
On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines

Abstract
1. Introduction
2. Methodology
2.1. Computational Details
2.2. Structural Descriptor of Electron Delocalization
2.3. Thermochemical Quantities
3. Results and Discussion
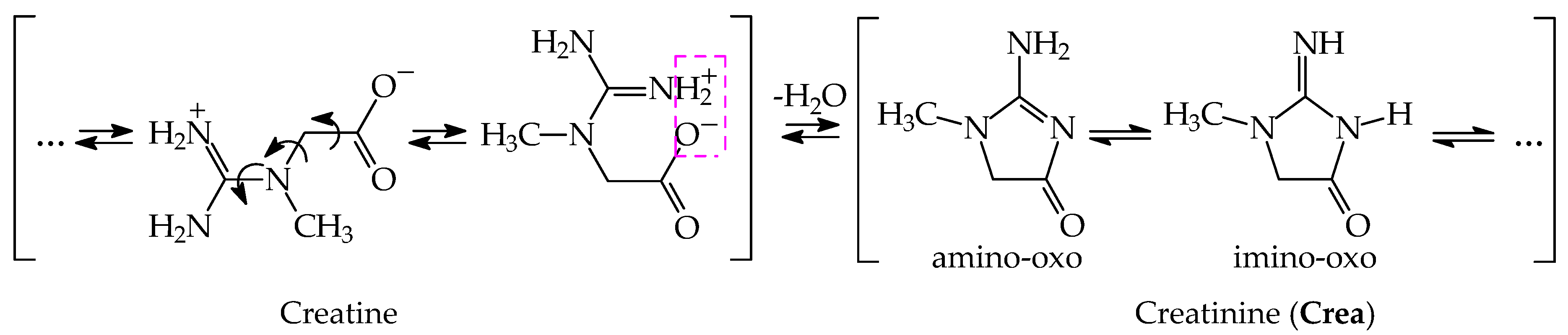
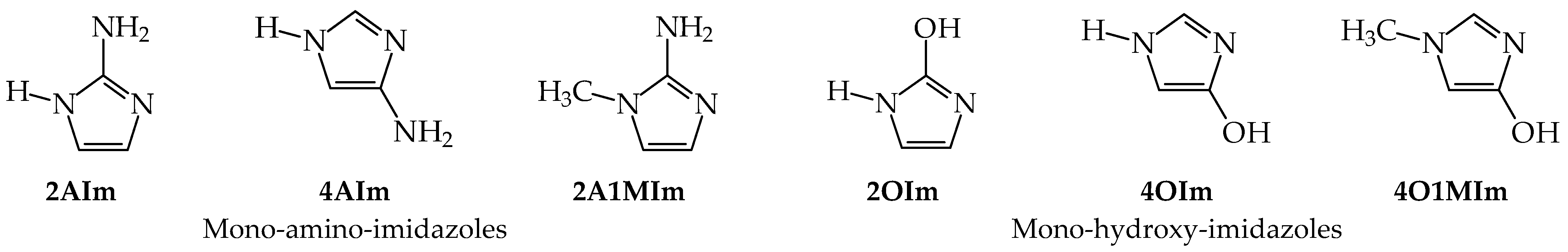
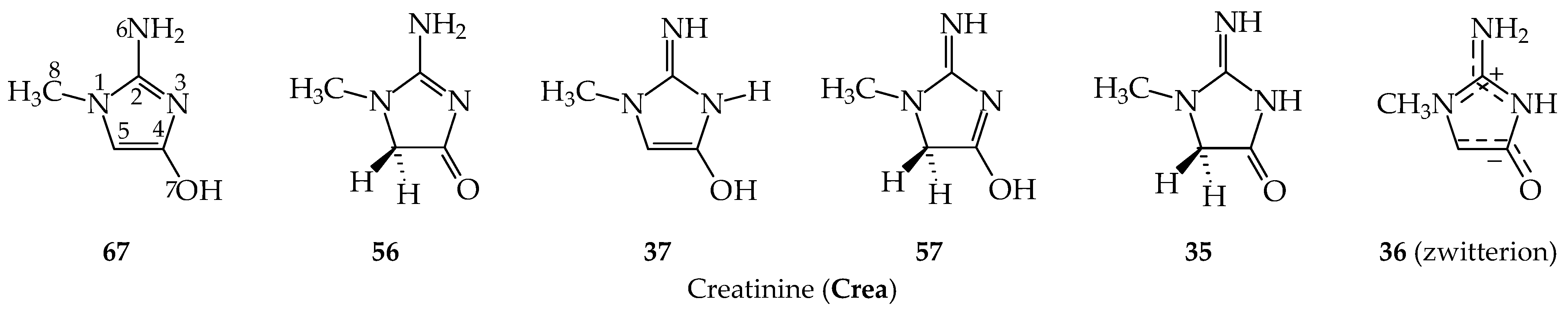
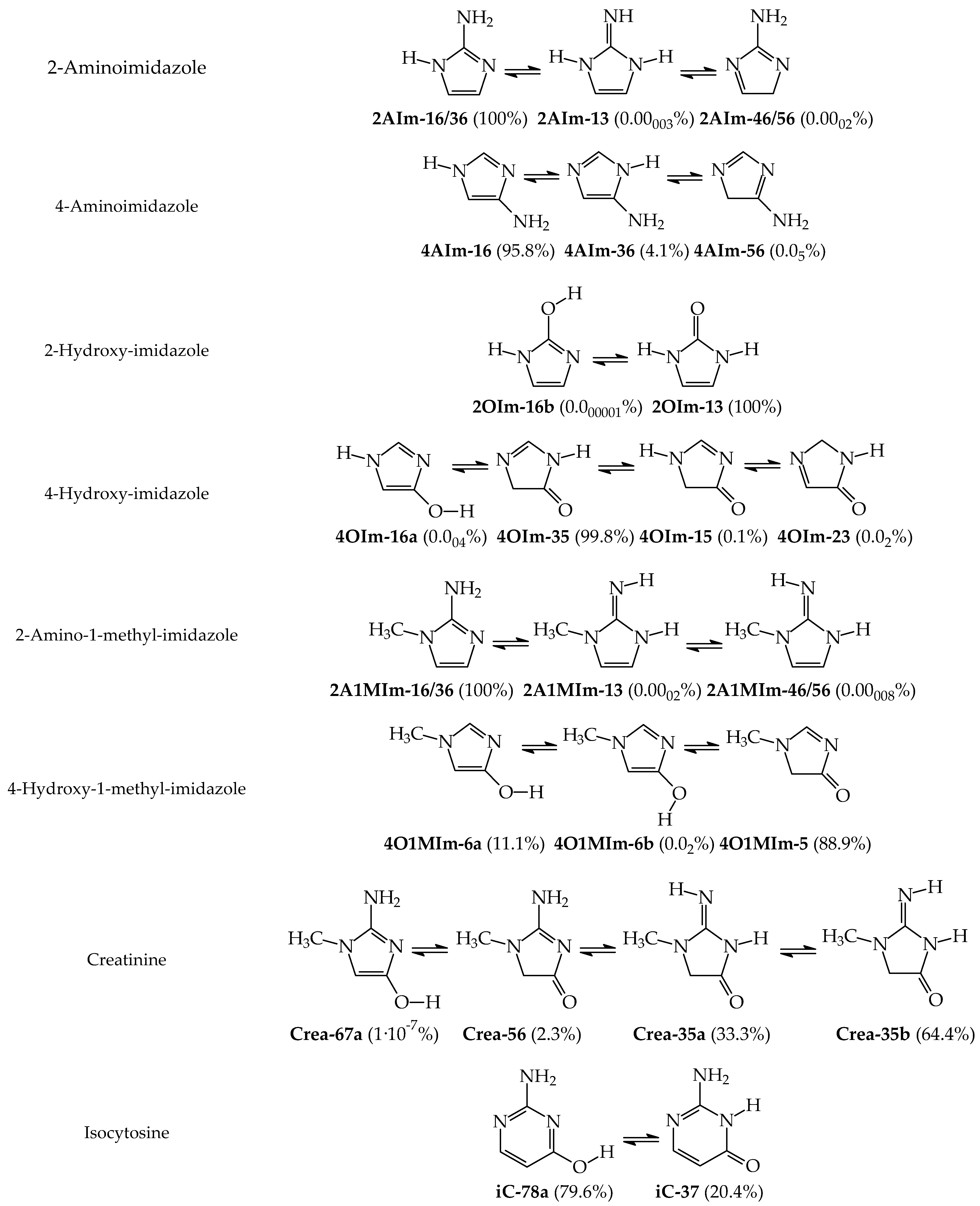
3.1. Possible Tautomers-Rotamers for Model Azoles and Creatinine
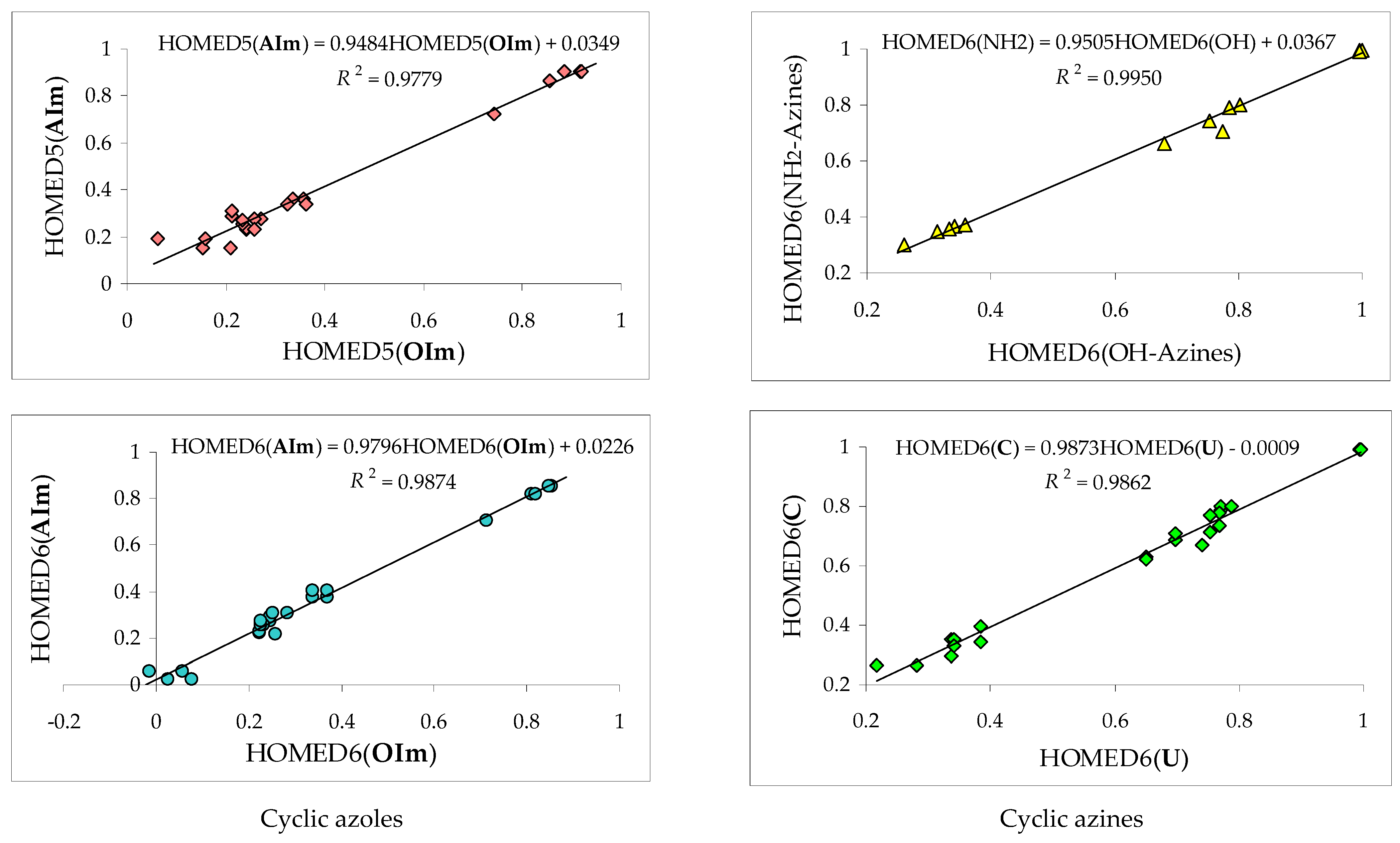
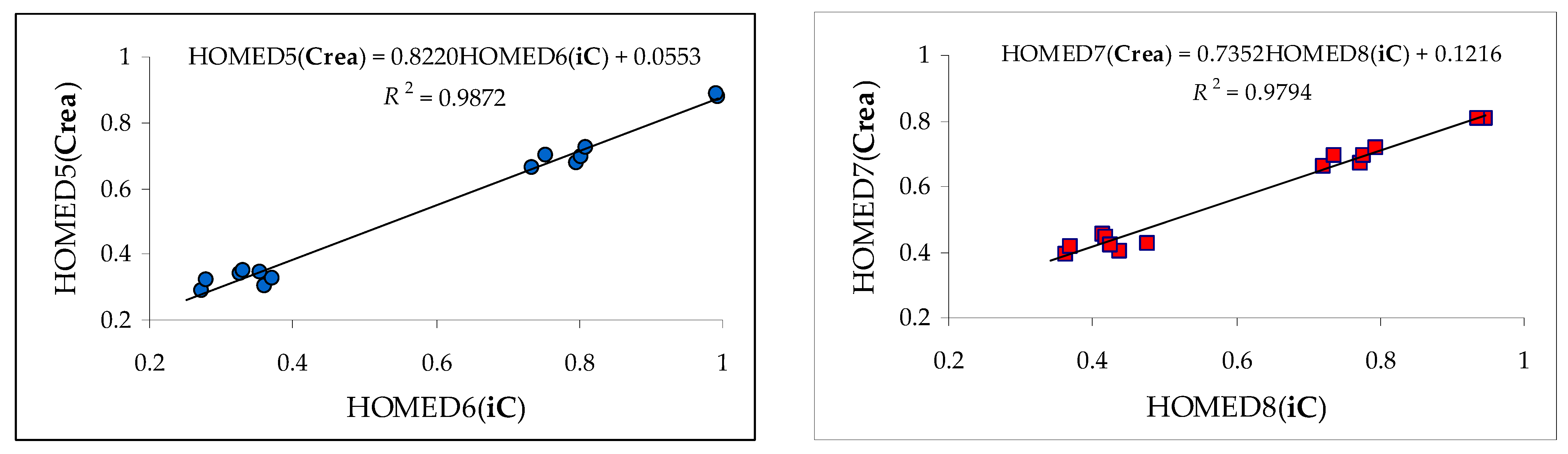
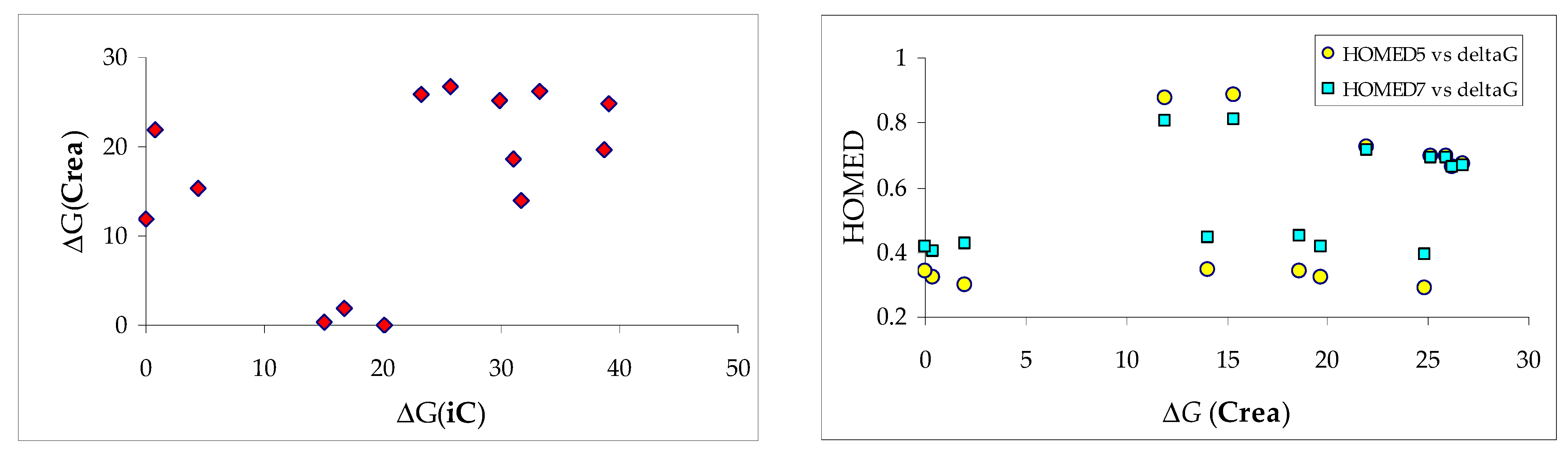
3.2. Variations of Structural-Descriptor HOMED for Isomers of Investigated Derivatives
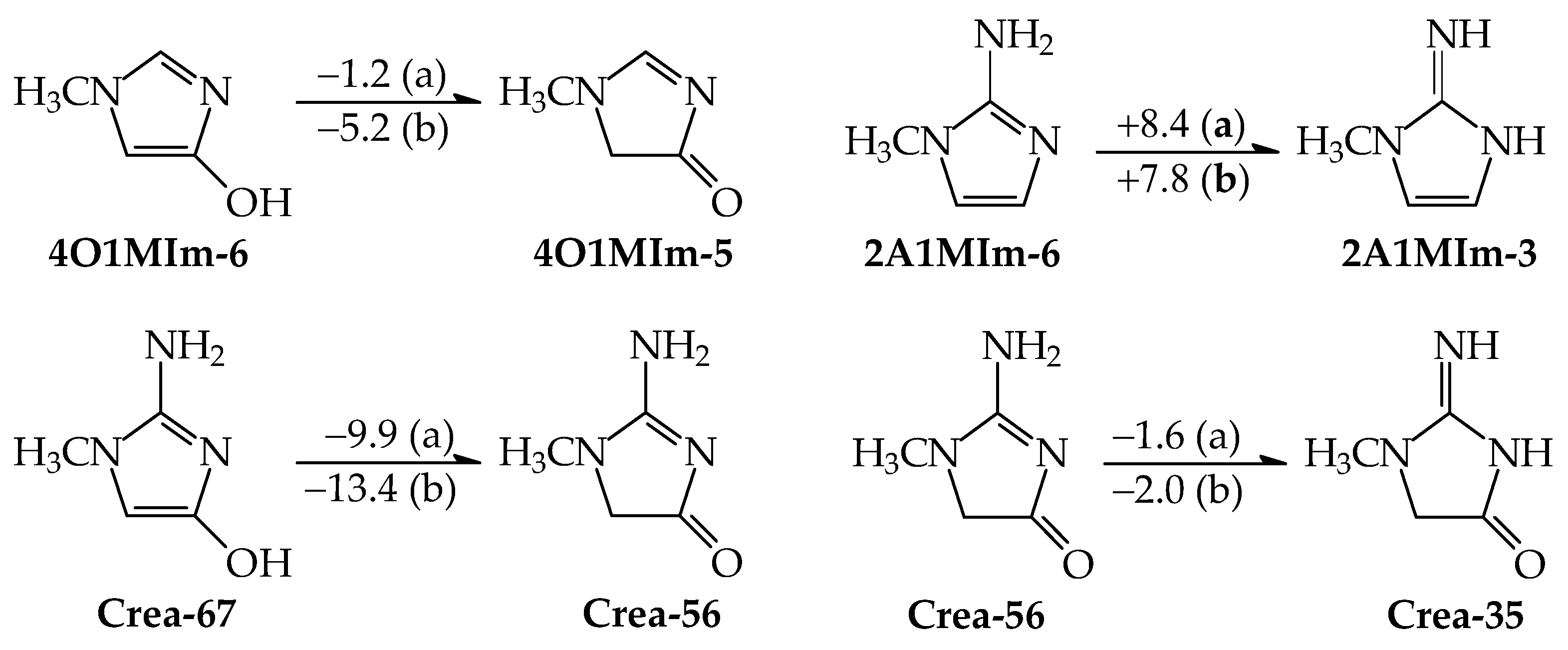
3.3. Thermochemistry of Isomeric Conversions for Title Compounds
3.4. Search for Origins of Tautomeric Preferences in Creatinine
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology. Adv. Heterocycl. Chem. 2000, 76, 184. [Google Scholar]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric Equilibria in Relation to pi-Electron Delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef] [PubMed]
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. The Tautomerism of Heterocycles: Five-Membered Rings with Two or More Heteroatoms. Adv. Heterocycl. Chem. 2006, 76, 157–323. [Google Scholar]
- Stanovnik, B.; Tišler, M.; Katritzky, A.R.; Denisko, O.V. The Tautomerism of Heterocycles: Substituent Tautomerism of Six-Membered Ring Heterocycles. Adv. Heterocycl. Chem. 2006, 91, 1–134. [Google Scholar]
- Lempert, C. The Chemistry of the Glycocyamidines. Chem. Rev. 1959, 59, 667–736. [Google Scholar] [CrossRef]
- Matsumoto, K.; Rapoport, H. Preparation and Properties of Some Acyl-Guanidines. J. Org. Chem. 1968, 33, 552–558. [Google Scholar] [CrossRef]
- Hunter, A. Creatine and Creatinine; Monographs on Biochemistry, Longmans, Green & Co.: London, UK, 1928. [Google Scholar]
- Cannan, R.K.; Shore, A. The Creatine-Creatinine Equilibrium. The Apparent Dissociation Constants of Creatine and Creatinine. Biochem. J. 1928, 22, 920–929. [Google Scholar] [CrossRef]
- Taylor, E.H. Clinical Chemistry; John Wiley and Sons: New York, NY, USA, 1989; pp. 58–62. [Google Scholar]
- Gangopadhyay, D.; Sharma, P.; Singh, S.K.; Deckert, V.; Popp, J.; Singh, R.K. Surface Enhanced Raman Scattering Based Reaction Monitoring of in vitro Decyclization of Creatinine → Creatine. RSC Adv. 2016, 6, 58943–58949. [Google Scholar] [CrossRef]
- Kenyon, G.L.; Rowley, G.L. Tautomeric Preferences among Glycocyamidines. J. Am. Chem. Soc. 1971, 93, 5552–5560. [Google Scholar] [CrossRef]
- Das, G. Rotational Aspects of Non-Ionized creatine in the Gas Phase. Monatsh. Chem. 2014, 145, 1431–1441. [Google Scholar] [CrossRef]
- Valadbeigi, Y.; Ilbeigi, V.; Tabrizchi, M. Effect of Mono- and Di-hydratation on the Stability and Tautomerisms of Different Tautomers of Creatinine: A Thermodynamic and Mechanistic Study. Comput. Theor. Chem. 2015, 1061, 27–35. [Google Scholar] [CrossRef]
- Léon, I.; Tasinato, N.; Spada, L.; Alonso, E.R.; Mata, S.; Balbi, A.; Puzzarini, C.; Alonso, J.L.; Barone, V. Looking for the Elusive Imine Tautomer of Creatinine: Different States of Aggregation Studied by Quantum Chemistry and Molecular Spectroscopy. ChemPlusChem 2021, 86, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Narimani, R.; Esmaeili, M.; Rasta, S.H.; Khosroshahi, H.T.; Mobed, A. Trend in Creatinine Determining Methods: Conventional Methods to Molecular-Based Methods. Anal. Sci. Adv. 2021, 2, 308–325. [Google Scholar] [CrossRef]
- Finlay, M.R. Quackery and Cookery: Justus von Liebig’s Extract of Meat and the Theory of Nutrition in the Victorian Age. Bull. Hist. Med. 1992, 66, 404–418. [Google Scholar] [PubMed]
- du Pré, S.; Mendel, H. The Crystal Structure of Creatinine. Acta Cryst. 1955, 8, 311–313. [Google Scholar] [CrossRef]
- Grzybowski, A.K.; Datta, S.P. The Ionisation Constant of the Protonated Form of Creatinine. J. Chem. Soc. 1964, 187–196. [Google Scholar] [CrossRef]
- Srinivasan, R.; Stewart, R. The Catalysis of Proton Exchange in Creatinine by General Acids and General Bases. Can. J. Chem. 1975, 53, 224–231. [Google Scholar] [CrossRef]
- Dietrich, R.F.; Marletta, M.A.; Kenyon, G.L. Carbon-13 Nuclear Magnetic Resonance Studies of Creatine, Creatinine and Some of their analogs. Org. Magn. Reson. 1980, 13, 79–88. [Google Scholar] [CrossRef]
- Butler, A.R.; Glidewell, C. Creatinine: An Examination of Its Structure and Some of Its Reactions by Synergistic Use of MNDO Calculations and Nuclear Magnetic Resonance Spectroscopy. J. Chem. Soc. Perkin Trans. 2 1985, 9, 1465–1467. [Google Scholar] [CrossRef]
- Reddick, R.E.; Kenyon, G.L. Syntheses and NMR Studies of Specifically Labeled [2-15N]Phosphocreatine, [2-15N]Creatinine, and Related 15N-Labeled Compounds. J. Am. Chem. Soc. 1987, 109, 4380–4387. [Google Scholar] [CrossRef]
- Trendafilova, N.; Kurbakova, A.P.; Efimenko, I.A.; Mitewa, M.; Bontchev, P.R. Infrared Spectra of Pt(I1) Creatinine Complexes. Normal Coordinate Analysis of Creatinine and Pt(Creat)2(NO2)2. Spectrochim. Acta Part A 1991, 47, 577–584. [Google Scholar] [CrossRef]
- Arakali, A.V.; McCloskey, J.; Parthasarathy, R.; Alderfer, J.L.; Chheda, G.B.; Srikrishnan, T. Study of Creatinine and Its 5-Alkoxy Analogs: Structure and Conformational Studies in the Solid and Solution States by X-Ray Crystallography, NMR, UV and Mass Spectrometry. Nucleosides Nucleotides Nucleic Acids 1997, 16, 2193–2218. [Google Scholar] [CrossRef]
- Bell, T.W.; Hou, Z.; Luo, Y.; Drew, M.G.B.; Chapoteau, E.; Czech, B.P.; Kumar, A. Detection of Creatinine by a Designed Receptor. Science 1995, 269, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Craw, J.S.; Greatbanks, S.B.; Hillier, I.H.; Harrison, M.J.; Burton, N.A. Solvation and Solid State Effects on the Structure of the Tautomers of Creatinine. J. Chem. Phys. 1997, 106, 6612–6617. [Google Scholar] [CrossRef]
- Craw, S.J.; Hillier, I.H.; Morris, G.A.; Vincent, M.A. Theoretical and Experimental Studies of the Barrier to Amine Rotation in Creatinine: Influence of Solvation Models and Explicit Solvation. Mol. Phys. 1997, 92, 421–428. [Google Scholar] [CrossRef]
- Kotsyubynskyy, D.; Molchanov, S.; Gryff-Keller, A. Creatinine and Creatininium Cation in DMSO-d6 Solution. Structure and Restricted Internal Rotation of NH2 Group. Magn. Res. Chem. 2004, 42, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, C.; Bayari, S.H. Vibrational and DFT Studies of Creatinine and Its Metal Complexes. Hacet. J. Biol. Chem. 2010, 38, 107–118. [Google Scholar]
- Vikram, K.; Mishra, S.; Srivastava, S.K.; Singh, R.K. Low Temperature Raman and DFT Study of Creatinine. J. Mol. Struct. 2012, 1012, 141–150. [Google Scholar] [CrossRef]
- Gao, J.; Hu, Y.; Li, S.; Zhang, Y.; Chen, X. Tautomeric Equilibrium of Creatinine and Creatininium Cation in Aqueous Solution Explored by Raman Spectroscopy and Density Functional Theory Calculations. Chem. Phys. 2013, 410, 81–89. [Google Scholar] [CrossRef]
- Raczyńska, E.D. On Prototropy and Bond Length Alternation in Neutral and Ionized Pyrimidine Bases and Their Model Azines in Vacuo. Molecules 2023, 28, 7282. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D. Quantum-Chemical Studies on the Favored and Rare Isomers of Isocytosine. Comput. Theor. Chem. 2017, 1121, 58–67. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Makowski, M. Effects of Positive and Negative Ionization on Prototropy in Pyrimidine Bases: An Unusual Case of Isocytosine. J. Phys. Chem. A 2018, 122, 7863–7879. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Marzin, C.; Katritzky, A.R.; Linda, P. The Tautomerism of Heterocycles; Academic Press: Cambridge, MA, USA, 1976; pp. 1–655. [Google Scholar]
- Öğretir, C.; Yarligan, S. AMl, PM3 and MNDO Study of the Tautomerism of 2-, 4- or 5-Imidazolones and Their Thio- and Azo- Analogs. J. Mol. Struct. (Theochem) 1996, 366, 227–231. [Google Scholar] [CrossRef]
- Kurzepa, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical Studies on Tautomerism and IR Spectra of C-5 Substituted Imidazoles. J. Mol. Struct. 2001, 565–566, 107–113. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, Y.; Xie, D.-Q.; Yan, G.-S. Ab initio Calculation and Monte Carlo Simulation on Tautomerism and Proton Transfer of 2-Hydroxyimidazole in Gas Phase and Water. Acta Chim. Sin. 2005, 63, 791–796. [Google Scholar]
- Raczyńska, E.D. Application of the Extended HOMED (Harmonic Oscillator Model of Aromaticity) Index to Simple and Tautomeric Five-Membered Heteroaromatic Cycles with C, N, O, P, and S Atoms. Symmetry 2019, 11, 146. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecular Orbital Theory; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.v.R.; Pople, J.A. Ab Initio Molecular Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Raczyńska, E.D.; Makowski, M.; Hallmann, M.; Kamińska, B. Geometric and Energetic Consequences of Prototropy for Adenine and Its Structural Models—A Review. RSC Adv. 2005, 5, 36587. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in determining the absolute proton affinities of neutral organic molecules in the gas phase and their interpretation: A theoretical account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef] [PubMed]
- Leito, I.; Koppel, I.A.; Koppel, I.; Kaupmees, K.; Tshepelevitsh, S.; Saame, J. Basicity limits of neutral organic superbases. Angew. Chem. Int. Ed. 2015, 54, 9262–9265. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Kamińska, B. Structural and Thermochemical Consequences of Prototropy and Ionization for the Biomolecule Xanthine in Vacuo. J. Chem. Thermodyn. 2022, 171, 106788. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Trucks, G.W.; Pople, J.A. Gaussian-2 Theory for Molecular Energies of First- and Second-Row Compounds. J. Chem. Phys. 1991, 94, 7221–7230. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Pople, J.A. Gaussian-2 Theory Using Reduced Møller-Plesset Orders. J. Chem. Phys. 1993, 98, 1293–1298. [Google Scholar] [CrossRef]
- Smith, B.J.; Radom, L. Assigning Absolute Values to Proton Affinities: A Differentiation Between Competing Scales. J. Am. Chem. Soc. 1993, 115, 4885–4888. [Google Scholar] [CrossRef]
- Raczyńska, E.D. Electron Delocalization and Relative Stabilities for the Favored and Rare Tautomers of Hydroxyazines in the Gas Phase—A Comparison with Aminoazines. Comput. Theor. Chem. 2014, 1042, 8–15. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian-03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Kruszewski, J.; Krygowski, T.M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Kruszewski, J. Aromaticity of Thiophene, Pyrrole and Furan in Terms of Aromaticity Indices and Hammett σ Constants. Bull. Acad. Pol. Sci. Chim. 1974, 22, 871–876. [Google Scholar]
- Krygowski, T.M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inform. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Perrin, C.L.; Agranat, I.; Bagno, A.; Braslavsky, S.E.; Fernandes, P.A.; Gal, J.-F.; Lloyd-Jones, G.C.; Mayr, H.; Murdoch, J.R.; Nudelman, N.S.; et al. Glossary of Terms Used in Physical Organic Chemistry (IUPAC Recommendations 2021). Pure Appl. Chem. 2022, 94, 353–534. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Makowski, M.; Zientara-Rytter, K.; Kolczyńska, K.; Stępniewski, T.M.; Hallmann, M. Quantum-Chemical Studies on the Favored and Rare Tautomers of Neutral and Redox Adenine. J. Phys. Chem. A 2013, 117, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Liebman, J.F. The annular tautomerism of imidazoles and pyrazoles: The possible existence of nonaromatic forms. Struct. Chem. 2006, 17, 439–444. [Google Scholar] [CrossRef]
- Raczyńska, E.D. Quantum-chemical studies of the consequences of one-electron oxidation and one-electron reduction for imidazole in the gas phase and water. Comput. Theor. Chem. 2012, 993, 73–79. [Google Scholar] [CrossRef]
- Smith, B.J.; Radom, L. Calculating of Proton Affinities Using the G2(MP2,SVP) Procedure. J. Phys. Chem. 1995, 99, 6468–6471. [Google Scholar] [CrossRef]
- East, A.L.L.; Smith, B.J.; Radom, L. Entropies and Free Energies of Protonation and Proton Transfer Reactions. J. Am. Chem. Soc. 1997, 119, 9014–9020. [Google Scholar] [CrossRef]
- Kabli, S.; van Beelen, E.S.E.; Ingemann, S.; Henriksen, L.; Hammerum, S. The Proton Affinities of Saturated and Unsaturated Heterocyclic Molecules. Int. J. Mass Spectrom. 2006, 249–250, 370–378. [Google Scholar] [CrossRef]
- Bouchoux, G. Gas Phase Basicities of Polyfunctional Molecules. Part 3: Amino Acids. Mass Spectrom. Rev. 2012, 31, 391–435. [Google Scholar] [CrossRef]
- Kolboe, S. Proton Affinity Calculations with High Level Methods. J. Chem. Theory Comput. 2014, 10, 3123–3128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Reference Molecule | Ro (Å) | α5 | α7 | α6 |
|---|---|---|---|---|---|
| CC | C6H6 | 1.3943 | 78.34 | 80.90 | 88.09 |
| CN | C3N3H3 | 1.3342 | 81.98 | 84.52 | 91.60 |
| CO | C(OH)3+ | 1.2811 | 67.84 | 69.74 | 75.00 |
| Isomer of X1MIm | Isomers of XIm | Presence of C-sp3 in the Ring | ΔHOMED5 | ΔHOMED6 |
|---|---|---|---|---|
| 2A1MIm-6 | 2AIm-16 | no | 0.010 | 0.001 |
| 2A1MIm-3a | 2AIm-13 | no | 0.007 | 0.010 |
| 2A1MIm-3b | 2AIm-13 | no | 0.005 | 0.009 |
| 2A1MIm-5a | 2AIm-15a | yes | 0.025 | 0.028 |
| 2A1MIm-5b | 2AIm-15b | yes | 0.026 | 0.030 |
| 4O1MIm-6a | 4OIm-16a | no | 0.004 | 0.003 |
| 4O1MIm-6b | 4OIm-16b | no | 0.004 | 0.003 |
| 4O1MIm-5 | 4OIm-15 | yes | 0.014 | 0.015 |
| Pair of Isomers for Proton Transfer | Type of Proton Transfer | δG(Me) | |
|---|---|---|---|
| X1MIm | XIm | ||
| 2A1MIm-6→2A1MIm-3a | 2AIm-16/36→2AIm-13 | N6→N3 | −0.5 |
| 2A1MIm-6→2A1MIm-3b | 2AIm-16/36→2AIm-13 | N6→N3 | −1.1 |
| 2A1MIm-3a→2A1MIm-5a | 2AIm-13→2AIm-15a/34b | N3→C5 | 0.7 |
| 2A1MIm-3b→2A1MIm-5b | 2AIm-13→2AIm-15b/34a | N3→C5 | 0.3 |
| 2A1MIm-6→2A1MIm-5a | 2AIm-16/36→2AIm-15a/34b | N6→C5 | 0.2 |
| 2A1MIm-6→2A1MIm-5b | 2AIm-16/36→2AIm-15b/34a | N6→C5 | −0.8 |
| 4O1MIm-5→4O1MIm-6a | 4OIm-15→4OIm-16a | C5→O6 | −0.8 |
| 4O1MIm-5→4O1MIm-6b | 4OIm-15→4OIm-16b | C5→O6 | −0.7 |
| Pair of Isomers a and b | δG(=NH) | Pair of Isomers a and b | δG(–OH) |
|---|---|---|---|
| 2AIm-15a/34b→2AIm-15b/34a | −3.9 | 2OIm-46a/56b→2OIm-46b/56a | −0.8 |
| 2A1MIm-3a→2A1MIm-3b | −0.6 | 4OIm-16a→4OIm-16b | 3.8 |
| 2A1MIm-5a→2A1MIm-5b | −4.9 | 4OIm-26a→4OIm-26b | 4.8 |
| 4AIm-23a→4AIm-23b | −0.6 | 4OIm-46a→4OIm-46b | 0.6 |
| 4AIm-15a→4AIm-15b | 4.4 | 4OIm-56a→4OIm-56b | 5.1 |
| 4AIm-35a→4AIm-35b | −1.6 | 4O1MIm-6a→4O1MIm-6b | 3.9 |
| Crea-37aa→Crea-37ba | −0.5 | Crea-67a→Crea67b | 3.5 |
| Crea-37ab→Crea-37bb | −0.7 | Crea-37aa→Crea-37ab | −0.8 |
| Crea-57aa→Crea-57ba | −4.5 | Crea-37ba→Crea-37bb | −1.1 |
| Crea-57ab→Crea-57bb | −5.2 | Crea-57aa→Crea-57ab | 6.3 |
| Crea-35a→Crea-35b | −0.4 | Crea-57ba→Crea-57bb | 5.6 |
| iC-38aa→iC-38ba | 7.5 | iC-78a→iC-78b | 4.4 |
| iC-38ab→iC-38bb | 6.6 | iC-38aa→iC-38ab | −2.4 |
| iC-58aa→iC-58ba | 0.6 | iC-38ba→iC-38bb | −3.3 |
| iC-58ab→iC-58bb | −0.4 | iC-58aa→iC-58ab | 8.0 |
| iC-35a→iC-35b | 5.1 | iC-58ba→iC-58bb | 7.0 |
| Pair of Isomers for Proton Transfer | Proton Transfer | δG(4-OH) | δG(2-NH2) | |
|---|---|---|---|---|
| Crea | 2A1MIm | |||
| Crea-67a→Crea-37aa | 2A1MIm-6→2A1MIm-3a | N6→N3 | 6.47 (a) | |
| Crea-67a→Crea-37ba | 2A1MIm-6→2A1MIm-3b | N6→N3 | 6.56 (a) | |
| Crea-67b→Crea-37ab | 2A1MIm-6→2A1MIm-3a | N6→N3 | 2.17 (b) | |
| Crea-67b→Crea-37bb | 2A1MIm-6→2A1MIm-3b | N6→N3 | 2.04 (b) | |
| Crea-37a→Crea-57aa | 2A1MIm-3a→2A1MIm-5a | N3→C5 | −16.67 (a) | |
| Crea-37a→Crea-57ba | 2A1MIm-3b→2A1MIm-5b | N3→C5 | −16.39 (a) | |
| Crea-37b→Crea-57ab | 2A1MIm-3a→2A1MIm-5a | N3→C5 | −9.53 (b) | |
| Crea-37b→Crea-57bb | 2A1MIm-3b→2A1MIm-5b | N3→C5 | −9.69 (b) | |
| Crea-67a→Crea-57aa | 2A1MIm-6→2A1MIm-5a | N6→C5 | −10.20 (a) | |
| Crea-67a→Crea-57ba | 2A1MIm-6→2A1MIm-5b | N6→C5 | −9.83 (a) | |
| Crea-67b→Crea-57ab | 2A1MIm-6→2A1MIm-5a | N6→C5 | −7.36 (b) | |
| Crea-67b→Crea-57bb | 2A1MIm-6→2A1MIm-5b | N6→C5 | −7.65 (b) | |
| Crea-67a→Crea-56 | 4O1MIm-6a→4O1MIm-5 | O6→C5 | −8.69 | |
| Crea-67b→Crea-56 | 4O1MIm-6b→4O1MIm-5 | O6→C5 | −8.21 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raczyńska, E.D. On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines. Symmetry 2024, 16, 98. https://doi.org/10.3390/sym16010098
Raczyńska ED. On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines. Symmetry. 2024; 16(1):98. https://doi.org/10.3390/sym16010098
Chicago/Turabian StyleRaczyńska, Ewa Daniela. 2024. "On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines" Symmetry 16, no. 1: 98. https://doi.org/10.3390/sym16010098
APA StyleRaczyńska, E. D. (2024). On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines. Symmetry, 16(1), 98. https://doi.org/10.3390/sym16010098