
Identification of Novel Drugs Targeting Cell Cycle Regulators for the Treatment of High-Grade Serous Ovarian Cancer via Integrated Bioinformatics Analysis


Abstract
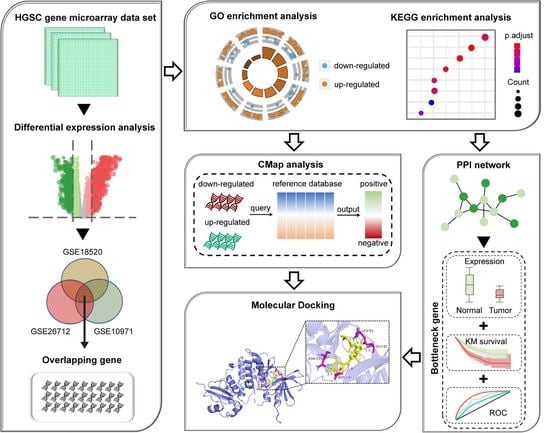
:
1. Introduction
2. Materials and Methods
2.1. Data Collection and DEGs Identification
2.2. Functional Enrichment Analyses
2.3. CMap-Based Screening of Potential Drug Molecules
2.4. PPI Network Construction and Analysis
2.5. ROC Curve Analysis
2.6. Kaplan–Meier Survival Analysis
2.7. Molecular Docking between Candidate Drug and Bottleneck Protein
2.8. Statistical Analysis
3. Results
3.1. Identification of the DEGs Involved in HGSC Pathogenesis
3.2. Functional Annotation of the Overlapping DEGs
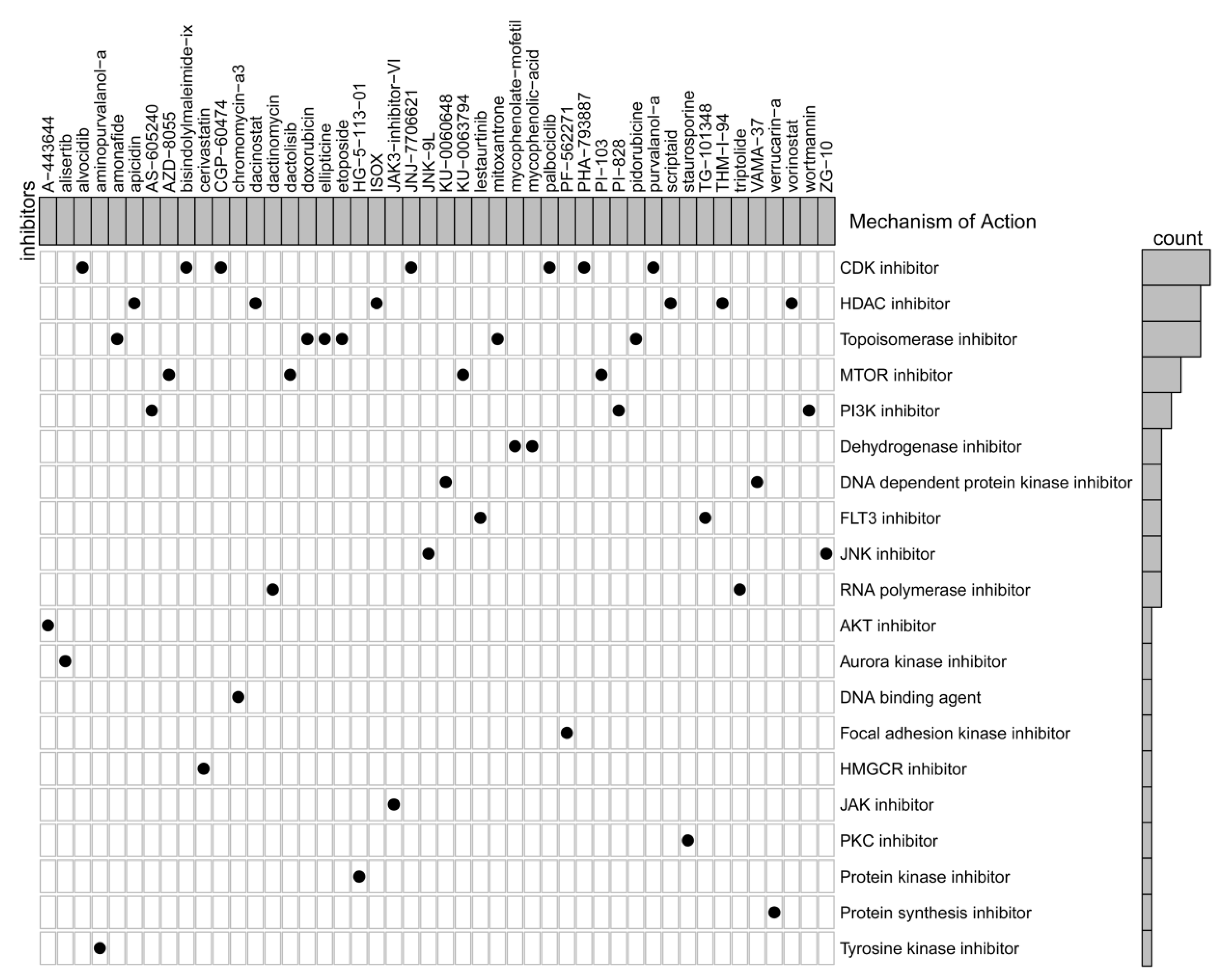
3.3. Screening of Candidate Small-Molecule Drugs
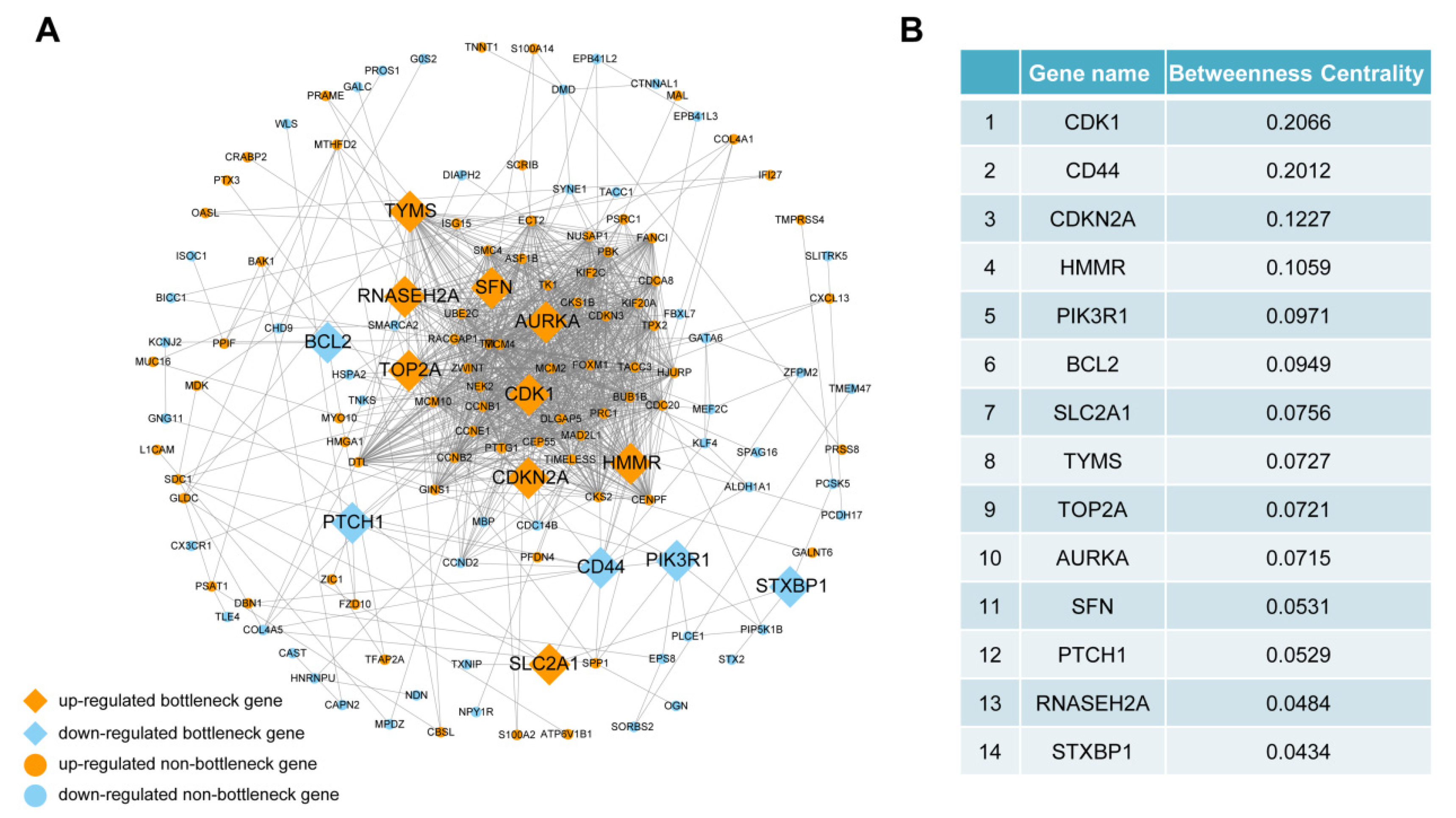
3.4. PPI Network Construction and Bottleneck Gene Identification
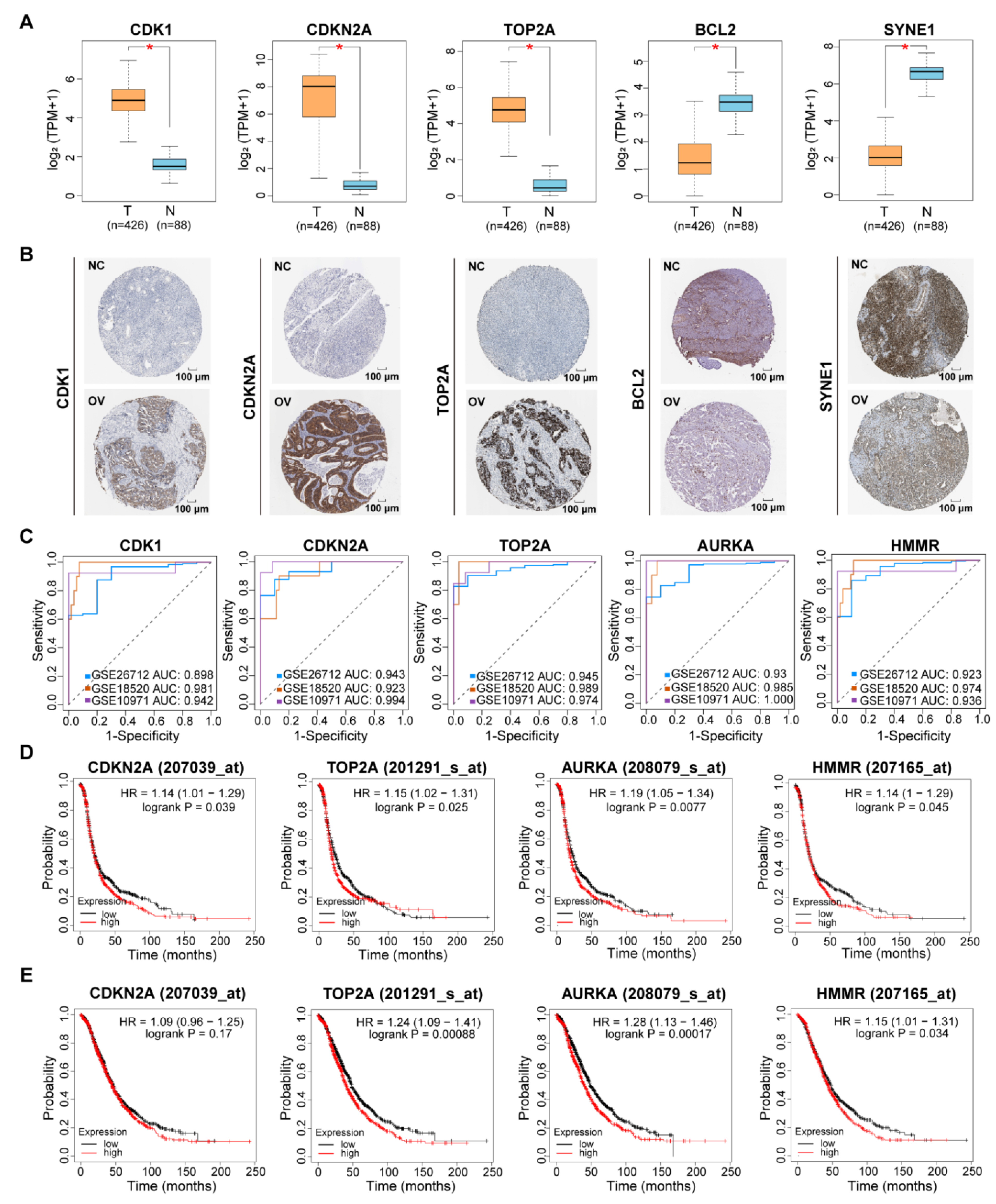
3.5. Functional Characterization of Bottleneck Genes for HGSC Diagnosis and Prognosis
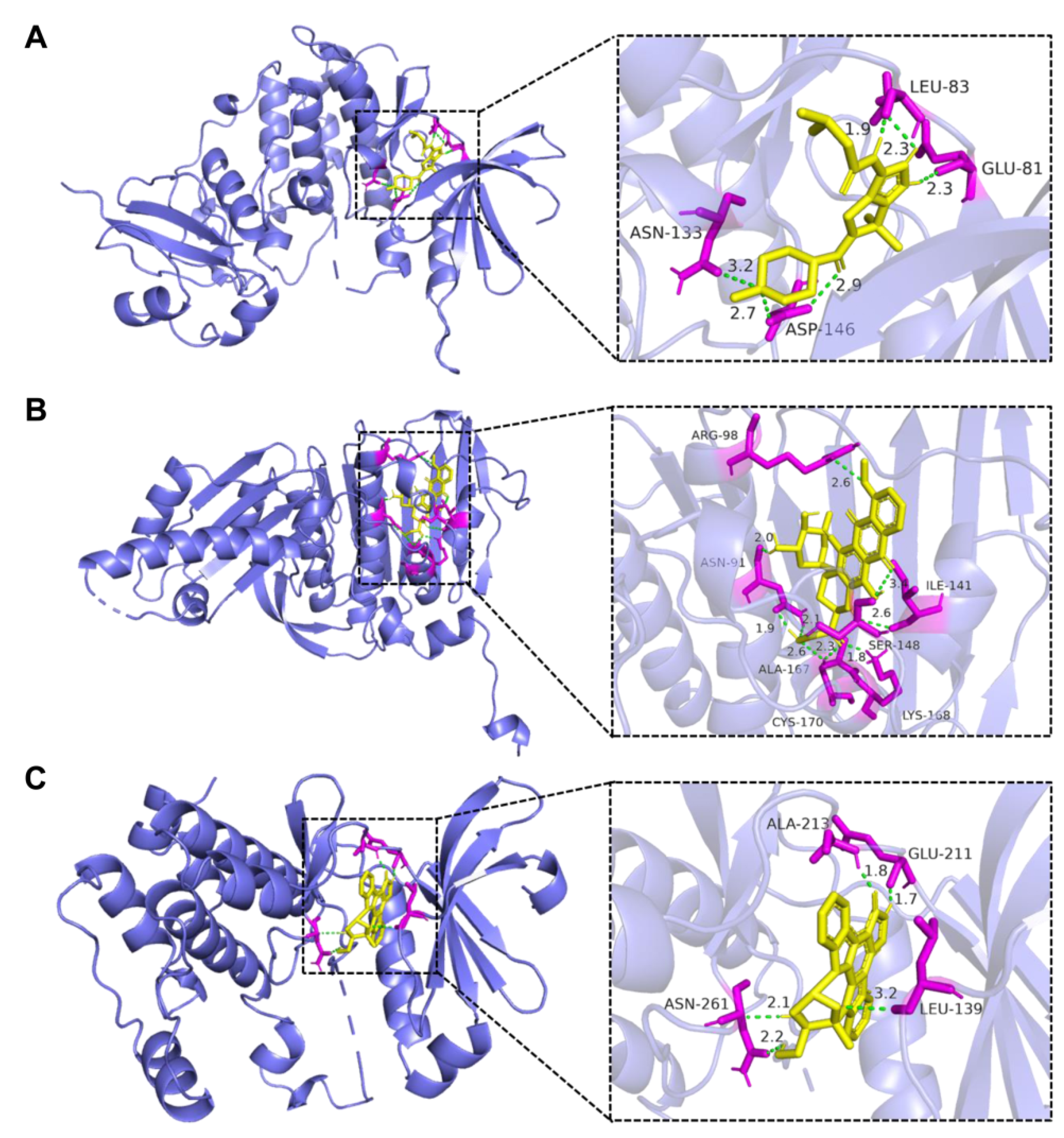
3.6. Interaction between Candidate Drug and Bottleneck Gene through Molecular Docking
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sah, S.; Ma, X.; Botros, A.; Gaul, D.A.; Yun, S.R.; Park, E.Y.; Kim, O.; Moore, S.G.; Kim, J.; Fernandez, F.M. Space- and Time-Resolved Metabolomics of a High-Grade Serous Ovarian Cancer Mouse Model. Cancers 2022, 14, 2262. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Pan, J.B.; Shah, P.; Ao, M.H.; Thomas, S.N.; Liu, Y.; Chen, L.J.; Schnaubelt, M.; Clark, D.J.; Rodriguez, H.; et al. Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 2020, 33, 108276. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2016, 17, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, W.; Lu, X.; Ma, S.; Dong, L.; Zou, B. Weighted gene co-expression network analysis and connectivity map identifies lovastatin as a treatment option of gastric cancer by inhibiting HDAC2. Gene 2019, 681, 15–25. [Google Scholar] [CrossRef]
- Zou, D.; Bai, J.; Lu, E.; Yang, C.; Liu, J.; Wen, Z.; Liu, X.; Jin, Z.; Xu, M.; Jiang, L. Identification of Novel Drug Candidate for Epithelial Ovarian Cancer via In Silico Investigation and In Vitro Validation. Front. Oncol. 2021, 11, 745590. [Google Scholar] [CrossRef]
- De Donato, M.; Babini, G.; Mozzetti, S.; Buttarelli, M.; Ciucci, A.; Arduini, G.; De Rosa, M.C.; Scambia, G.; Gallo, D. KLF7: A new candidate biomarker and therapeutic target for high-grade serous ovarian cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Huang, D.; Chowdhury, S.; Wang, H.; Savage, S.R.; Ivey, R.G.; Kennedy, J.J.; Whiteaker, J.R.; Lin, C.; Hou, X.; Oberg, A.L. Multiomic analysis identifies CPT1A as a potential therapeutic target in platinum-refractory, high-grade serous ovarian cancer. Cell Rep. Med. 2021, 2, 10047s1. [Google Scholar] [CrossRef]
- Robert, A.M.; Robert, C.S.; Robert, L. Symmetry breaking in biological systems. From molecules to tissues. Struct. Chem. 2007, 18, 899–907. [Google Scholar] [CrossRef]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Rezaei-Tavirani, M.; Goliaei, B.; Peyvandi, A.A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 2014, 7, 17. [Google Scholar] [PubMed]
- Athanasios, A.; Charalampos, V.; Vasileios, T. Protein-protein interaction (PPI) network: Recent advances in drug discovery. Curr. Drug Metab. 2017, 18, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Nithya, C.; Kiran, M.; Nagarajaram, H.A. Comparative analysis of Pure Hubs and Pure Bottlenecks in Human Protein-protein Interaction Networks. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bonome, T.; Levine, D.A.; Shih, J.; Randonovich, M.; Pise-Masison, C.A.; Bogomolniy, F.; Ozbun, L.; Brady, J.; Barrett, J.C.; Boyd, J. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008, 68, 5478–5486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, S.C.; Bonome, T.; Vathipadiekal, V.; Bell, A.; Johnson, M.E.; Park, D.-C.; Hao, K.; Yip, D.K.; Donninger, H.; Ozbun, L. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: Microfibril-associated glycoprotein 2. Cancer Cell 2009, 16, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tone, A.A.; Begley, H.; Sharma, M.; Murphy, J.; Rosen, B.; Brown, T.J.; Shaw, P.A. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin. Cancer Res. 2008, 14, 4067–4078. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. In Data Mining in Proteomics; Humana Press: Totowa, NJ, USA, 2011; pp. 291–303. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lánczky, A.; Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Qu, X.A.; Rajpal, D.K. Applications of Connectivity Map in drug discovery and development. Drug Discov. Today 2012, 17, 1289–1298. [Google Scholar] [CrossRef]
- Gallion, H.H.; Liu, P.; Alberts, D.E.; O’Toole, R.V.; O’Sullivan, J.; Mills, G.; Smith, H.O.; Hynes, H.E. Phase II trial of amonafide in previously treated patients with advanced ovarian cancer: A Southwest Oncology Group study. Gynecol. Oncol. 1992, 46, 230–232. [Google Scholar] [CrossRef]
- Ho, G.F. Palbociclib in the treatment of recurrent ovarian cancer. Gynecol. Oncol. Rep. 2020, 34, 100626. [Google Scholar]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Copeland, L.J.; Kavanagh, J.J.; Cangir, A.; Junco, G.D.; Saul, P.B.; Stringer, C.A.; Freedman, R.S.; Edwards, C.L.; Wharton, J.T. Treatment of malignant nondysgerminomatous germ cell tumors of the ovary with vincristine, dactinomycin, and cyclophosphamide. Cancer 1985, 56, 2756–2761. [Google Scholar] [CrossRef]
- Ueda, T.; Takai, N.; Nishida, M.; Nasu, K.; Narahara, H. Apicidin, a novel histone deacetylase inhibitor, has profound anti-growth activity in human endometrial and ovarian cancer cells. Int. J. Mol. Med. 2007, 19, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Sanchez-Martinez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Kathiravan, M.K.; Khilare, M.M.; Nikoomanesh, K.; Chothe, A.S.; Jain, K.S. Topoisomerase as target for antibacterial and anticancer drug discovery. J. Enzym. Inhib. Med. Chem. 2013, 28, 419–435. [Google Scholar] [CrossRef]
- Wu, B.; Yang, W.; Fu, Z.; Xie, H.; Guo, Z.; Liu, D.; Ge, J.; Zhong, S.; Liu, L.; Liu, J. Selected using bioinformatics and molecular docking analyses, PHA-793887 is effective against osteosarcoma. Aging 2021, 13, 16425. [Google Scholar] [CrossRef]
- Villanueva, P.J.; Gutierrez, D.A.; Contreras, L.; Parra, K.; Segura-Cabrera, A.; Varela-Ramirez, A.; Aguilera, R.J. The Antimalarial Drug Pyronaridine Inhibits Topoisomerase II in Breast Cancer Cells and Hinders Tumor Progression In Vivo. Clin. Cancer Drugs 2021, 8, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Kairalla, J.A.; Hilden, J.M.; Dreyer, Z.E.; Carroll, A.J.; Heerema, N.A.; Wang, C.; Devidas, M.; Gore, L.; Salzer, W.L. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children’s Oncology Group trial AALL0631. Leukemia 2021, 35, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Brasca, M.G.; Albanese, C.; Alzani, R.; Amici, R.; Avanzi, N.; Ballinari, D.; Bischoff, J.; Borghi, D.; Casale, E.; Croci, V. Optimization of 6, 6-dimethyl pyrrolo [3, 4-c] pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg. Med. Chem. 2010, 18, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; Earl, H.M.; Hiller, L.; Dunn, J.A.; Bathers, S.; Grieve, R.J.; Spooner, D.A.; Agrawal, R.K.; Fernando, I.N.; Brunt, A.M. Epirubicin and cyclophosphamide, methotrexate, and fluorouracil as adjuvant therapy for early breast cancer. N. Engl. J. Med. 2006, 355, 1851–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabbir, M.; Stuart, R. Lestaurtinib, a multitargeted tyrosinse kinase inhibitor: From bench to bedside. Expert Opin. Investig. Drugs 2010, 19, 427–436. [Google Scholar] [CrossRef]
- Minturn, J.E.; Evans, A.E.; Villablanca, J.G.; Yanik, G.A.; Park, J.R.; Shusterman, S.; Groshen, S.; Hellriegel, E.T.; Bensen-Kennedy, D.; Matthay, K.K. Phase I trial of lestaurtinib for children with refractory neuroblastoma: A new approaches to neuroblastoma therapy consortium study. Cancer Chemother. Pharmacol. 2011, 68, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165715. [Google Scholar]
- Gao, Y.; Zhao, H.; Ren, M.; Chen, Q.; Li, J.; Li, Z.; Yin, C.; Yue, W. TOP2A promotes tumorigenesis of high-grade serous ovarian cancer by regulating the TGF-β/Smad pathway. J. Cancer 2020, 11, 4181. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Wood, D.J.; Korolchuk, S.; Tatum, N.J.; Wang, L.Z.; Endicott, J.A.; Noble, M.E.M.; Martin, M.P. Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem. Biol. 2019, 26, 121–130.e5. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.Q.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Name | Score | Description |
|---|---|---|---|
| 1 | purvalanol-a | −99.75 | CDK inhibitor |
| 2 | aminopurvalanol-a | −99.72 | Tyrosine kinase inhibitor |
| 3 | mycophenolic-acid | −99.22 | Dehydrogenase inhibitor |
| 4 | Amonafide | −99.19 | Topoisomerase inhibitor |
| 5 | PI-103 | −98.87 | MTOR inhibitor |
| 6 | PI-828 | −98.85 | PI3K inhibitor |
| 7 | AS-605240 | −98.63 | PI3K inhibitor |
| 8 | AZD-8055 | −98.52 | MTOR inhibitor |
| 9 | Ellipticine | −98.48 | Topoisomerase inhibitor |
| 10 | Palbociclib | −98.41 | CDK inhibitor |
| 11 | KU-0060648 | −98.34 | DNA dependent protein kinase inhibitor |
| 12 | JAK3-inhibitor-VI | −98.27 | JAK inhibitor |
| 13 | VAMA-37 | −98.27 | DNA dependent protein kinase inhibitor |
| 14 | Vorinostat | −98.27 | HDAC inhibitor |
| 15 | Staurosporine | −98.2 | PKC inhibitor |
| 16 | CGP-60474 | −98.17 | CDK inhibitor |
| 17 | ZG-10 | −98.1 | JNK inhibitor |
| 18 | TG-101348 | −98.1 | FLT3 inhibitor |
| 19 | ISOX | −98.1 | HDAC inhibitor |
| 20 | PHA-793887 | −98.06 | CDK inhibitor |
| 21 | bisindolylmaleimide-ix | −98.06 | CDK inhibitor |
| 22 | Dactinomycin | −97.99 | RNA polymerase inhibitor |
| 23 | JNJ-7706621 | −97.96 | CDK inhibitor |
| 24 | PF-562271 | −97.89 | Focal adhesion kinase inhibitor |
| 25 | Wortmannin | −97.87 | PI3K inhibitor |
| 26 | Cerivastatin | −97.85 | HMGCR inhibitor |
| 27 | mycophenolate-mofetil | −97.79 | Dehydrogenase inhibitor |
| 28 | chromomycin-a3 | −97.74 | DNA binding agent |
| 29 | HG-5-113-01 | −97.74 | Protein kinase inhibitor |
| 30 | THM-I-94 | −97.74 | HDAC inhibitor |
| 31 | Apicidin | −97.64 | HDAC inhibitor |
| 32 | Lestaurtinib | −97.6 | FLT3 inhibitor |
| 33 | Pidorubicine | −97.6 | Topoisomerase inhibitor |
| 34 | Doxorubicin | −97.53 | Topoisomerase inhibitor |
| 35 | Dactolisib | −97.51 | MTOR inhibitor |
| 36 | Etoposide | −97.5 | Topoisomerase inhibitor |
| 37 | Mitoxantrone | −97.5 | Topoisomerase inhibitor |
| 38 | Dacinostat | −97.46 | HDAC inhibitor |
| 39 | Triptolide | −97.39 | RNA polymerase inhibitor |
| 40 | A-443644 | −97.32 | AKT inhibitor |
| 41 | verrucarin-a | −97.25 | Protein synthesis inhibitor |
| 42 | Scriptaid | −97.22 | HDAC inhibitor |
| 43 | Alisertib | −97.18 | Aurora kinase inhibitor |
| 44 | JNK-9L | −97.18 | JNK inhibitor |
| 45 | KU-0063794 | −97.14 | MTOR inhibitor |
| 46 | Alvocidib | −97.04 | CDK inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Zuo, J.; Shen, Y.; Yan, D.; Chen, J.; Qi, X. Identification of Novel Drugs Targeting Cell Cycle Regulators for the Treatment of High-Grade Serous Ovarian Cancer via Integrated Bioinformatics Analysis. Symmetry 2022, 14, 1403. https://doi.org/10.3390/sym14071403
Zhao Y, Zuo J, Shen Y, Yan D, Chen J, Qi X. Identification of Novel Drugs Targeting Cell Cycle Regulators for the Treatment of High-Grade Serous Ovarian Cancer via Integrated Bioinformatics Analysis. Symmetry. 2022; 14(7):1403. https://doi.org/10.3390/sym14071403
Chicago/Turabian StyleZhao, Yuanchun, Jiachen Zuo, Yiming Shen, Donghui Yan, Jiajia Chen, and Xin Qi. 2022. "Identification of Novel Drugs Targeting Cell Cycle Regulators for the Treatment of High-Grade Serous Ovarian Cancer via Integrated Bioinformatics Analysis" Symmetry 14, no. 7: 1403. https://doi.org/10.3390/sym14071403
APA StyleZhao, Y., Zuo, J., Shen, Y., Yan, D., Chen, J., & Qi, X. (2022). Identification of Novel Drugs Targeting Cell Cycle Regulators for the Treatment of High-Grade Serous Ovarian Cancer via Integrated Bioinformatics Analysis. Symmetry, 14(7), 1403. https://doi.org/10.3390/sym14071403