
LIBGRPP: A Library for the Evaluation of Molecular Integrals of the Generalized Relativistic Pseudopotential Operator over Gaussian Functions

 , , , , and
, , , , and 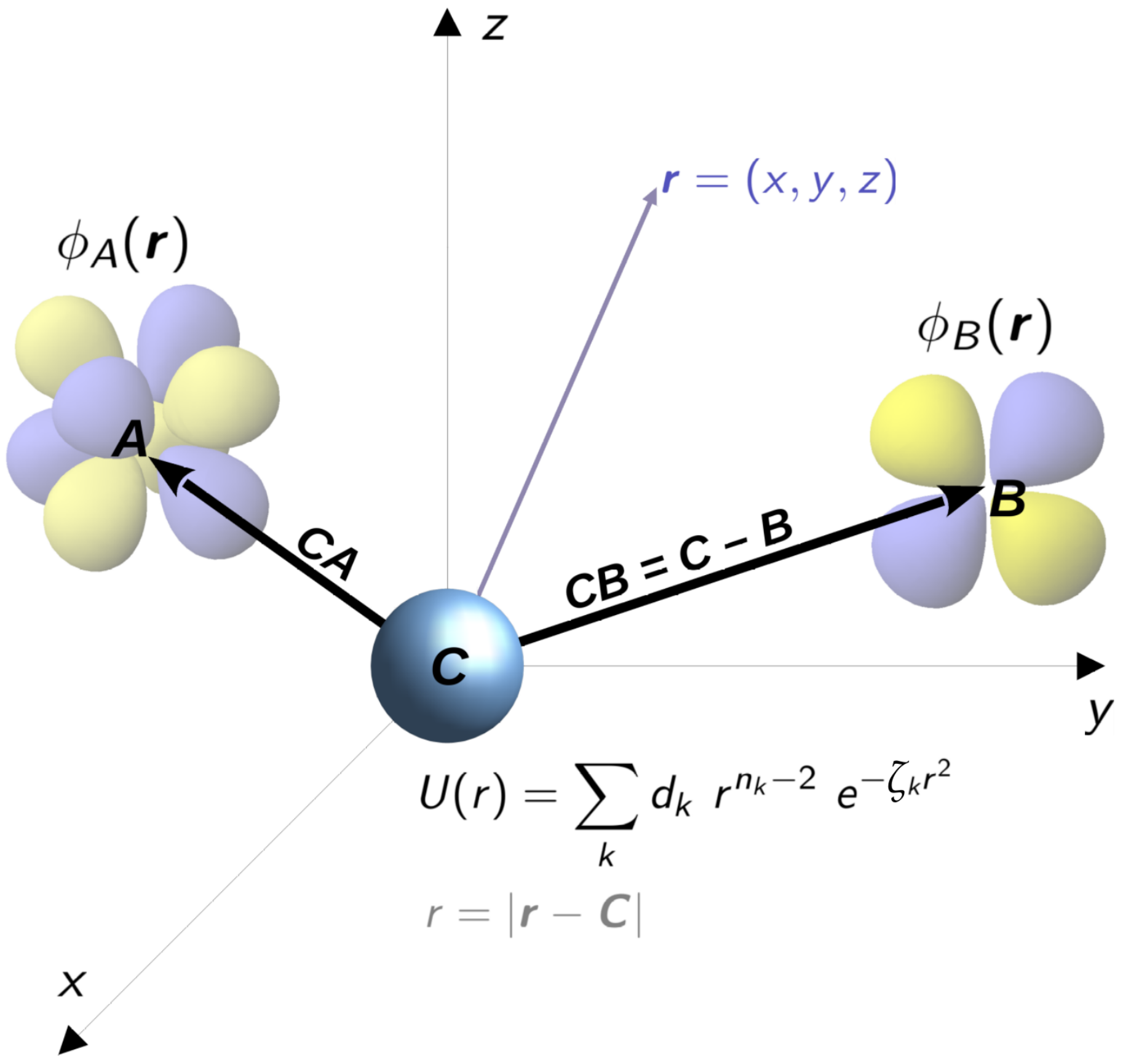{kind=link}
{kind=link}
{kind=link}
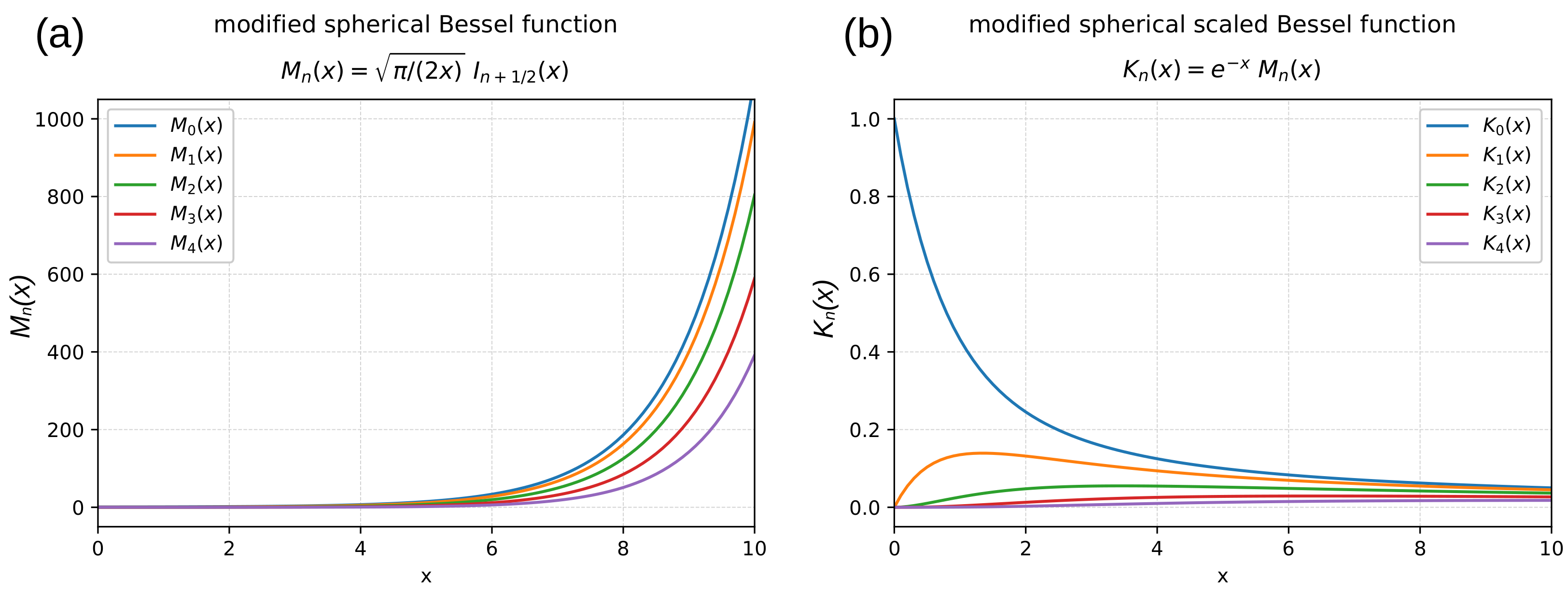
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Theory
2.1. Generalized Relativistic Pseudopotentials
- An approximate nature of the many-electron Hamiltonian used to evaluate atomic spinors, which in turn define the potentials . The construction of modern GRPPs is based on atomic four-component all-electron calculations with the Dirac–Coulomb–Breit Hamiltonian, employing Fermi nuclear charge distribution, and accounting for the quantum electrodynamic correction [28] by means of the Lamb shift model potential [18,64];
- The neglect of correlations between excluded and explicitly treated electrons and inner core polarization and smoothing of pseudo wavefunctions in the inner core area. The corresponding errors naturally decrease while reducing the number of excluded electronic shells (so-called tiny-core and empty-core versions of GRPPs [28,58]);
- A roughly approximate mean-field-like simulation of Breit interactions between the explicitly treated electrons by the corresponding contributions to one-electron GRPPs. In principle, this factor can limit the feasibility of core size reduction for heavy atoms.
2.2. Scalar-Relativistic Part: Integrals over the Local Potential
2.3. Scalar-Relativistic Part: Integrals with Angular Projectors
2.4. Integrals over the Effective Spin-Orbit Interaction Operator
2.5. Integrals over Non-Local Terms of GRPP
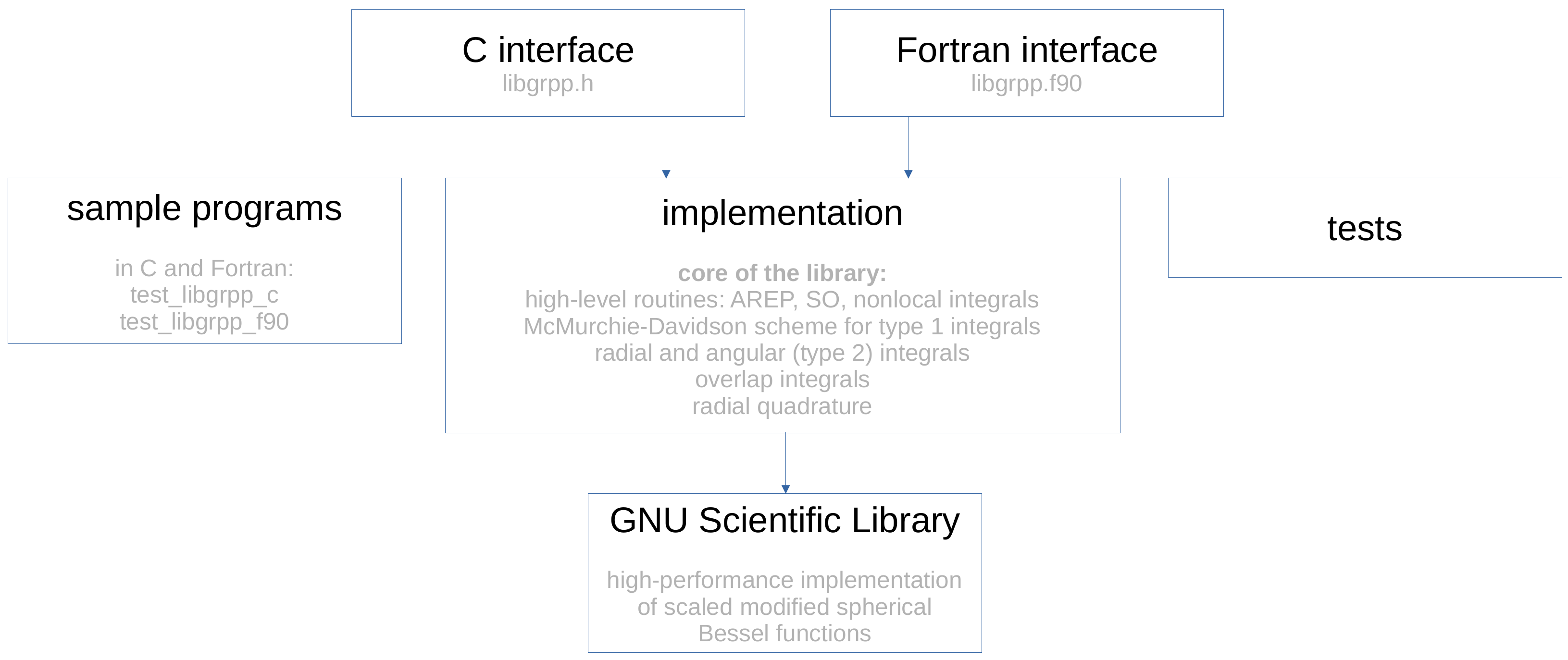
3. The LIBGRPP Library
4. Pilot Applications
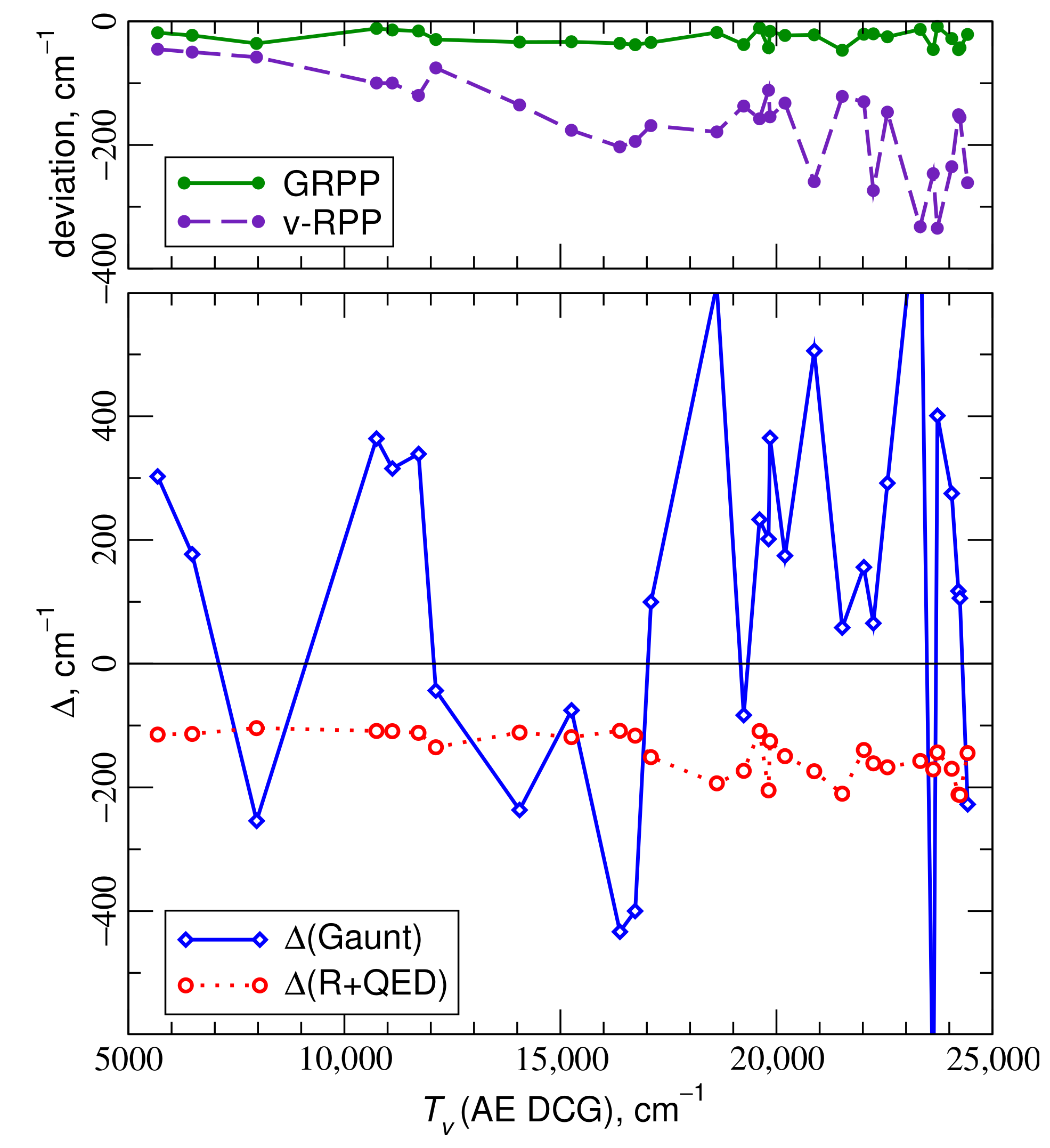
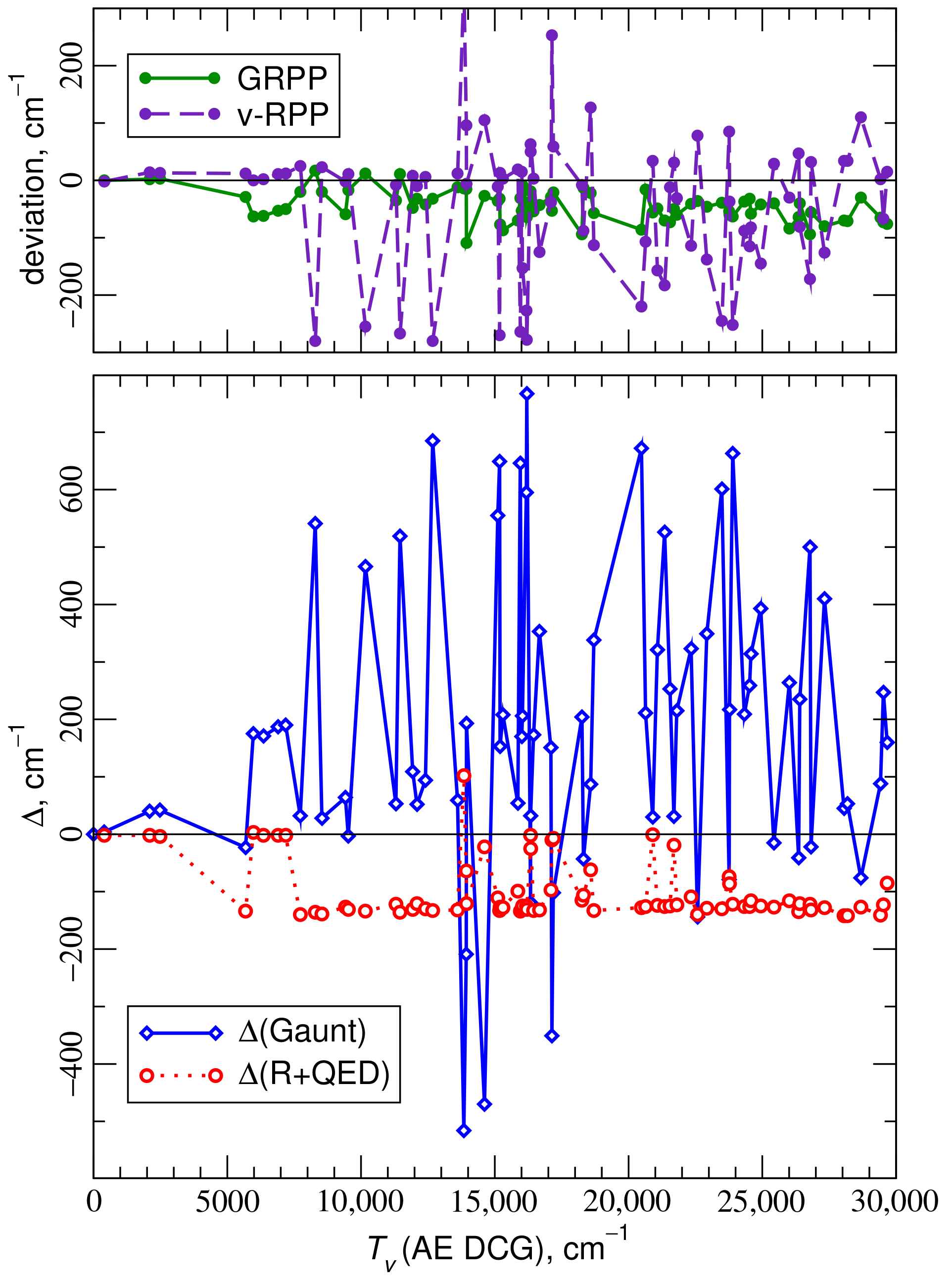
4.1. Electronic States of the ThO Molecule
4.2. Electronic States of the UO Molecule
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FS-RCCSD | Fock space relativistic coupled cluster method with single and double excitations |
| GRPP | Generalized relativistic pseudopotential |
| IH | Intermediate Hamiltonian |
| QED | Quantum electrodynamics |
| SO | Spin-orbit |
| v-RPP | valence (semilocal) part of GRPP |
Appendix A. Analytic Gradients of GRPP Integrals
Appendix B. Obara-Saika Recurrence Relations for the Local Part of the GRPP Operator
Appendix C. Analytic Evaluation of One-Center RPP Integrals
References
- Eliav, E.; Kaldor, U. Study of actinides by relativistic coupled cluster methods. In Computational Methods in Lanthanide and Actinide Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; Chapter 2; pp. 23–54. [Google Scholar] [CrossRef]
- Eliav, E.; Fritzsche, S.; Kaldor, U. Electronic structure theory of the superheavy elements. Nucl. Phys. 2015, 944, 518–550. [Google Scholar] [CrossRef]
- Eliav, E.; Borschevsky, A.; Kaldor, U. High-accuracy relativistic coupled-cluster calculations for the heaviest elements. In Handbook of Relativistic Quantum Chemistry; Liu, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 825–855. [Google Scholar] [CrossRef]
- Eliav, E.; Borschevsky, A.; Zaitsevskii, A.; Oleynichenko, A.V.; Kaldor, U. Relativistic Fock-space coupled cluster method: Theory and recent applications. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Dyall, K.; Faegri, K. Introduction to Relativistic Quantum Chemistry; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Sikkema, J.; Visscher, L.; Saue, T.; Iliaš, M. The molecular mean-field approach for correlated relativistic calculations. J. Chem. Phys. 2009, 131, 124116. [Google Scholar] [CrossRef] [PubMed]
- Saue, T. Relativistic Hamiltonians for chemistry: A primer. Chem. Phys. Chem. 2011, 12, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Bratsev, V.F.; Deyneka, G.B.; Tupitsyn, I.I. Application of the Hartree-Fock method to calculation of relativistic atomic wave functions. Bull. Acad. Sci. USSR Phys. Ser. 1977, 41, 173–182. [Google Scholar]
- Parpia, F.A.; Froese Fischer, C.; Grant, I.P. GRASP92: A package for large-scale relativistic atomic structure calculations. Comput. Phys. Commun. 1996, 94, 249–271. [Google Scholar] [CrossRef]
- Kozlov, M.; Porsev, S.; Safronova, M.S.; Tupitsyn, I. CI-MBPT: A package of programs for relativistic atomic calculations based on a method combining configuration interaction and many-body perturbation theory. Comput. Phys. Commun. 2015, 195, 199–213. [Google Scholar] [CrossRef]
- Kahl, E.V.; Berengut, J.C. AMBiT: A programme for high-precision relativistic atomic structure calculations. Comput. Phys. Commun. 2019, 238, 232–243. [Google Scholar] [CrossRef]
- Fritzsche, S. A fresh computational approach to atomic structures, processes and cascades. Comput. Phys. Commun. 2019, 240, 1–14. [Google Scholar] [CrossRef]
- Visscher, L.; Visser, O.; Aerts, P.J.C.; Merenga, H.; Nieuwpoort, W.C. Relativistic quantum chemistry: The MOLFDIR program package. Comput. Phys. Commun. 1994, 81, 120–144. [Google Scholar] [CrossRef]
- Liu, W.; Wang, F.; Li, L. The Beijing Density Functional (BDF) program package: Methodologies and applications. J. Theor. Comput. Chem. 2003, 2, 257–272. [Google Scholar] [CrossRef]
- Van Wüllen, C. A quasirelativistic two-component density Functional and Hartree-Fock program. In Progress in Physical Chemistry Volume 3; Oldenbourg Wissenschaftsverlag GmbH: Munich, Germany, 2010; pp. 123–136. [Google Scholar] [CrossRef]
- Saue, T.; Bast, R.; Gomes, A.S.P.; Jensen, H.J.A.; Visscher, L.; Aucar, I.A.; Di Remigio, R.; Dyall, K.G.; Eliav, E.; Fasshauer, E.; et al. The DIRAC code for relativistic molecular calculations. J. Chem. Phys. 2020, 152, 204104. [Google Scholar] [CrossRef]
- Repisky, M.; Komorovsky, S.; Kadek, M.; Konecny, L.; Ekström, U.; Malkin, E.; Kaupp, M.; Ruud, K.; Malkina, O.L.; Malkin, V.G. ReSpect: Relativistic spectroscopy DFT program package. J. Chem. Phys. 2020, 152, 184101. [Google Scholar] [CrossRef]
- Shabaev, V.M.; Tupitsyn, I.I.; Yerokhin, V.A. Model operator approach to the Lamb shift calculations in relativistic many-electron atoms. Phys. Rev. A 2013, 88, 012513. [Google Scholar] [CrossRef]
- Eliav, E.; Kaldor, U. Relativistic Four-Component Multireference Coupled Cluster Methods: Towards A Covariant Approach. In Recent Progress in Coupled Cluster Methods: Theory and Applications; Cársky, P., Paldus, J., Pittner, J., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 113–144. [Google Scholar] [CrossRef]
- Thierfelder, C.; Schwerdtfeger, P. Quantum electrodynamic corrections for the valence shell in heavy many-electron atoms. Phys. Rev. A 2010, 82, 062503. [Google Scholar] [CrossRef]
- Roberts, B.M.; Dzuba, V.A.; Flambaum, V.V. Quantum electrodynamics corrections to energies, transition amplitudes, and parity nonconservation in Rb, Cs, Ba+, Tl, Fr, and Ra+. Phys. Rev. A 2013, 87, 054502. [Google Scholar] [CrossRef]
- Lindgren, I. Relativistic Many-Body Theory. A New Field-Theoretical Approach, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Sunaga, A.; Saue, T. Towards highly accurate calculations of parity violation in chiral molecules: Relativistic coupled-cluster theory including QED-effects. Mol. Phys. 2021, 119, e1974592. [Google Scholar] [CrossRef]
- Sunaga, A.; Salman, M.; Saue, T. 4-component relativistic Hamiltonian with effective QED potentials for molecular calculations. J. Chem. Phys. 2022, 157, 164101. [Google Scholar] [CrossRef]
- Kaygorodov, M.Y.; Usov, D.P.; Eliav, E.; Kozhedub, Y.S.; Malyshev, A.V.; Oleynichenko, A.V.; Shabaev, V.M.; Skripnikov, L.V.; Titov, A.V.; Tupitsyn, I.I.; et al. Ionization potentials and electron affinities of Rg, Cn, Nh, and Fl superheavy elements. Phys. Rev. A 2022, 105, 062805. [Google Scholar] [CrossRef]
- Petrov, A.N.; Mosyagin, N.S.; Titov, A.V.; Tupitsyn, I.I. Accounting for the Breit interaction in relativistic effective core potential calculations of actinides. J. Phys. B 2004, 37, 4621–4637. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Chubukov, D.V.; Shakhova, V.M. The role of QED effects in transition energies of heavy-atom alkaline earth monofluoride molecules: A theoretical study of Ba+, BaF, RaF, and E120F. J. Chem. Phys. 2021, 155, 144103. [Google Scholar] [CrossRef]
- Zaitsevskii, A.; Mosyagin, N.S.; Oleynichenko, A.V.; Eliav, E. Generalized relativistic small-core pseudopotentials accounting for quantum electrodynamic effects: Construction and pilot applications. arXiv 2022, arXiv:2208.12296. [Google Scholar] [CrossRef]
- Knecht, S.; Repisky, M.; Jensen, H.J.A.; Saue, T. Exact two-component Hamiltonians for relativistic quantum chemistry: Two-electron picture-change corrections made simple. J. Chem. Phys. 2022, 157, 114106. [Google Scholar] [CrossRef] [PubMed]
- Seijo, L.; Barandiarán, Z. Relativistic ab-initio model potential calculations for molecules and embedded clusters. Theor. Comput. Chem. 2004, 14, 417–475. [Google Scholar] [CrossRef]
- Abarenkov, I.V.; Heine, V. The model potential for positive ions. Philos. Mag. 1965, 12, 529–537. [Google Scholar] [CrossRef]
- Christiansen, P.A.; Lee, Y.S.; Pitzer, K.S. Improved ab initio effective core potentials for molecular calculations. J. Chem. Phys. 1979, 71, 4445–4450. [Google Scholar] [CrossRef]
- Titov, A.V.; Mosyagin, N.S. Generalized relativistic effective core potential: Theoretical grounds. Int. J. Quantum Chem. 1999, 71, 359–401. [Google Scholar] [CrossRef]
- Schwerdtfeger, P. The pseudopotential approximation in electronic structure theory. Chem. Phys. Chem. 2011, 12, 3143–3155. [Google Scholar] [CrossRef]
- Dolg, M.; Cao, X. Relativistic pseudopotentials: Their development and scope of applications. Chem. Rev. 2012, 112, 403–480. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Zaitsevskii, A.V.; Skripnikov, L.V.; Titov, A.V. Generalized relativistic effective core potentials for actinides. Int. J. Quantum Chem. 2016, 116, 301–315. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Zaitsevskii, A.V.; Titov, A.V. Generalized relativistic effective core potentials for superheavy elements. Int. J. Quantum Chem. 2020, 120, e26076. [Google Scholar] [CrossRef]
- Lee, Y.S.; Ermler, W.C.; Pitzer, K.S. Ab initio effective core potentials including relativistic effects. I. Formalism and applications to the Xe and Au atoms. J. Chem. Phys. 1977, 67, 5861–5876. [Google Scholar] [CrossRef]
- Hafner, P.; Schwarz, W. Molecular spinors from the quasi-relativistic pseudopotential approach. Chem. Phys. Lett. 1979, 65, 537–541. [Google Scholar] [CrossRef]
- Pitzer, R.M.; Winter, N.W. Spin-orbit (core) and core potential integrals. Int. J. Quantum Chem. 1991, 40, 773–780. [Google Scholar] [CrossRef]
- Pacios, L.F.; Christiansen, P.A. Ab initio relativistic effective potentials with spin-orbit operators. I. Li through Ar. J. Chem. Phys. 1985, 82, 2664–2671. [Google Scholar] [CrossRef]
- Hurley, M.M.; Pacios, L.F.; Christiansen, P.A.; Ross, R.B.; Ermler, W.C. Ab initio relativistic effective potentials with spin-orbit operators. II. K through Kr. J. Chem. Phys. 1986, 84, 6840–6853. [Google Scholar] [CrossRef]
- LaJohn, L.A.; Christiansen, P.A.; Ross, R.B.; Atashroo, T.; Ermler, W.C. Ab initio relativistic effective potentials with spin–orbit operators. III. Rb through Xe. J. Chem. Phys. 1987, 87, 2812–2824. [Google Scholar] [CrossRef]
- Ross, R.B.; Powers, J.M.; Atashroo, T.; Ermler, W.C.; LaJohn, L.A.; Christiansen, P.A. Ab initio relativistic effective potentials with spin–orbit operators. IV. Cs through Rn. J. Chem. Phys. 1990, 93, 6654–6670. [Google Scholar] [CrossRef]
- Pacios, L.F.; Olcina, V.B. Modified Ar core ab initio relativistic effective potentials for transition metals Sc through Cu. J. Chem. Phys. 1991, 95, 441–450. [Google Scholar] [CrossRef]
- Ermler, W.C.; Ross, R.B.; Christiansen, P.A. Ab initio relativistic effective potentials with spin-orbit operators. VI. Fr through Pu. Int. J. Quantum Chem. 1991, 40, 829–846. [Google Scholar] [CrossRef]
- Ross, R.B.; Gayen, S.; Ermler, W.C. Ab initio relativistic effective potentials with spin-orbit operators. V. Ce through Lu. J. Chem. Phys. 1994, 100, 8145–8155. [Google Scholar] [CrossRef]
- Wildman, S.A.; DiLabio, G.A.; Christiansen, P.A. Accurate relativistic effective potentials for the sixth-row main group elements. J. Chem. Phys. 1997, 107, 9975–9979. [Google Scholar] [CrossRef]
- Nash, C.S.; Bursten, B.E.; Ermler, W.C. Ab initio relativistic potentials with spin-orbit operators. VII. Am through element 118. J. Chem. Phys. 1997, 106, 5133–5142, Erratum in J. Chem. Phys. 1999, 111, 2347. [Google Scholar] [CrossRef]
- Cundari, T.R.; Stevens, W.J. Effective core potential methods for the lanthanides. J. Chem. Phys. 1993, 98, 5555–5565. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Dolg, M.; Cao, X. Accurate relativistic small-core pseudopotentials for actinides. Energy adjustment for uranium and first applications to uranium hydride. J. Phys. Chem. A 2009, 113, 12573–12581. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Zaitsevskii, A.; Titov, A.V. Shape-consistent relativistic effective potentials of small atomic cores. Int. Rev. At. Mol. Phys. 2010, 1, 63–72. [Google Scholar]
- Mosyagin, N.S. Generalized relativistic effective core potentials for lanthanides. Nonlinear Phenom. Complex Syst. 2017, 20, 111–132. [Google Scholar]
- Tupitsyn, I.I.; Mosyagin, N.S.; Titov, A.V. Generalized relativistic effective core potential. I. Numerical calculations for atoms Hg through Bi. J. Chem. Phys. 1995, 103, 6548–6555. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Titov, A.V.; Latajka, Z. Generalized relativistic effective core potential: Gaussian expansions of potentials and pseudospinors for atoms Hg through Rn. Int. J. Quantum Chem. 1997, 63, 1107–1122. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Titov, A.V.; Eliav, E.; Kaldor, U. Generalized relativistic effective core potential and relativistic coupled cluster calculation of the spectroscopic constants for the HgH molecule and its cation. J. Chem. Phys. 2001, 115, 2007–2013. [Google Scholar] [CrossRef]
- Mosyagin, N.; Oleynichenko, A.; Zaitsevskii, A.; Kudrin, A.; Pazyuk, E.; Stolyarov, A. Ab initio relativistic treatment of the a3Π − X1 Σ+, a′3Σ+ − X1Σ+ and A1Π − X1Σ+ systems of the CO molecule. J. Quant. Spectrosc. Radiat. Transf. 2021, 263, 107532. [Google Scholar] [CrossRef]
- Zaitsevskii, A.; Skripnikov, L.V.; Mosyagin, N.S.; Isaev, T.; Berger, R.; Breier, A.A.; Giesen, T.F. Accurate ab initio calculations of RaF electronic structure appeal to more laser-spectroscopical measurements. J. Chem. Phys. 2022, 156, 044306. [Google Scholar] [CrossRef]
- Titov, A.V.; Petrov, A.N.; Panin, A.I.; Khait, Y.G. MOLGEP Code for Calculation of Matrix Elements with GRECP (St.-Petersburg, 1999).
- McMurchie, L.E.; Davidson, E.R. Calculation of integrals over ab initio pseudopotentials. J. Comput. Phys. 1981, 44, 289–301. [Google Scholar] [CrossRef]
- Park, Y.C.; Lee, Y.S. Two-component spin-orbit effective core potential calculations with an all-electron relativistic program DIRAC. Bull. Korean Chem. Soc. 2012, 33, 803–808. [Google Scholar] [CrossRef]
- Mitin, A.V.; Van Wüllen, C. Two-component relativistic density-functional calculations of the dimers of the halogens from bromine through element 117 using effective core potential and all-electron methods. J. Chem. Phys. 2006, 124, 064305. [Google Scholar] [CrossRef]
- Shabaev, V.M.; Tupitsyn, I.I.; Yerokhin, V.A. QEDMOD: Fortran program for calculating the model Lamb-shift operator. Comput. Phys. Commun. 2018, 223, 69. [Google Scholar] [CrossRef]
- Generalized Relativistic Pseudopotentials. Available online: http://qchem.pnpi.spb.ru/recp (accessed on 2 January 2023).
- Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic-Structure Theory; Wiley: New York, NY, USA, 2000. [Google Scholar] [CrossRef]
- Van Wüllen, C. Numerical instabilities in the computation of pseudopotential matrix elements. J. Comput. Chem. 2005, 27, 135–141. [Google Scholar] [CrossRef]
- McMurchie, L.E.; Davidson, E.R. One- and two-electron integrals over Cartesian Gaussian functions. J. Comput. Phys. 1978, 26, 218–231. [Google Scholar] [CrossRef]
- Jensen, J.O.; Carrieri, A.H.; Vlahacos, C.P.; Zeroka, D.; Hameka, H.F.; Merrow, C.N. Evaluation of one-electron integrals for arbitrary operators V(r) over Cartesian Gaussians: Application to inverse-square distance and Yukawa operators. J. Comput. Chem. 1993, 14, 986–994. [Google Scholar] [CrossRef]
- Gao, B.; Thorvaldsen, A.J.; Ruud, K. GEN1INT: A unified procedure for the evaluation of one-electron integrals over Gaussian basis functions and their geometric derivatives. Int. J. Quantum Chem. 2011, 111, 858–872. [Google Scholar] [CrossRef]
- Skylaris, C.K.; Gagliardi, L.; Handy, N.C.; Ioannou, A.G.; Spencer, S.; Willetts, A.; Simper, A.M. An efficient method for calculating effective core potential integrals which involve projection operators. Chem. Phys. Lett. 1998, 296, 445–451. [Google Scholar] [CrossRef]
- Flores-Moreno, R.; Alvarez-Mendez, R.J.; Vela, A.; Köster, A.M. Half-numerical evaluation of pseudopotential integrals. J. Comput. Chem. 2006, 27, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.E.; Knowles, P.J. Improved radial grids for quadrature in molecular density-functional calculations. J. Chem. Phys. 1996, 104, 9848–9858. [Google Scholar] [CrossRef]
- Lindh, R.; Malmqvist, P.Å.; Gagliardi, L. Molecular integrals by numerical quadrature. I. Radial integration. Theor. Chim. Acta 2001, 106, 178–187. [Google Scholar] [CrossRef]
- Song, C.; Wang, L.P.; Sachse, T.; Preiss, J.; Presselt, M.; Martinez, T.J. Efficient implementation of effective core potential integrals and gradients on graphical processing units. J. Chem. Phys. 2015, 143, 014114. [Google Scholar] [CrossRef]
- McKenzie, S.C.; Epifanovsky, E.; Barca, G.M.J.; Gilbert, A.T.B.; Gill, P.M.W. Efficient method for calculating effective core potential integrals. J. Phys. Chem. A 2018, 122, 3066–3075. [Google Scholar] [CrossRef]
- Shaw, R.A.; Hill, J.G. Prescreening and efficiency in the evaluation of integrals over ab initio effective core potentials. J. Chem. Phys. 2017, 147, 074108. [Google Scholar] [CrossRef]
- McKenzie, S.C. Efficient Computation of Integrals in Modern Correlated Methods. Ph.D. Thesis, Faculty of Science, University of Sydney, Sydney, Australia, 2020. [Google Scholar]
- Galassi, M.; Davies, J.; Theiler, J.; Gough, B.; Jungman, G. GNU Scientific Library-Reference Manual, Third Edition, for GSL Version 1.12, 3rd ed.; Network Theory Ltd.: London, UK, 2009. [Google Scholar]
- Obara, S.; Saika, A. Efficient recursive computation of molecular integrals over Cartesian Gaussian functions. J. Chem. Phys. 1986, 84, 3963–3974. [Google Scholar] [CrossRef]
- Gomes, A.S.P.; Saue, T.; Visscher, L.A.; Jensen, H.J.; Bast, R.; Aucar, I.A.; Bakken, V.; Dyall, K.G.; Dubillard, S.; Ekstroem, U.; et al. DIRAC, a Relativistic Ab Initio Electronic Structure Program, Release DIRAC19. 2019. Available online: http://diracprogram.org (accessed on 17 December 2022).
- Skripnikov, L.V.; Petrov, A.N.; Mosyagin, N.S.; Ezhov, V.F.; Titov, A.V. Ab initio calculation of the spectroscopic properties of TlF−. Opt. Spectrosc. 2009, 106, 790–792. [Google Scholar] [CrossRef]
- Isaev, T.A.; Mosyagin, N.S.; Petrov, A.N.; Titov, A.V. In search of the electron electric dipole moment: Relativistic correlation calculations of the P,T-violation effect in the ground state of HI+. Phys. Rev. Lett. 2005, 95, 163004. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Isaev, T.A.; Titov, A.V. Is E112 a relatively inert element? Benchmark relativistic correlation study of spectroscopic constants in E112H and its cation. J. Chem. Phys. 2006, 124, 224302. [Google Scholar] [CrossRef]
- Kudashov, A.D.; Petrov, A.N.; Skripnikov, L.V.; Mosyagin, N.S.; Isaev, T.A.; Berger, R.; Titov, A.V. Ab initio study of radium monofluoride (RaF) as a candidate to search for parity- and time-and-parity–violation effects. Phys. Rev. A 2014, 90, 052513. [Google Scholar] [CrossRef]
- Oleynichenko, A.V.; Zaitsevskii, A.; Eliav, E. Towards high performance relativistic electronic structure modelling: The EXP-T program package. In Proceedings of the Supercomputing; Voevodin, V., Sobolev, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; Volume 1331, pp. 375–386. [Google Scholar] [CrossRef]
- Oleynichenko, A.V.; Zaitsevskii, A.; Skripnikov, L.V.; Eliav, E. Relativistic Fock space coupled cluster method for many-electron systems: Non-perturbative account for connected triple excitations. Symmetry 2020, 12, 1101. [Google Scholar] [CrossRef]
- Oleynichenko, A.; Zaitsevskii, A.; Eliav, E. EXP-T, an Extensible Code for Fock Space Relativistic Coupled Cluster Calculations. 2022. Available online: http://www.qchem.pnpi.spb.ru/expt (accessed on 17 December 2022).
- Dewberry, C.T.; Etchison, K.C.; Cooke, S.A. The pure rotational spectrum of the actinide-containing compound thorium monoxide. Phys. Chem. Chem. Phys. 2007, 9, 4895. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the actinides Ac–Lr. Theor. Chem. Acc. 2007, 117, 491–500. [Google Scholar] [CrossRef]
- De Jong, W.A.; Harrison, R.J.; Dixon, D.A. Parallel Douglas-Kroll energy and gradients in NWChem: Estimating scalar relativistic effects using Douglas-Kroll contracted basis sets. J. Chem. Phys. 2001, 114, 48. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef]
- Gagliardi, L.; Heaven, M.C.; Krogh, J.W.; Roos, B.O. The electronic spectrum of the UO2 molecule. J. Am. Chem. Soc. 2004, 127, 86–91. [Google Scholar] [CrossRef]
- Infante, I.; Eliav, E.; Vilkas, M.J.; Ishikawa, Y.; Kaldor, U.; Visscher, L. A Fock space coupled cluster study on the electronic structure of the UO2, UO2+, U4+, and U5+ species. J. Chem. Phys. 2007, 127, 124308. [Google Scholar] [CrossRef]
- Li, W.L.; Su, J.; Jian, T.; Lopez, G.V.; Hu, H.S.; Cao, G.J.; Li, J.; Wang, L.S. Strong electron correlation in UO2−: A photoelectron spectroscopy and relativistic quantum chemistry study. J. Chem. Phys. 2014, 140, 094306. [Google Scholar] [CrossRef] [PubMed]
- Czekner, J.; Lopez, G.V.; Wang, L.S. High resolution photoelectron imaging of UO− and UO2− and the low-lying electronic states and vibrational frequencies of UO and UO2. J. Chem. Phys. 2014, 141, 244302. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.; Konings, R.J.M.; Gibson, J.K.; Infante, I.; Gagliardi, L. Quantum chemical calculations and experimental investigations of molecular actinide oxides. Chem. Rev. 2015, 115, 1725–1759. [Google Scholar] [CrossRef] [PubMed]
- Parpia, F.A.; Mohanty, A.K. Relativistic basis-set calculations for atoms with Fermi nuclei. Phys. Rev. A 1992, 46, 3735–3745. [Google Scholar] [CrossRef] [PubMed]
- Visscher, L.; Dyall, K.G. Dirac-Fock atomic electronic structure calculations using different nuclear charge distributions. At. Data Nucl. Data Tables 1997, 67, 207–224. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N.S.; Bogdanov, N.A.; Booth, G.H.; Chen, J.; Cui, Z.H.; et al. Recent developments in the PySCF program package. J. Chem. Phys. 2020, 153, 024109. [Google Scholar] [CrossRef]
- Lomachuk, Y.V.; Maltsev, D.A.; Mosyagin, N.S.; Skripnikov, L.V.; Bogdanov, R.V.; Titov, A.V. Compound-tunable embedding potential: Which oxidation state of uranium and thorium as point defects in xenotime is favorable? Phys. Chem. Chem. Phys. 2020, 22, 17922–17931. [Google Scholar] [CrossRef]
- Maltsev, D.A.; Lomachuk, Y.V.; Shakhova, V.M.; Mosyagin, N.S.; Skripnikov, L.V.; Titov, A.V. Compound-tunable embedding potential method and its application to calcium niobate crystal CaNb2O6 with point defects containing tantalum and uranium. Phys. Rev. B 2021, 103, 205105. [Google Scholar] [CrossRef]
- Shakhova, V.M.; Maltsev, D.A.; Lomachuk, Y.V.; Mosyagin, N.S.; Skripnikov, L.V.; Titov, A.V. Compound-tunable embedding potential method: Analysis of pseudopotentials for Yb in YbF2, YbF3, YbCl2 and YbCl3 crystals. Phys. Chem. Chem. Phys. 2022, 24, 19333–19345. [Google Scholar] [CrossRef]
- Komornicki, A.; Ishida, K.; Morokuma, K.; Ditchfield, R.; Conrad, M. Efficient determination and characterization of transition states using ab-initio methods. Chem. Phys. Lett. 1977, 45, 595–602. [Google Scholar] [CrossRef]
- Kitaura, K.; Obara, S.; Morokuma, K. Energy gradient with the effective core potential approximation in the ab initio MO method and its application to the structure of Pt(H)2(PH3)2. Chem. Phys. Lett. 1981, 77, 452–454. [Google Scholar] [CrossRef]
- Breidung, J.; Thiel, W.; Komornicki, A. Analytical second derivatives for effective core potentials. Chem. Phys. Lett. 1988, 153, 76–81. [Google Scholar] [CrossRef]
- Russo, T.V.; Martin, R.L.; Hay, P.J.; Rappe, A.K. Vibrational frequencies of transition metal chloride and oxo compounds using effective core potential analytic second derivatives. J. Chem. Phys. 1995, 102, 9315–9321. [Google Scholar] [CrossRef]
- Cui, Q.; Musaev, D.G.; Svensson, M.; Morokuma, K. Analytical second derivatives for effective core potential. Application to transition structures of Cp2Ru2(μ-H)4 and to the mechanism of reaction Cu + CH2N2. J. Phys. Chem. 1996, 100, 10936–10944. [Google Scholar] [CrossRef]
- Bode, B.M.; Gordon, M.S. Fast computation of analytical second derivatives with effective core potentials: Application to Si8C12, Ge8C12, and Sn8C12. J. Chem. Phys. 1999, 111, 8778–8784. [Google Scholar] [CrossRef]
- Gradshteyn, I.S.; Ryzhik, I.M. Table of Integrals, Series, and Products; Academic Press: Cambridge, MA, USA, 2007. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oleynichenko, A.V.; Zaitsevskii, A.; Mosyagin, N.S.; Petrov, A.N.; Eliav, E.; Titov, A.V. LIBGRPP: A Library for the Evaluation of Molecular Integrals of the Generalized Relativistic Pseudopotential Operator over Gaussian Functions. Symmetry 2023, 15, 197. https://doi.org/10.3390/sym15010197
Oleynichenko AV, Zaitsevskii A, Mosyagin NS, Petrov AN, Eliav E, Titov AV. LIBGRPP: A Library for the Evaluation of Molecular Integrals of the Generalized Relativistic Pseudopotential Operator over Gaussian Functions. Symmetry. 2023; 15(1):197. https://doi.org/10.3390/sym15010197
Chicago/Turabian StyleOleynichenko, Alexander V., Andréi Zaitsevskii, Nikolai S. Mosyagin, Alexander N. Petrov, Ephraim Eliav, and Anatoly V. Titov. 2023. "LIBGRPP: A Library for the Evaluation of Molecular Integrals of the Generalized Relativistic Pseudopotential Operator over Gaussian Functions" Symmetry 15, no. 1: 197. https://doi.org/10.3390/sym15010197
APA StyleOleynichenko, A. V., Zaitsevskii, A., Mosyagin, N. S., Petrov, A. N., Eliav, E., & Titov, A. V. (2023). LIBGRPP: A Library for the Evaluation of Molecular Integrals of the Generalized Relativistic Pseudopotential Operator over Gaussian Functions. Symmetry, 15(1), 197. https://doi.org/10.3390/sym15010197