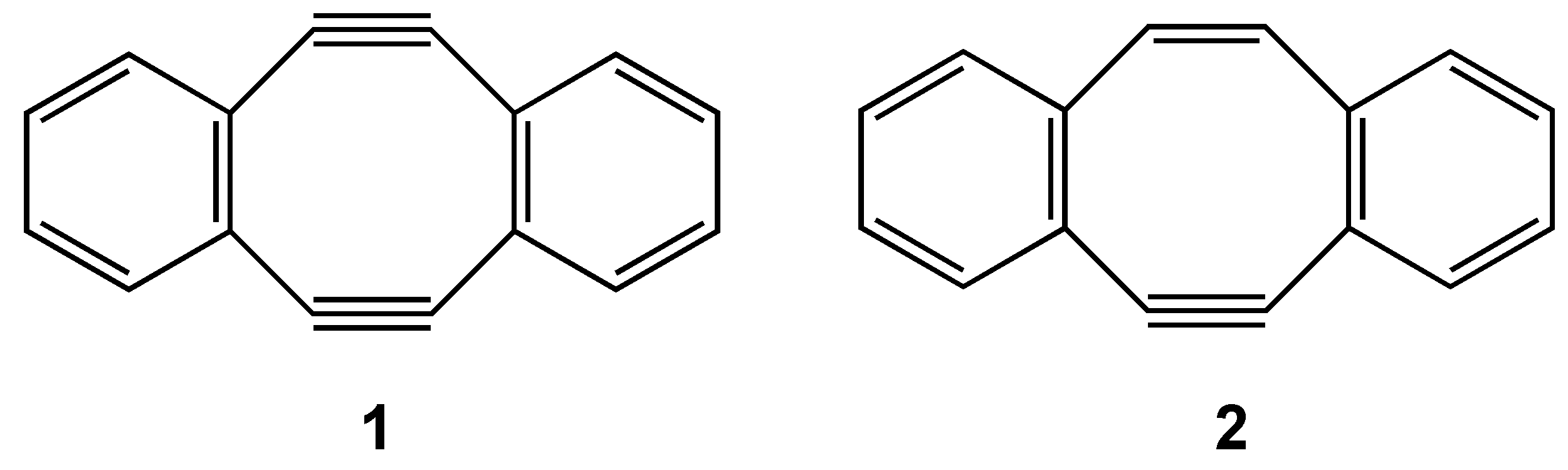
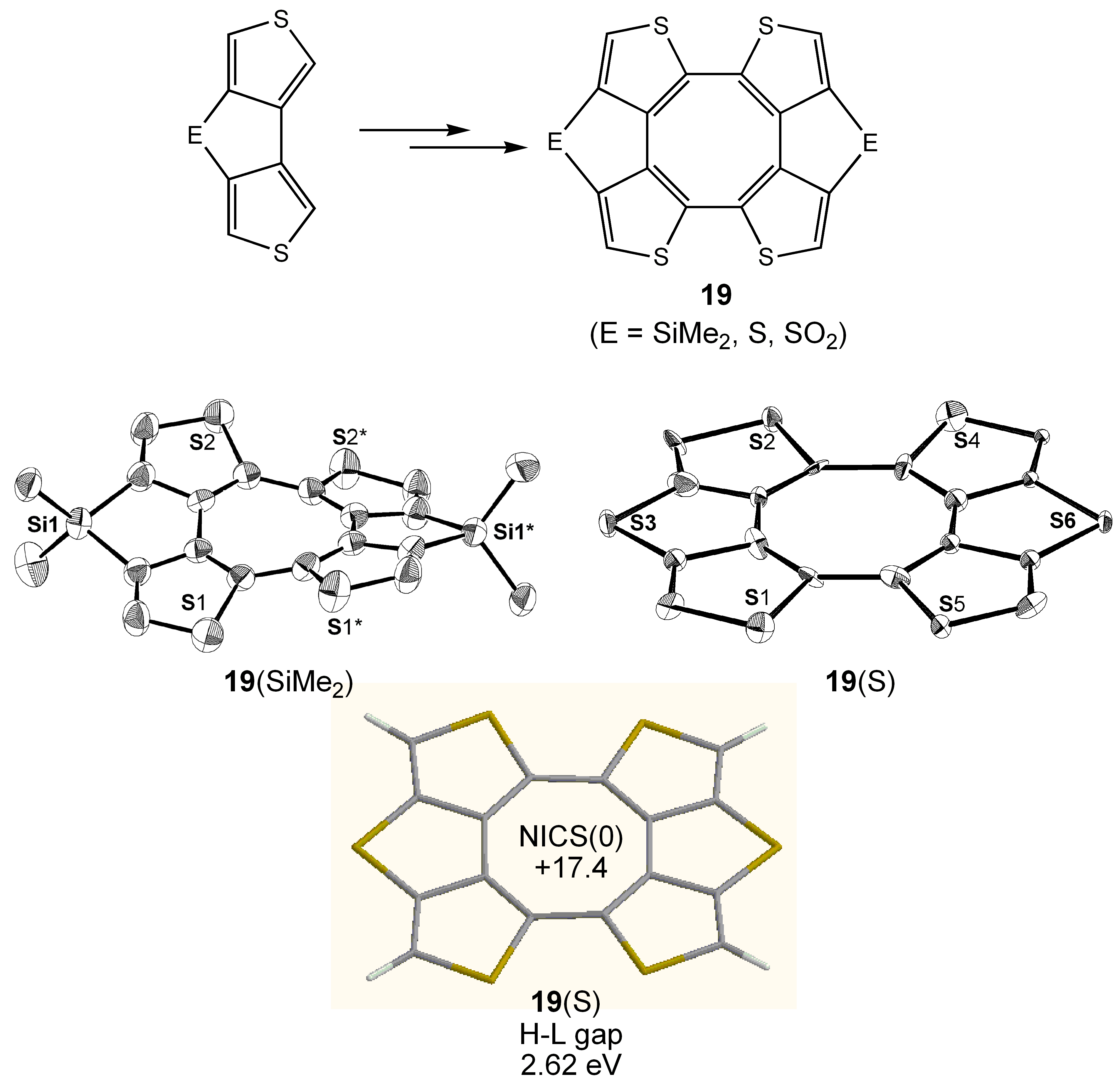
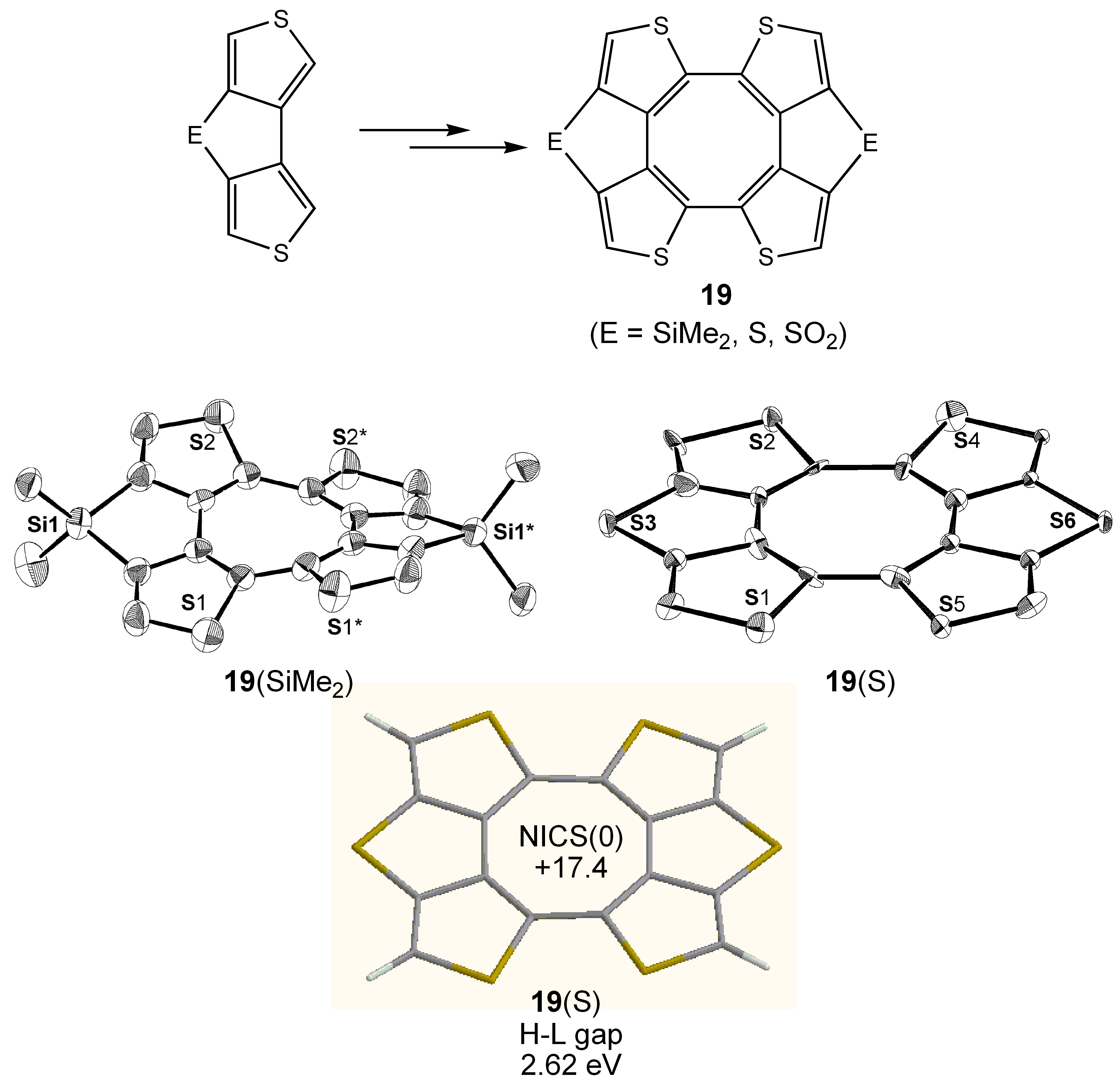
3.5. Planar COT Annelated with Thiophene Rings.
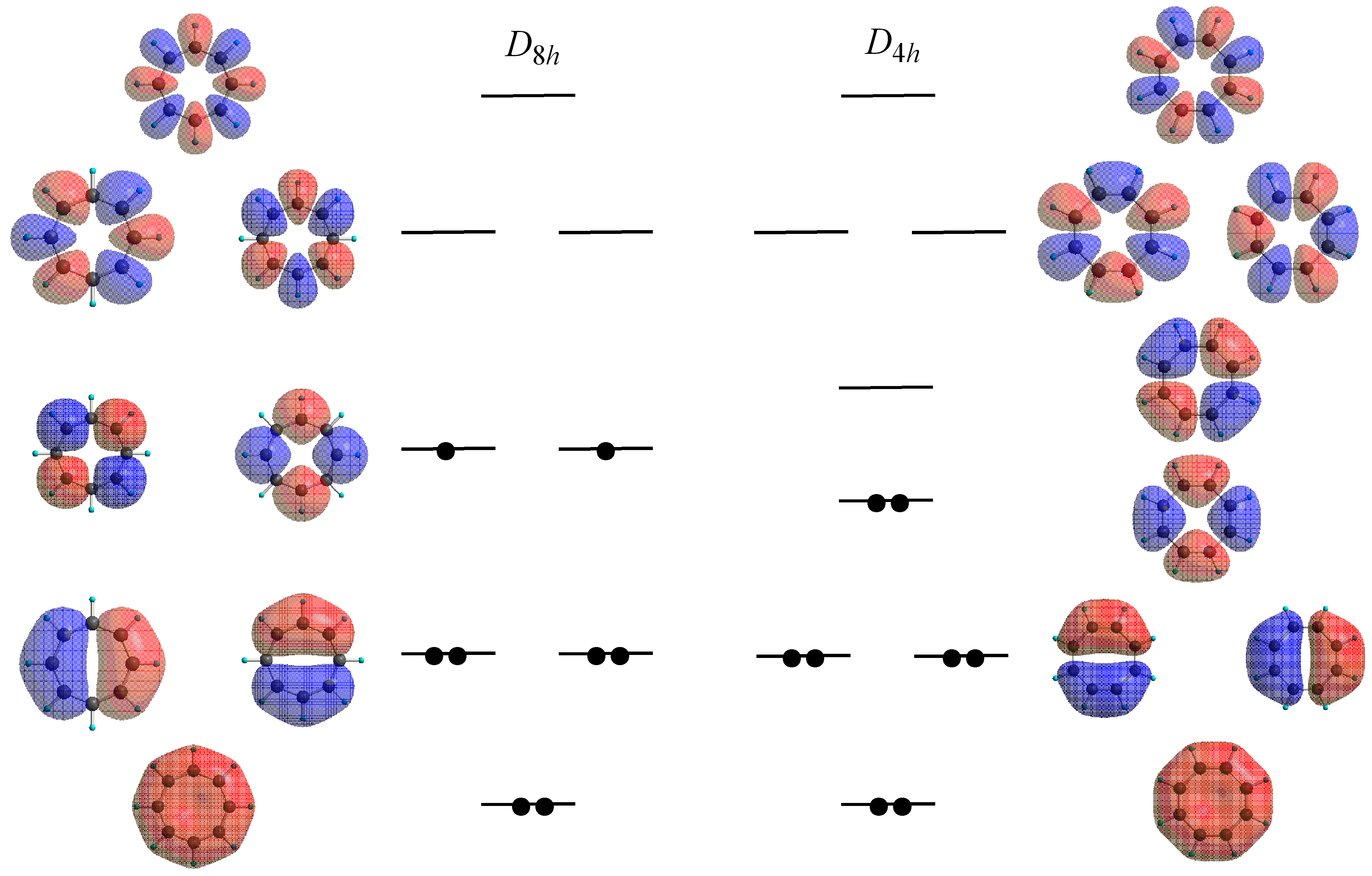
4. Stacking of Planar COT Rings
5. Conclusions
Acknowledgements
References
- Willstätter, R.; Waser, E. Über Cyclo-octatetraen. Chem. Ber. 1911, 44, 3423–3445. [Google Scholar] [CrossRef]
- Willstätter, R.; Heidelberger, M. Zur Kenntnis des Cyclo-octatetraens. Chem Ber. 1913, 46, 517–527. [Google Scholar] [CrossRef]
- Karle, I.L. An Electron Diffraction Investigation of Cyclooctatetraene and Benzene. J. Chem. Phys. 1952, 20, 65–70. [Google Scholar] [CrossRef]
- Bastiansen, O.; Hedberg, L.; Hedberg, K. Reinvestigation of the Molecular Structure of 1,3,5,7- Cyclooctatetraene by Electron Diffraction. J. Chem. Phys. 1957, 27, 1311–1317. [Google Scholar] [CrossRef]
- Traetteberg, M. The Molecular Structure of 1,3,5,7-Cyclo-octatetraene. Acta Chem. Scand. 1966, 20, 1724–1726. [Google Scholar] [CrossRef]
- Bordner, J.; Parker, R.G.; Stanford, R.H. The Crystal Structure of Octamethylcycloocta-tetraene. Acta Crystallogr. Sect. B 1972, 28, 1069–1075. [Google Scholar] [CrossRef]
- Hückel, E. Quantentheoretische Beiträge zum Benzolproblem. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Bally, T. Cyclobutadiene: The Antiaromatic Paradigm? Angew. Chem. Int. Ed. 2006, 45, 6616–6619. [Google Scholar] [CrossRef]
- Wiberg, K.B. Antiaromaticity in Monocyclic Conjugated Carbon Rings. Chem. Rev. 2001, 101, 1317–1331. [Google Scholar] [CrossRef]
- Allen, A.D.; Tidwell, T.T. Antiaromaticity in Open-Shell Cyclopropenyl to Cycloheptatrienyl Cations, Anions, Free Radicals, and Radical Ions. Chem. Rev. 2001, 101, 1333–1348. [Google Scholar] [CrossRef]
- Breslow, R.; Brown, J.; Gajewski, J.J. Antiaromaticity of Cyclopropenyl Anions. J. Am. Chem. Soc. 1967, 89, 4383–4390. [Google Scholar] [CrossRef]
- Breslow, R. Antiaromaticity. Acc. Chem. Res. 1973, 6, 393–398. [Google Scholar] [CrossRef]
- Breslow, R. Small Antiaromatic Rings. Angew. Chem. Int. Ed. Engl. 1968, 7, 565–570. [Google Scholar] [CrossRef]
- Hrovat, D.A.; Borden, W.T. CASSCF Calculations Find that a D8h Geometry is the Transition State for Double Bond Shifting in Cyclooctatetraene. J. Am. Chem. Soc. 1992, 114, 5879–5881. [Google Scholar] [CrossRef]
- Borden, W.T.; Davidson, E.R. Effects of electron repulsion in conjugated hydrocarbon diradicals. J. Am. Chem. Soc. 1977, 99, 4587–4594. [Google Scholar] [CrossRef]
- Borden, W.T.; Iwamura, H.; Berson, J.A. Violations of Hund’s Rule in Non-Kekule Hydrocarbons: Theoretical Prediction and Experimental Verification. Acc. Chem. Res. 1994, 27, 109–116. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Harget, A.J.; Haselbach, E. Cyclooctatetraene and Ions Derived from It. J. Am. Chem. Soc. 1969, 91, 7521–7523. [Google Scholar] [CrossRef]
- Ermer, O.; Klärner, F.-G.; Wette, M. Planarization of Unsaturated Rings. Cycloheptatriene with a Planar Seven-Membered Ring. J. Am. Chem. Soc. 1986, 108, 4908–4911. [Google Scholar] [CrossRef]
- Paquette, L. A.; Trova, M. P.; Luo, J.; Clough, A. E.; Anderson, L. B. Synthesis and Dynamic Behavior of (1,5)Cyclooctatetraenophanes. Effect of Distal Atom Bridging on Racemization Rates and Electrochemical Reducibility. J. Am. Chem. Soc. 1990, 112, 228–239. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Luo, J.; Cottrell, C.E.; Clough, A.E.; Anderson, L.B. Is Pseudorotation the Operational Pathway for Bond Shifting within [8]Annulenes? Probe of Planarization Requirements by 1,3-Annulation of the Cyclooctatetraene Ring. Kinetic Analysis of Racemization and 2-D NMR Quantitation of π-Bond Alternation and Ring Inversion as a Function of Polymethylene Chain Length. J. Am. Chem. Soc. 1990, 112, 239–253. [Google Scholar]
- Paquette, L.A. The Current View of Dynamic Change within Cyclooctatetraenes. Acc. Chem. Res. 1993, 26, 57–62. [Google Scholar] [CrossRef]
- Anet, F.A.L.; Bourn, A.J.R.; Lin, Y.S. Ring Inversion and Bond Shift in Cyclooctatetraene Derivatives. J. Am. Chem. Soc. 1964, 86, 3576–3577. [Google Scholar] [CrossRef]
- Oth, J.F.M. Conformational Mobility and Fast Bond Shift in the Annulenes. Pure Appl. Chem. 1971, 25, 573–622. [Google Scholar] [CrossRef]
- Kato, S.; Lee, H.S.; Gareyev, R.; Wenthold, P.G.; Lineberger, W.C.; DePuy, C.H.; Bierbaum, V.M. Experimental and Computational Studies of the Structures and Energetics of Cyclooctatetraene and Its Derivatives. J. Am. Chem. Soc. 1997, 119, 7863–7864. [Google Scholar] [CrossRef]
- Wenthold, P.G.; Hrovat, D.A.; Borden, W.T.; Lineberger, W.C. Transition-State Spectroscopy of Cyclooctatetraene. Science 1996, 272, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Anet, F.A.L. The Rate of Bond Change in Cycloöctatetraene. J. Am. Chem. Soc. 1962, 84, 671–672. [Google Scholar] [CrossRef]
- Paquette, L.A. Ring Inversion and Bond Shifting Energetics in Substituted Chiral Cyclooctatetraenes. Pure Appl. Chem. 1982, 54, 987–1004. [Google Scholar] [CrossRef]
- Komatsu, K.; Nishinaga, T.; Aonuma, S.; Hirosawa, C.; Takeuchi, K.; Lindner, H.J.; Richter, J. Synthesis, Structure, and Reduction of the Cyclooctatetraene Tetra-annelated with Bicyclo[2.2.2]-octene Frameworks. Tetrahedron Lett. 1991, 32, 6767–6770. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Seminario, J.M. Antiaromaticity in Relation to 1,3,5,7-Cyclooctatetraene Structures. Int. J. Quantum Chem. 1994, 50, 273–277. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Bach, R.D.; Laiter, S. Isodesmic and homodesmotic stabilization energies of [n]annulenes and their relevance to aromaticity and antiaromaticity: is absolute antiaromaticity possible? J. Mol. Struct. THEOCHEM 1997, 417, 123–129. [Google Scholar] [CrossRef]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Corminboeuf, C.; Seifert, G. The Magnetic Shielding Function of Molecules and Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3889–3910. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.A.N.F.; Mallion, R.B. Aromaticity and Ring Currents. Chem. Rev. 2001, 101, 1349–1384. [Google Scholar] [CrossRef] [PubMed]
- Dauben, H.J., Jr.; Wilson, J.D.; Laity, J.L. Diamagnetic Susceptibility Eexaltation as a Criterion of Aromaticity. J. Am. Chem. Soc. 1968, 90, 811–813. [Google Scholar] [CrossRef]
- Dauben, H.J., Jr.; Wilson, J.D.; Laity, J.L. Diamagnetic Susceptibility Exaltation in Hydrocarbons. J. Am. Chem. Soc. 1969, 91, 1991–1998. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.v.E. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of Magnetic Response Properties using a Continuous Set of Gauge Transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Coriani, S.; Lazzeretti, P.; Malagoli, M.; Zanasi, R. On CHF Calculations of Second-Order Magnetic Properties using the Method of Continuous Ttransformation of Origin of the Current Density. Theor. Chim. Acta 1994, 89, 181–192. [Google Scholar] [CrossRef]
- A related review was reported in 2001. Klärner, F.-G. About the Antiaromaticity of Planar Cyclooctatetraene. Angew. Chem. Int. Ed. 2001, 40, 3977–3981. [Google Scholar]; for reviews on the chemistry of cyclooctatetraene, see: Schröder, G. Cyclooctatetraen; Verlag Chemie: Weinheim, Germany, 1965. [Google Scholar]; Weinheim Fray, G.I.; Saxton, R.G. The Chemistry of Cyclooctatetraene and Its Derivatives; Cambridge University Press: New York, NY, USA, 1978. [Google Scholar]; Nishinaga, T. Cyclooctatetraenes. Sci. Synth. 2009, 45a, 383–406. [Google Scholar]
- Gogonea, V.; Schleyer, P.v.R.; Schreiner, P.R. Consequences of Triplet Aromaticity in 4nπ-Electron Annulenes: Calculation of Magnetic Shieldings for Open-Shell Species. Angew. Chem. Int. Ed. 1998, 37, 1945–1948. [Google Scholar] [CrossRef]
- Karadakov, P.B. Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Pindelska, E.; Cyrański, M.K.; Häfelinger, G. Planarization of 1,3,5,7-Cyclooctatetraene as a Result of a Partial Rehybridization at Carbon Atoms: an MP2/6-31G* and B3LYP/6-311G** Study. Chem. Phys. Lett. 2002, 359, 158–162. [Google Scholar] [CrossRef]
- Ohmae, T.; Nishinaga, T.; Wu, M.; Iyoda, M. Cyclic Tetrathiophenes Planarized by Silicon and Sulfur Bridges Bearing Antiaromatic Cyclooctatetraene Core: Syntheses, Structures, and Properties. J. Am. Chem. Soc. 2010, 132, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. [Google Scholar]
- Aihara, J. Aromaticity-Based Theory of Pericyclic Reactions. Bull. Chem. Soc. Jpn. 1978, 51, 1788–1792. [Google Scholar] [CrossRef]
- Jug, K.; Malar, E.J.P. Geometry of Triplets and Dianions of Aromatic and Antiaromatic Systems. J. Mol. Struct. (THEOCHEM) 1987, 153, 221–226. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Four- and Two-Electron Rules for Diatropic and Paratropic Ring Currents in Monocyclic π-Systems. Chem. Commun. 2001, 2220–2221. [Google Scholar] [CrossRef]
- Steiner, E.; Soncini, A.; Fowler, P.W. Full Spectral Decomposition of Ring Currents. J. Phys. Chem. A 2006, 110, 12882–12886. [Google Scholar] [CrossRef] [PubMed]
- Havenith, R.A.; Fowler, P.W.; Jenneskens, L.W. Persistence of Paratropic Ring Currents in Nonplanar, Tub-Shaped Geometries of 1,3,5,7-Cyclooctatetraene. Org. Lett. 2006, 8, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.N.C.; Garratt, P.J.; Sondheimer, F. Unsaturated Eight-Membered Ring Compounds. XI. Synthesis of sym-Dibenzo-1,5-cyclooctadiene-3,7-diyne and sym-Dibenzo-1,3,5- cyclooctatrien-7-yne, Presumably Planar Conjugated Eight-Membered Ring Compounds. J. Am. Chem. Soc. 1974, 96, 5604–5605. [Google Scholar]
- Wong, H.N.C.; Sondheimer, F. Synthesis and Reactions of 5,6,11,12-Tetradehydrodibenzo[a,e]cyclooctene and 5,6-Didehydrodibenzo[a,e]cyclooctene. Tetrahedron 1981, 37, 99–109. [Google Scholar] [CrossRef]
- Huang, N.Z.; Sondheimer, F. The Planar Dehydro[8]annulenes. Acc. Chem. Res. 1982, 15, 96–102. [Google Scholar] [CrossRef]
- Destro, R.; Pilati, T.; Simonetta, M. Crystal structure of 5,6,11,12-tetradehydrodibenzo[a,e]cyclooctene (sym-dibenzo-1,5-cyclooctadiene-3,7-diyne). J. Am. Chem. Soc. 1975, 97, 658–659. [Google Scholar] [CrossRef]
- Matzger, A.J.; Vollhardt, K.P.C. Benzocyclynes Adhere to Hückel’s Rule by the Ring Current Criterion in Experiment (1H NMR) and Theory (NICS). Tetrahedron Lett. 1998, 39, 6791–6794. [Google Scholar] [CrossRef]
- Elix, J.A.; Sargent, M.V.; Sondheimer, F. Bicyclo[6.2.0]deca-1,3,5,7-tetraene. J. Am. Chem. Soc. 1967, 89, 180. [Google Scholar]
- Elix, J.A.; Sargent, M.V.; Sondheimer, F. Unsaturated 8-Membered Ring Compounds. VIII. Photochemistry of 7,8-Dimethylene-1,3,5-cyclooctatrienes. Synthesis of Bicyclo[6.2.0]deca- 1,3,5,7-tetraene. J. Am. Chem. Soc. 1970, 92, 969–973. [Google Scholar]
- Pirrung, M.C.; Krishnamurthy, N.; Nunn, D. S.; McPhail, A. T. Synthesis, Structure, and Properties of a 2-(Trimethylsilyl)cyclobutenocyclooctatetraene. J. Am. Chem. Soc. 1991, 113, 4910–4917. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.Z.; Cottrell, C.E. Flattening of the Cyclooctatetraene Ring by Annulation. J. Am. Chem. Soc. 1987, 109, 3730–3734. [Google Scholar] [CrossRef]
- Oda, M.; Oikawa, H. The Synthesis of Bicyclo[6.2.0]decapentaene. Tetrahedron Lett. 1980, 21, 107–110. [Google Scholar] [CrossRef]
- Kabuto, C.; Oda, M. Crystal and Molecular Structure of 9,10-Diphenylbicyclo[6.2.0]decapentaene a 10 π Aromatic Compound. Tetrahedron Lett. 1980, 21, 103–106. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; Lugli, F.; Fowler, P.W.; Steiner, E. Ring Current Patterns in Annelated Bicyclic Polyenes. J. Phys. Chem. A 2002, 106, 5703–5708. [Google Scholar]
- Kiesewetter, M.K.; Reiter, R.C.; Stevenson, C.D. The Second Cyclopropannulene: Cycloprop-[8]annulene. J. Am. Chem. Soc. 2005, 127, 1118–1119. [Google Scholar] [CrossRef] [PubMed]
- Dürr, H.; Klauck, G.; Peters, K.; von Schnering, H.G. A Novel Planar Antiaromatic Dibenzo[8]annulene. Angew. Chem., Int. Ed. Engl. 1983, 22, 332–333. [Google Scholar] [CrossRef]
- Ermer, O.; Klärner, F.-G.; Wette, M. Planarization of Unsaturated Rings. Cycloheptatriene with a Planar Seven-Membered Ring. J. Am. Chem. Soc. 1986, 108, 4908–4911. [Google Scholar]
- Klärner, F.-G.; Ehrhardt, R.; Bandmann, H.; Boese, R.; Bläser, D.; Houk, K.N.; Beno, B.R. Pressure-Induced Cycloadditions of Dicyanoacetylene to Strained Arenes: The Formation of Cyclooctatetraene, 9,10-Dihydronaphthalene, and Azulene Derivatives; A Degenerate [1,5] Sigmatropic Shift - Comparison between Theory and Experiment. Chem. Eur. J. 1999, 5, 2119–2132. [Google Scholar] [CrossRef]
- Wilcox, C.F., Jr.; Uetrecht, J.P.; Grohman, K.K. Preparation of Cycloocta[def]biphenylene, a Novel Benzenoid Antiaromatic Hydrocarbon. J. Am. Chem. Soc. 1972, 94, 2532–2533. [Google Scholar] [CrossRef]
- Wilcox, C.F., Jr.; Uetrecht, J.P.; Grantham, G.D.; Grohmann, K.G. Synthesis and Properties of Cycloocta[def]biphenylene, a Stable Benzenoid Paratropic Hydrocarbon. J. Am. Chem. Soc. 1975, 97, 1914–1920. [Google Scholar] [CrossRef]
- Willner, I.; Rabinovitz, M. Cycloocta[def]fluorene: a Planar Cyclooctatetraene Derivative. Paratropicity of Hydrocarbon and Anion. J. Org. Chem. 1980, 45, 1628–1633. [Google Scholar] [CrossRef]
- Hellwinkel, D.; Reiff, G. Cyclooctatetraene Systems Flattened by Steric Constraints. Angew. Chem., Int. Ed. Engl. 1970, 9, 527–528. [Google Scholar] [CrossRef]
- Rathore, R.; Abdelwahed, S.H. Soluble Cycloannulated Tetroxa[8]circulane Derivatives: Synthesis, Optical and Electrochemical Properties, and Generation of Their Robust Cation-Radical Salts. Tetrahedron Lett. 2004, 45, 5267–5270. [Google Scholar] [CrossRef]
- Soulen, R.L.; Choi, S.K.; Park, J.D. Copper Coupling of 1-Chloro-2-iodo- and 1,2-Diiodo-perfluorocycloalkenes. J. Fluorine Chem. 1973/74, 3, 141–150. [Google Scholar] [CrossRef]
- Einstein, F.W.B.; Willis, A.C.; Cullen, W.R.; Soulen, R.L. Perfluorotetracyclobutacyclo-octatetraene; a Planar Eight-Memberedring System; X-ray Crystal Structure. J. Chem. Soc. Chem. Commun. 1981, 526–528. [Google Scholar] [CrossRef]
- Baldridge, K.K.; Siegel, J.S. Quantum Mechanical Designs toward Planar Delocalized Cyclooctatetraene: A New Target for Synthesis. J. Am. Chem. Soc. 2001, 123, 1755–1759. [Google Scholar] [CrossRef]
- Shelton, G.R.; Hrovat, D.A.; Wei, H.; Borden, W.T. Why Does Perfluorination Render Bicyclo[2.2.0]hex-1(4)-ene Stable toward Dimerization? Calculations Provide the Answers. J. Am. Chem. Soc. 2006, 128, 12020–12027. [Google Scholar] [CrossRef]
- Fowler, P.W.; Havenith, R.W.A.; Jenneskens, L.W.; Soncini, A.; Steiner, E. Paratropic Delocalized Ring Currents in Flattened Cyclooctatetraene Systems with Bond Alternation. Angew. Chem. Int. Ed. 2002, 41, 1558–1560. [Google Scholar] [CrossRef]
- Britton, W.E.; Ferraris, J.P.; Soulen, R.L. Electrochemistry of Perfluorotetracyclobuta-1,3,5,7-cyclooctatetraene, a Powerful Neutral Organic Oxidant. J. Am. Chem. Soc. 1982, 104, 5322–5325. [Google Scholar] [CrossRef]
- Matsuura, A.; Komatsu, K. Efficient Synthesis of Benzene and Planar Cyclooctatetraene Fully Annelated with Bicyclo[2.1.1]hex-2-ene. J. Am. Chem. Soc. 2001, 123, 1768–1769. [Google Scholar] [CrossRef]
- Nishinaga, T.; Uto, T.; Inoue, R.; Matsuura, A.; Treitel, N.; Rabinoviz, M.; Komatsu, K. Antiaromaticity and Reactivity of a Planar Cyclooctatetraene Fully Annelated with Bicyclo[2.1.1]hexane Units. Chem. Eur. J. 2008, 14, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, T.; Uto, T.; Komatsu, K. Novel Cyclooctatetraene Radical Cation Planarized by Full Annelation with Bicyclo[2.1.1]hexene Units. Org. Lett. 2004, 6, 4611–4614. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, T.; Komatsu, K.; Sugita, N.; Lindner, H.J.; Richter, J. First X-ray Structure of a Cyclooctatetraene Cation Radical: the Hexachloroantimonate of the Tetrakis(bicyclo[2.2.2]octeno)cyclooctatetraene Cation Radical. J. Am. Chem. Soc. 1993, 115, 11642–11643. [Google Scholar] [CrossRef]
- Nakamura, Y.; Aratani, N.; Shinokubo, H.; Takagi, A.; Kawai, T.; Matsumoto, T.; Yoon, Z.S.; Kim, D.Y.; Ahn, T.K.; Kim, D.; Muranaka, A.; Kobayashi, N.; Osuka, A. A Directly Fused Tetrameric Porphyrin Sheet and Its Anomalous Electronic Properties That Arise from the Planar Cyclooctatetraene Core. J. Am. Chem. Soc. 2006, 128, 4119–4127. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Aratani, N.; Furukawa, K.; Osuka, A. Synthesis and Characterizations of Free Base and Cu(II) Complex of a Porphyrin Sheet. Tetrahedron 2008, 64, 11433–11439. [Google Scholar] [CrossRef]
- Nakamura, Y.; Aratani, N.; Osuka, A. Experimental and Theoretical Investigations into the Paratropic Ring Current of a Porphyrin Sheet. Chem. Asian. J. 2007, 2, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P. v. R.; Manoharan, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.; van Eikema Hommes, N. J. R. Dissected Nucleus-Independent Chemical Shift Analysis of π-Aromaticityand Antiaromaticity. Org. Lett. 2001, 3, 2465–2468. [Google Scholar] [CrossRef]
- Corminboeuf, C.; Schleyer, P.v.R.; Warner, P. Are Antiaromatic Rings Stacked Face-to-Face Aromatic? Org. Lett. 2007, 9, 3263–3266. [Google Scholar] [CrossRef]
- Rzepa, H.S. Möbius Aromaticity and Delocalization. Chem. Rev. 2005, 105, 3697–3715. [Google Scholar] [CrossRef]
- Yoon, Z.S.; Osuka, A.; Kim, D. Möbius Aromaticity and Antiaromaticity in Expanded Porphyrins. Nature Chem. 2009, 1, 113–122. [Google Scholar] [CrossRef]
- Bean, D.E.; Fowler, P. W. Stacked-Ring Aromaticity: An Orbital Model. Org. Lett. 2008, 10, 5573–5576. [Google Scholar] [CrossRef] [PubMed]
- Aihara, J.-i. Origin of Stacked-Ring Aromaticity. J. Phys. Chem. A 2009, 113, 7945–7952. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| molecule | state | method (geometry) | C–C bond length (Å) | method (magnetic properties) | NICS(0) (ppm) | ref. |
|---|---|---|---|---|---|---|
| C8H8 (D2d) | S0 | B3LYP/6-311+G** | 1.340 1.472 | GIAO/HF/6-31+G* | +3.0 | [42] |
| HF/6-31G** | 1.344 1.479 | GIAO/HF/6-311+G* | +1.9 | [43] | ||
| GIAO/CASSCF(8,8) /6-311+G* | +1.2 | [43] | ||||
| C8H8 (D4h) | S0 | B3LYP/6-31G* | N.R. | GIAO/HF/6-31+G* | +30.1 | [38] |
| HF/6-31G** | 1.351 1.472 | GIAO/HF/6-311+G* | +29.3 | [43] | ||
| GIAO/CASSCF(8,8) /6-311+G* | +16.1 | [43] | ||||
| C8H8 (D8h) | S0 | CASSCF(8,8) /6-31G** | 1.408 | GIAO/CASSCF(8,8) /6-311+G* | +40.7 | [43] |
| T1 | B3LYP/6-311+G** | 1.403 | GIAO/HF/6-31+G* | –12.4 | [42] | |
| CASSCF(8,8) /6-31G** | 1.406 | GIAO/HF/6-311+G* | –12.1 | [43] | ||
| GIAO/CASSCF(8,8) /6-311+G* | –8.9 | [43] |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nishinaga, T.; Ohmae, T.; Iyoda, M. Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene. Symmetry 2010, 2, 76-97. https://doi.org/10.3390/sym2010076
Nishinaga T, Ohmae T, Iyoda M. Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene. Symmetry. 2010; 2(1):76-97. https://doi.org/10.3390/sym2010076
Chicago/Turabian StyleNishinaga, Tohru, Takeshi Ohmae, and Masahiko Iyoda. 2010. "Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene" Symmetry 2, no. 1: 76-97. https://doi.org/10.3390/sym2010076
APA StyleNishinaga, T., Ohmae, T., & Iyoda, M. (2010). Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene. Symmetry, 2(1), 76-97. https://doi.org/10.3390/sym2010076