
Recent Deep Learning Methodology Development for RNA–RNA Interaction Prediction



Abstract
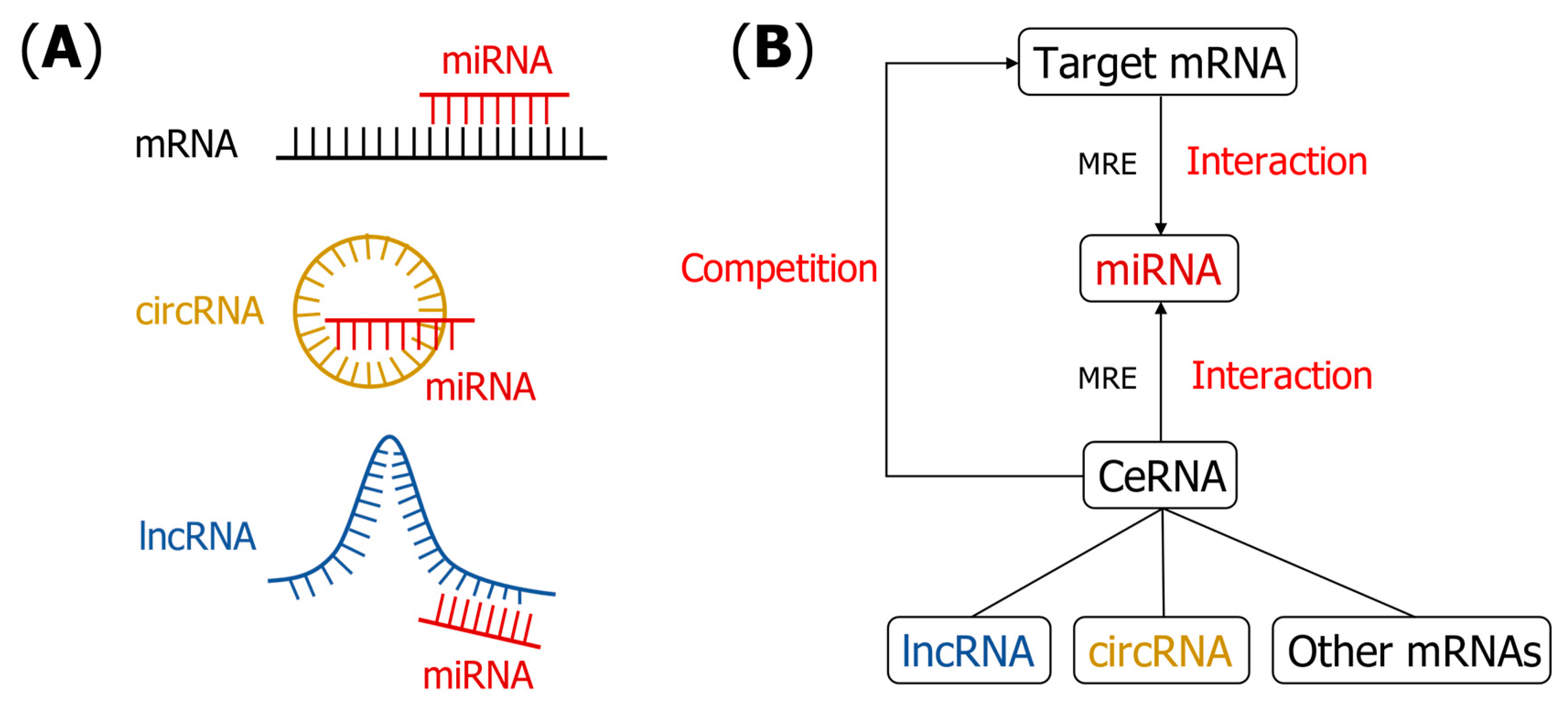
1. Introduction
2. Benchmark Datasets Used for Training Deep Models
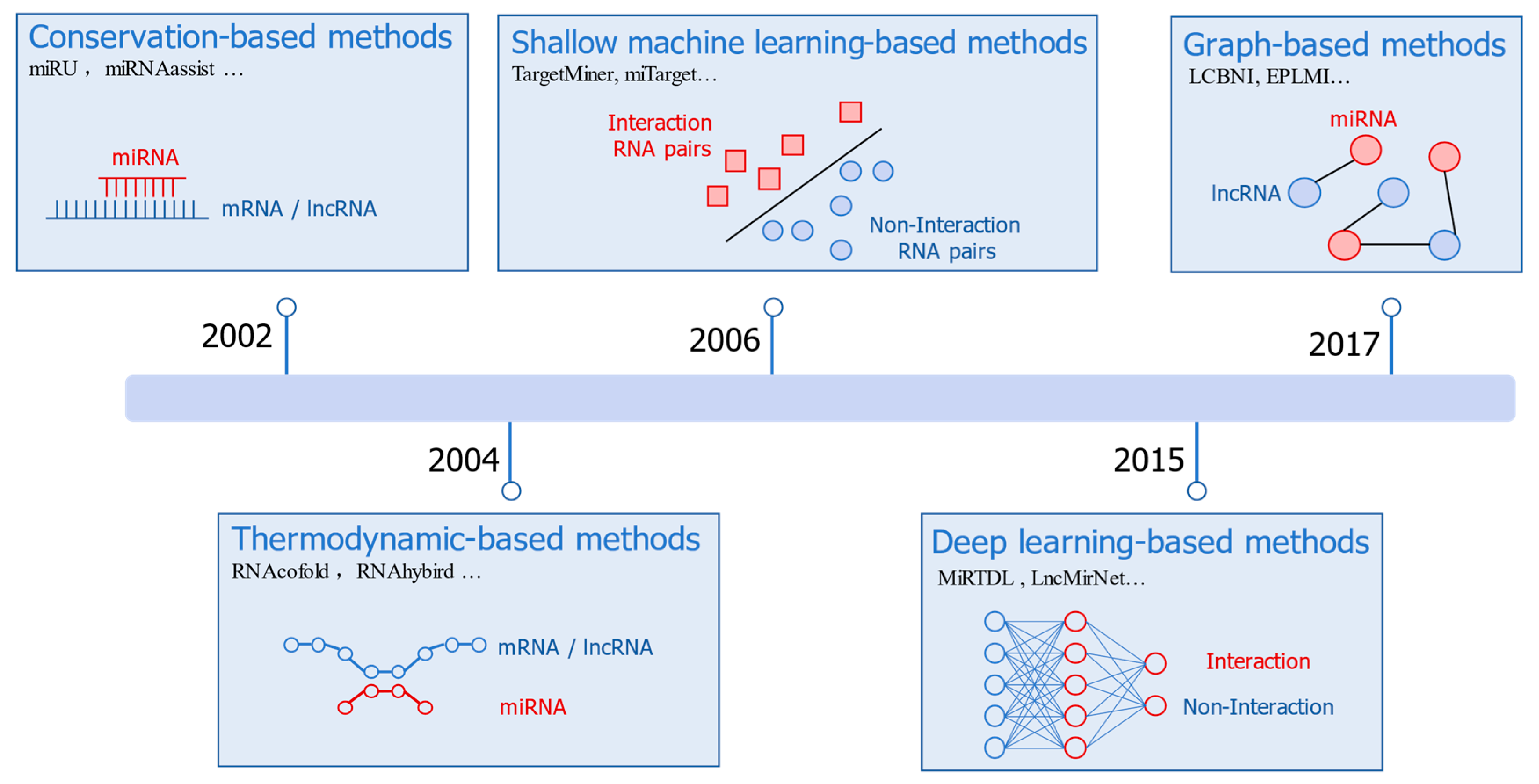
3. Deep-Learning-Based Methods for RRI Prediction
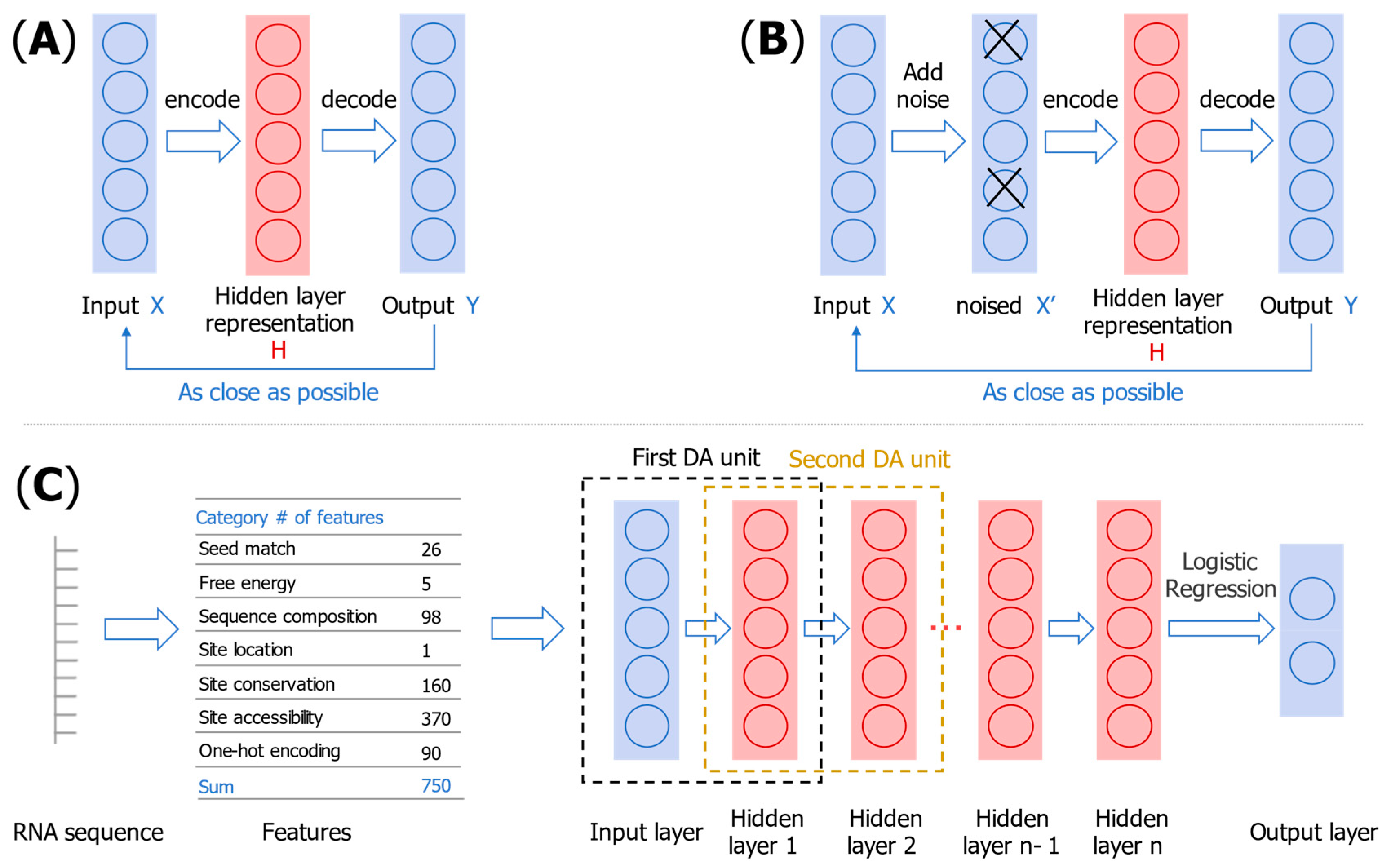
3.1. miRNA–mRNA Interaction Prediction
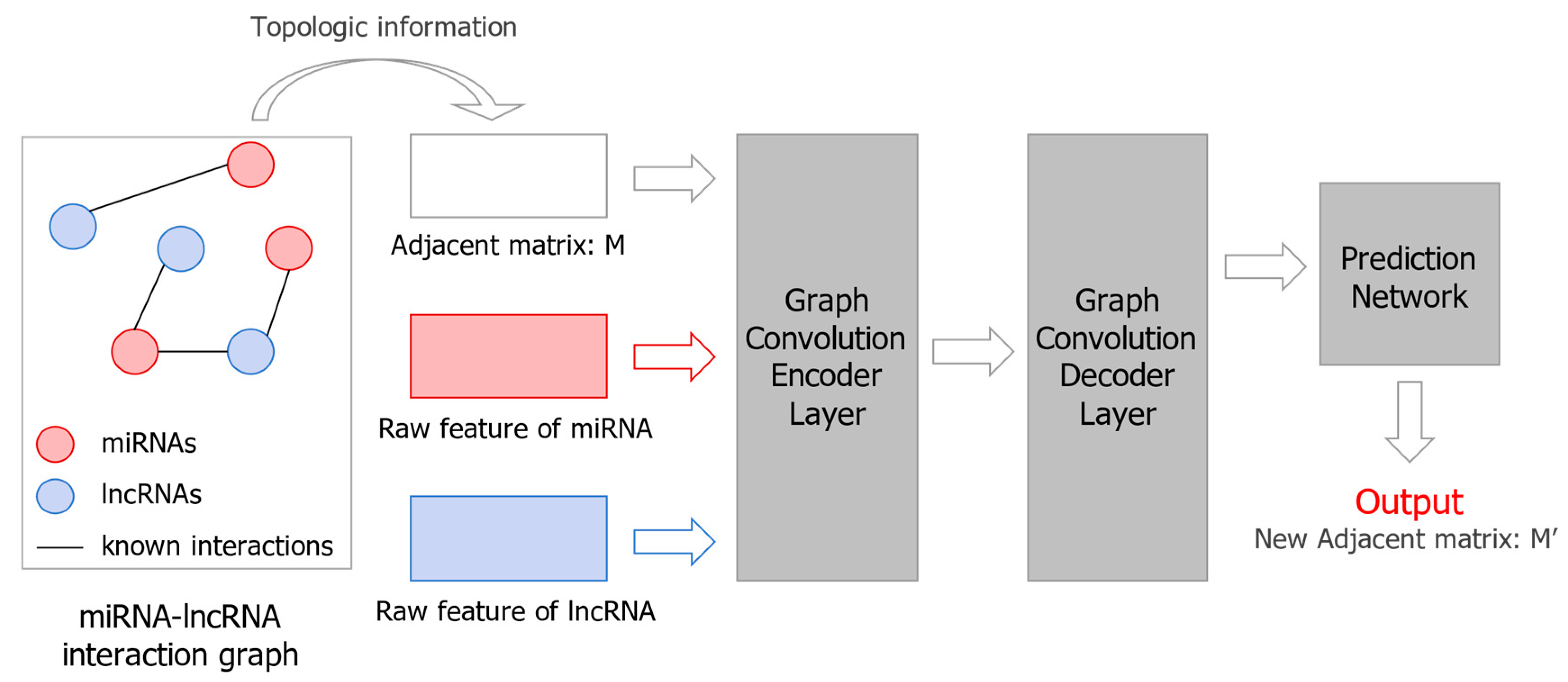
3.2. lncRNA–miRNA Interaction (LMI) Prediction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.J.; Tay, Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Greco, S.; Gorospe, M.; Martelli, F. Noncoding RNA in age-related cardiovascular diseases. J. Mol. Cell. Cardiol. 2015, 83, 142–155. [Google Scholar] [CrossRef]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microRNA targets. Nat. Struct. Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Pan, X.; Wenzel, A.; Jensen, L.J.; Gorodkin, J. Genome-wide identification of clusters of predicted microRNA binding sites as microRNA sponge candidates. PLoS ONE 2018, 13, e0202369. [Google Scholar] [CrossRef]
- Liang, M.; Hu, K.; He, C.; Zhou, J.; Liao, Y. Upregulated lncRNA Gm2044 inhibits male germ cell development by acting as miR-202 host gene. Anim. Cells Syst. 2019, 23, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic targeting of long non-coding RNAs in cancer. Trends Mol. Med. 2018, 24, 257–277. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Zahid, S.; Ashraf, M.A.B.; Qazi, M.H.; Asif, M.; Zaheer, A.; Arshad, M.; Raza, A.; Jamal, M.S. Non-coding RNAs in cancer diagnosis and therapy. Non-Coding RNA Res. 2016, 1, 69–76. [Google Scholar] [CrossRef]
- Leucci, E.; Patella, F.; Waage, J.; Holmstrøm, K.; Lindow, M.; Porse, B.; Kauppinen, S.; Lund, A.H. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013, 3, 2535. [Google Scholar] [CrossRef]
- Guo, G.; Liu, X.; Sun, F.; Cao, J.; Huo, N.; Wuda, B.; Xin, M.; Hu, Z.; Du, J.; Xia, R.; et al. Wheat miR9678 Affects Seed Germination by Generating Phased siRNAs and Modulating Abscisic Acid/Gibberellin Signaling. Plant Cell 2018, 30, 796–814. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell Mol Life Sci 2018, 75, 467–484. [Google Scholar] [CrossRef]
- Heinemann, U.; Roske, Y. Symmetry in Nucleic-Acid Double Helices. Symmetry 2020, 12, 737. [Google Scholar] [CrossRef]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Ascano, M.; Hafner, M.; Cekan, P.; Gerstberger, S.; Tuschl, T. Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 2012, 3, 159–177. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef]
- Xie, F.L.; Huang, S.Q.; Guo, K.; Xiang, A.L.; Zhu, Y.Y.; Nie, L.; Yang, Z.M. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007, 581, 1464–1474. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Mon. Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mitra, R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics 2009, 25, 2625–2631. [Google Scholar] [CrossRef]
- Kim, S.K.; Nam, J.W.; Rhee, J.K.; Lee, W.J.; Zhang, B.T. miTarget: microRNA target gene prediction using a support vector machine. BMC Bioinform. 2006, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Shuang, C.; Maozu, G.; Chunyu, W.; Xiaoyan, L.; Yang, L.; Xuejian, W. MiRTDL: A Deep Learning Approach for miRNA Target Prediction. IEEE ACM Trans. Comput. Biol. Bioinform. 2016, 13, 1161–1169. [Google Scholar]
- Yang, S.; Wang, Y.; Lin, Y.; Shao, D.; He, K.; Huang, L. LncMirNet: Predicting LncRNA–miRNA Interaction Based on Deep Learning of Ribonucleic Acid Sequences. Molecules 2020, 25, 4372. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhu, F.; Tian, G.; Wang, H. LCBNI: Link completion bipartite network inference for predicting new lncRNA-miRNA interactions. In Proceedings of the 2018 IEEE International Conference of Safety Produce Informatization (IICSPI), Miami, FL, USA, 10–12 April 2018; pp. 873–877. [Google Scholar]
- Huang, Y.-A.; Chan, K.C.; You, Z.-H. Constructing prediction models from expression profiles for large scale lncRNA–miRNA interaction profiling. Bioinformatics 2018, 34, 812–819. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef]
- Alkan, C.; Karakoç, E.; Nadeau, J.H.; Sahinalp, S.C.; Zhang, K. RNA-RNA interaction prediction and antisense RNA target search. J. Comput. Biol. 2006, 13, 267–282. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yada, T. miRNA-target prediction based on transcriptional regulation. BMC Genom. 2013, 14 (Suppl. 2), S3. [Google Scholar] [CrossRef]
- Marín, R.M.; Vanícek, J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011, 39, 19–29. [Google Scholar] [CrossRef]
- Jha, A.; Shankar, R. Employing machine learning for reliable miRNA target identification in plants. BMC Genom. 2011, 12, 636. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, J.; Ding, P. Inferring MicroRNA Targets Based on Restricted Boltzmann Machines. IEEE J. Biomed. Health Inform. 2019, 23, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guan, X.; Khan, M.T.; Xiong, Y.; Wei, D.Q. LMI-DForest: A deep forest model towards the prediction of lncRNA-miRNA interactions. Comput. Biol. Chem. 2020, 89, 107406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Deng, Y.; Liu, Q.; Ye, B.; Dai, Z.; Chen, Y.; Dai, X. Identifying Circular RNA and Predicting Its Regulatory Interactions by Machine Learning. Front. Genet. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Kalchbrenner, N.; Grefenstette, E.; Blunsom, P. A Convolutional Neural Network for Modelling Sentences. In Proceedings of the 52nd Annual Meeting of the Association for Computational Linguistics, Baltimore, MD, USA, 23–25 June 2014; Toutanova, K., Wu, H., Eds.; Assoc Computational Linguistics-Acl: Stroudsburg, PA, USA, 2014; Volume 1, pp. 655–665. [Google Scholar]
- Taigman, Y.; Yang, M.; Ranzato, M.; Wolf, L. DeepFace: Closing the Gap to Human-Level Performance in Face Verification. In Proceedings of the 2014 IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 23–28 June 2014; IEEE: New York, NY, USA; pp. 1701–1708. [Google Scholar]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.L.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Shen, H.-B. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 2018, 34, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2018; pp. 6410–6421. [Google Scholar]
- Lee, B.; Baek, J.; Park, S.; Yoon, S. deepTarget: End-to-end learning framework for microRNA target prediction using deep recurrent neural networks. In Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Seattle, WA, USA, 2–5 October 2016. [Google Scholar]
- Pla, A.; Zhong, X.; Rayner, S. miRAW: A deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. PLoS Comput. Biol. 2018, 14, e1006185. [Google Scholar] [CrossRef]
- Wen, M.; Cong, P.; Zhang, Z.; Lu, H.; Li, T. DeepMirTar: A deep-learning approach for predicting human miRNA targets. Bioinformatics 2018, 34, 3781–3787. [Google Scholar] [CrossRef]
- Huang, Y.A.; Huang, Z.A.; You, Z.H.; Zhu, Z.; Huang, W.Z.; Guo, J.X.; Yu, C.Q. Predicting lncRNA-miRNA Interaction via Graph Convolution Auto-Encoder. Front. Genet. 2019, 10, 758. [Google Scholar] [CrossRef]
- Zhang, P.; Meng, J.; Luan, Y.; Liu, C. Plant miRNA-lncRNA Interaction Prediction with the Ensemble of CNN and IndRNN. Interdiscip. Sci. 2020, 12, 82–89. [Google Scholar] [CrossRef]
- Kang, Q.; Meng, J.; Cui, J.; Luan, Y.; Chen, M. PmliPred: A method based on hybrid model and fuzzy decision for plant miRNA-lncRNA interaction prediction. Bioinformatics 2020, 36, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhao, X.; Barbazuk, W.B.; Lee, J.-H. miTAR: A hybrid deep learning-based approach for predicting miRNA targets. BMC Bioinform. 2021, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, B. Deep Learning-Based microRNA Target Prediction Using Experimental Negative Data. IEEE Access. 2020, 8, 197908–197916. [Google Scholar] [CrossRef]
- Zhang, Y.; Jia, C.; Kwoh, C.K. Predicting the interaction biomolecule types for lncRNA: An ensemble deep learning approach. Brief. Bioinform. 2020, 22, bbaa228. [Google Scholar] [CrossRef]
- Zhao, C.; Qiu, Y.; Zhou, S.; Liu, S.; Zhang, W.; Niu, Y. Graph embedding ensemble methods based on the heterogeneous network for lncRNA-miRNA interaction prediction. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Rabin, D.; Crothers, D. Analysis of RNA secondary structure by photochemical reversal of psoralen crosslinks. Nucleic Acids Res. 1979, 7, 689–703. [Google Scholar] [CrossRef][Green Version]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA–target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Liu, C.-J.; Gao, C.; Ma, Z.; Cong, R.; Zhang, Q.; Guo, A.-Y. lncRInter: A database of experimentally validated long non-coding RNA interaction. J. Genet. Genom. 2017, 44, 265–268. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. lncRNASNP2: An updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Shrestha, S.; Yang, C.-D.; Chang, N.-W.; Lin, Y.-L.; Liao, K.-W.; Huang, W.-C.; Sun, T.-H.; Tu, S.-J.; Lee, W.-H. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2. 0: A comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Oerlemans, A.; Lao, S.; Wu, S.; Lew, M.S. Deep learning for visual understanding: A review. Neurocomputing 2016, 187, 27–48. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Zheng, P.; Xu, S.-t.; Wu, X. Object detection with deep learning: A review. IEEE Trans. Neural Netw. Learn. Syst. 2019, 30, 3212–3232. [Google Scholar] [CrossRef]
- Mikolov, T.; Kombrink, S.; Burget, L.; Černocký, J.; Khudanpur, S. Extensions of recurrent neural network language model. In Proceedings of the 2011 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Prague, Czech Republic, 22–27 May 2011; pp. 5528–5531. [Google Scholar]
- Mikolov, T.; Karafiát, M.; Burget, L.; Černocký, J.; Khudanpur, S. Recurrent neural network based language model. In Proceedings of the Eleventh annual conference of the international speech communication association, Chiba, Japan, 26–30 September 2010. [Google Scholar]
- Wang, Y.; Yao, H.; Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing 2016, 184, 232–242. [Google Scholar] [CrossRef]
- Bengio, Y.; Yao, L.; Alain, G.; Vincent, P. Generalized denoising auto-encoders as generative models. arXiv 2013, arXiv:1305.6663. [Google Scholar]
- Du, B.; Xiong, W.; Wu, J.; Zhang, L.; Zhang, L.; Tao, D. Stacked convolutional denoising auto-encoders for feature representation. IEEE Trans. Cybern. 2016, 47, 1017–1027. [Google Scholar] [CrossRef]
- Lai, E.C. Predicting and validating microRNA targets. Genome Biol. 2004, 5, 1–6. [Google Scholar] [CrossRef][Green Version]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA–target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.h.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Wainberg, M.; Merico, D.; Delong, A.; Frey, B.J. Deep learning in biomedicine. Nat. Biotechnol. 2018, 36, 829–838. [Google Scholar] [CrossRef]
- Zheng, X.; Fu, X.; Wang, K.; Wang, M. Deep neural networks for human microRNA precursor detection. BMC Bioinform. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Pan, X.; Rijnbeek, P.; Yan, J.; Shen, H.-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genom. 2018, 19, 511. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. arXiv 2015, arXiv:1502.03167. [Google Scholar]
- Xuan, P.; Pan, S.; Zhang, T.; Liu, Y.; Sun, H. Graph convolutional network and convolutional neural network based method for predicting lncRNA-disease associations. Cells 2019, 8, 1012. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Wang, Z.; Huang, J.; Parthasarathy, S.; Moosavinasab, S.; Huang, Y.; Lin, S.M.; Zhang, W.; Zhang, P.; Sun, H. Graph embedding on biomedical networks: Methods, applications and evaluations. Bioinformatics 2020, 36, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Y.; Shen, H.B. Inferring Disease-Associated MicroRNAs Using Semi-supervised Multi-Label Graph Convolutional Networks. Iscience 2019, 20, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.H.; Baldwin, T. An empirical evaluation of doc2vec with practical insights into document embedding generation. arXiv 2016, arXiv:1607.05368. [Google Scholar]
- Ahmed, N.; Rossi, R.A.; Lee, J.; Willke, T.; Zhou, R.; Kong, X.; Eldardiry, H. Role-based Graph Embeddings. IEEE Trans. Knowl. Data Eng. 2020, 34, 2401–2415. [Google Scholar] [CrossRef]
- Miladi, M.; Montaseri, S.; Backofen, R.; Raden, M. Integration of accessibility data from structure probing into RNA-RNA interaction prediction. Bioinformatics 2019, 35, 2862–2864. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Huang, Y.-A.; Chan, K.C.; You, Z.-H.; Hu, P.; Wang, L.; Huang, Z.-A. Predicting microRNA–disease associations from lncRNA–microRNA interactions via Multiview Multitask Learning. Brief. Bioinform. 2021, 22, bbaa133. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, Y.; Peng, J.; Cheng, L. DeepLGP: A novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 2020, 36, 4466–4472. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA Type | Description | Length | Pairwise Interactions |
|---|---|---|---|
| mRNA | Carrier of genetic information | ~ nt | miRNA, lncRNA |
| miRNA | Micro non-coding RNA | about 22 nt | mRNA, lncRNA, circRNA |
| lncRNA | Long non-coding RNA | more than 200 nt | mRNA, miRNA |
| circRNA | RNA which forms a closed loop | more than 100 nt | miRNA |
| Methods | Characteristic | References |
|---|---|---|
| Conservation-based methods | Detecting complementary regions | miRU [26], miRNAassist [27] |
| Thermodynamic-based methods | Calculating the minimum free energy structure | RNAcofold [28], RNAhybird [29] |
| Shallow-machine-learning-based methods | Data-driven and feature extraction | TargetMiner [30], miTarget [31] |
| Deep-learning-based methods | Data-driven and learning the high-level discriminative features | MiRTDL [32], LncMirNet [33] |
| Graph-based methods | Network inference | LCBNI [34], EPLMI [35] |
| Name | Year | Method | Type | Website | Reference |
|---|---|---|---|---|---|
| MiRTDL | 2015 | CNN | miRNA–mRNA | [32] | |
| deepTarget | 2016 | RNN, AE | miRNA–mRNA | http://data.snu.ac.kr/pub/deepTarget/ | [51] |
| miRAW | 2018 | CNN | miRNA–mRNA | http://data.snu.ac.kr/pub/deepTarget/ | [52] |
| DeepMirTar | 2018 | SdAE | miRNA–mRNA | https://github.com/Bjoux2/DeepMirTar_SdA | [53] |
| GCLMI | 2019 | GCN, AE | lncRNA–miRNA | [54] | |
| CIRNN | 2019 | CNN, RNN | lncRNA–miRNA (Plant) | [55] | |
| PmliPred | 2020 | CNN, BiRNN | lncRNA–miRNA (Plant) | http://bis.zju.edu.cn/PmliPred/ | [56] |
| miTAR | 2020 | CNN, BiRNN | miRNA–mRNA | https://github.com/tjgu/miTAR | [57] |
| NONAME | 2020 | CNN | miRNA–mRNA | https://github.com/ailab-seoultech/deepTarget | [58] |
| LncMirNet | 2020 | CNN | lncRNA–miRNA | https://github.com/abcair/LncMirNet | [33] |
| lncIBTP | 2020 | CNN | lncRNA–RNA | https://drive.google.com/file/d/1w_2sthSYQXW3FfaF8YNgbu-hJUbHdzWp/view?usp=sharing | [59] |
| GEEL-FI | 2020 | DANN | lncRNA–miRNA | [60] |
| Name | Last Update | Type | URL | Reference |
|---|---|---|---|---|
| miRecords | 2013 | miRNA–mRNA | http://c1.accurascience.com/miRecords/ | [62] |
| ENCORI | 2014 | RNA–RNA | http://starbase.sysu.edu.cn/ | [63] |
| TarBase | 2017 | miRNA–mRNA | https://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex | [64] |
| lncRInter | 2017 | lncRNA–miRNA | http://bioinfo.life.hust.edu.cn/lncRInter/ | [65] |
| lncRNASNP2 | 2017 | lncRNA–miRNA | http://bioinfo.life.hust.edu.cn/lncRNASNP#!/mirna | [66] |
| miRTarBase | 2018 | lncRNA–miRNA | http://mirtarbase.cuhk.edu.cn/php/index.php | [67] |
| LncRNA2Target | 2018 | lncRNA-mRNA | http://123.59.132.21/lncrna2target/ | [68] |
| RNAInter | 2020 | RNA–RNA | http://www.rna-society.org/raid/ | [69] |
| Nucleotides Type | One-Hot Encoding | An Initial Embedding Representation |
|---|---|---|
| A | [1, 0, 0, 0] | [0.1, 0.3, 0.9, 0.5] |
| U | [0, 1, 0, 0] | [0.2, 0.7, −0.5,0.3] |
| C | [0, 0, 1, 0] | [−0.3, 0.5, 0.8, 0.6] |
| G | [0, 0, 0, 1] | [0.4, 0.1, −0.9, 0.7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Y.; Pan, X.; Shen, H.-B. Recent Deep Learning Methodology Development for RNA–RNA Interaction Prediction. Symmetry 2022, 14, 1302. https://doi.org/10.3390/sym14071302
Fang Y, Pan X, Shen H-B. Recent Deep Learning Methodology Development for RNA–RNA Interaction Prediction. Symmetry. 2022; 14(7):1302. https://doi.org/10.3390/sym14071302
Chicago/Turabian StyleFang, Yi, Xiaoyong Pan, and Hong-Bin Shen. 2022. "Recent Deep Learning Methodology Development for RNA–RNA Interaction Prediction" Symmetry 14, no. 7: 1302. https://doi.org/10.3390/sym14071302
APA StyleFang, Y., Pan, X., & Shen, H.-B. (2022). Recent Deep Learning Methodology Development for RNA–RNA Interaction Prediction. Symmetry, 14(7), 1302. https://doi.org/10.3390/sym14071302