Abstract
Genetic regulation of organisms involves complicated RNA–RNA interactions (RRIs) among messenger RNA (mRNA), microRNA (miRNA), and long non-coding RNA (lncRNA). Detecting RRIs is beneficial for discovering biological mechanisms as well as designing new drugs. In recent years, with more and more experimentally verified RNA–RNA interactions being deposited into databases, statistical machine learning, especially recent deep-learning-based automatic algorithms, have been widely applied to RRI prediction with remarkable success. This paper first gives a brief introduction to the traditional machine learning methods applied on RRI prediction and benchmark databases for training the models, and then provides a recent methodology overview of deep learning models in the prediction of microRNA (miRNA)–mRNA interactions and long non-coding RNA (lncRNA)–miRNA interactions.
1. Introduction
RNAs are important biological molecules, including messenger RNA (mRNA) and non-coding RNA (ncRNA) [1,2]. ncRNA has several types, such as microRNA (miRNA), circular RNA (circRNA), and long non-coding RNA (lncRNA). In the past decade, they have attracted widespread attention [3,4,5,6].
Generally speaking, mRNAs can be translated into proteins that maintain the life activities of organisms, and ncRNAs also play important roles in biological processes, such as genetic regulation, by interacting with other molecules, such as proteins or RNAs. The miRNAs are approximately 22-nucleotide (nt)-length ncRNAs, combining with partially complementary mRNA sequences, which are called miRNA response elements (MREs) [7], to regulate the translation process of mRNAs. circRNAs are usually closed loops formed by reverse splicing of pre-mRNA [8]. Previous studies have shown that some circRNAs could act as “miRNA sponges” (competitive inhibitors of miRNA) in the process of genetic regulation [9,10]. The lncRNAs are ncRNAs more than 200 nt in length. Some lncRNAs could act as precursors of miRNAs [11], and other lncRNAs could competitively bind to a miRNA and prevent it from binding to mRNAs or circRNAs [12,13]. Salmena et al. proposed a competing endogenous hypothesis [14], which interprets the interactions of mRNA, lncRNA, circRNA, and other ncRNAs with MRE as “communicate” interactions, and systematically explains the process of their mutual regulation, as illustrated in Figure 1. Recent studies have shown that some miRNAs and lncRNAs play crucial roles in cancer progression [15,16]; thus, predicting correlated RRIs provides a remarkable opportunity for targeted therapies for cancer. Additionally, there are some reported complicated interactions among RNAs. For example, miRNAs can directly target lncRNAs for certain biological functions, such as promoting the degradation of lncRNAs [17,18]. In addition, interactions exist among ncRNAs themselves, i.e., small nucleolar RNAs (snoRNAs) and ribosomal RNAs (rRNAs) [19]. The descriptions and pairwise interactions of the main RNA types involved in RRI are summarized in Table 1.
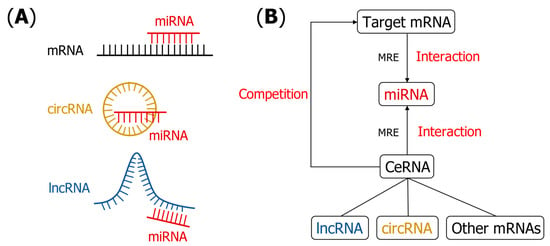
Figure 1.
(A) Schematic figure to show potential interaction patterns between miRNAs and other RNAs. (B) RNA–RNA interactions interpreted by ceRNA hypothesis. Target mRNA attracts the binding miRNAs through MRE. Other mRNAs, lncRNA and circRNA, competitively bind to miRNAs by MRE and block the miRNA–mRNA interaction. MRE plays the role of “words of communication” in the regulation process [14].

Table 1.
Descriptions and pairwise interactions of the main RNA types involved in RRI.
It is worth pointing out that RNA–RNA interaction (RRI) embodies a biological symmetry. Asymmetric single-stranded RNA molecules form local double-helical structures with helical symmetry [20] through complementary base pairing in their interactions, and these structural changes promote specific biological functions.
Although RRI can be determined with many different wet-lab experiments [21,22,23,24,25], they are time-consuming and costly in general. Bioinformatics-based models are a promising way to speed up the understanding of the functions of RNAs, whose predictions could provide useful clues and top-rated candidates for further experimental design and verification. The computational models can be applied in the following three fields at least: first, for a function unannotated RNA, its potential interacting partners in the RNA regulatory network could be mined with a large-scale screening manner from a large database, and thus its functions could be partially inferred from its annotated neighborhood; second, interaction motif patterns can be discovered with the automatic models, which are expected to be helpful to reversely infer the detailed biology mechanisms; third, the mined RRI knowledge as well as the interaction motif patterns could be further applied to design corresponding drugs to regulate the expression of disease-related genes.
In recent years, there have been many computational methods designed to predict the interactions between RNAs. These methods can be generally classified into three groups: traditional, conservation-based methods, data-driven, traditional, statistical-machine-learning-based methods, and recent deep-learning-based methods, as shown in Figure 2 and Table 2. From the perspective of the prediction purpose, these methods can be classified into two groups: site-level-based models and RNA-level-based models. The former focuses on predicting the specific binding sites on RNAs, while the latter only judges whether the input RNA pairs interact or not.
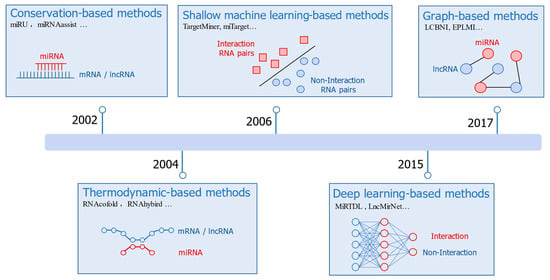
Figure 2.
A rough timeline for the development of RRI prediction methods.
Table 2.
The classification of recent computational RRI prediction methods.
For traditional, conservation-based methods, the initial and direct hypothesis about RRI prediction is to use sequence alignment tools such as BLAST [36]. If there is a complementary region in the two RNA sequences, then the two RNAs would be predicted potentially interacting with a high probability. This hypothesis would result in many false positives since complementary regions in RNA pairs will occur frequently, but not all of them will form true interactions. On the other hand, some interacting RNAs may only have partial complementarity between the two RNA sequences [37], which further increases the difficulty of the sequence-alignment-based approach.
Later methods interpret RNA interactions from the thermodynamic perspective by calculating the minimum free energy (MFE) structure of the RNA complex, which extends the RRI prediction problem to the generalization problem of the RNA secondary structure folding and prediction. RNAcofold [28] concatenated the two RNA sequences to calculate its secondary structure with MFE. RNAhybrid [29] calculated the MFE structure under a constraint of abandoning the interactions inside the two RNA molecules. Alkan et al. [38] hypothesized that the complex structure would be restricted with no internal pseudoknots, crossing interactions, or zigzags to avoid the prohibitive computational resources needed for interactions prediction between two long RNA molecules.
In addition to base pairing, conservation score [39] and site accessibility [40] are also introduced for predicting miRNA–mRNA interactions. Graph-based methods [34,35] are developed for predicting lncRNA–miRNA interactions, which predict the potential edges in lncRNA–miRNA interaction networks with lncRNAs and miRNAs as the nodes and the known interactions as the existing edges.
Data-driven models have witnessed rapid progress in recent years with more and more experimental data being deposited into the database. Machine-learning-based methods, e.g., support vector machines (SVMs) [30,31,41], are frequently used in early stages. Other different machine learning models have also been used. For instance, IMTRBM [42] applied restricted Boltzmann machines to predict miRNA–mRNA interactions. LMI-DForest [43] applied a deep forest model to predict lncRNA–miRNA interactions. The circMRT [44] is an ensemble-machine-learning-based method to predict the regulatory information of circRNAs. A common feature for these machine-learning-based methods is that they rely on hand-designed features; thus, expert knowledge is important for the model development. Since the molecular mechanism understanding on the RRI is far from complete, it is a challenging task to collect all the discriminative features.
This paper mainly focuses on the recent deep-learning-based methodology development in RRI prediction. A typical characteristic of deep-learning-based models is that they can better and automatically learn the high-level discriminative features from data. They have achieved remarkable results in many fields, such as natural language processing [45], face recognition [46], and computational biology [47]. Recently, deep-learning-based methods are applied in many aspects of molecular biology, including protein–protein interactions (PPIs) prediction [48], RNA-binding proteins (RBPs) identification [49], and drug design [50]. To date, there have been a few deep-learning-based methods proposed for RRI prediction, and some of them are summarized in Table 3. These existing methods can be classified into two groups according to their purposes, i.e., lncRNA–miRNA interaction prediction and miRNA–mRNA interaction prediction. To the best of our knowledge, deep-learning-based methods for predicting the interactions with circRNA or other ncRNAs are still very rare. A potential reason could be labeled training dataset sizes for them are small, which limits the development of deep models.
Table 3.
Some deep learning methods developed for RRI prediction tasks. All the websites were accessed before 1 February 2021.
2. Benchmark Datasets Used for Training Deep Models
With the development of high-throughput RNA sequencing and crosslinking methods such as HITS-CLIP [21], PAR-CLIP [22], PARIS [25], and CLASH [23], numerous RRI data can be obtained. For example, the PARIS method employed the psoralen-derivative 4′-aminomethyltrioxsalen [61] (AMT) to fix and crosslink RNA duplexes and specifically identified, base-paired RNA fragments through RNA purification. These RRI data can serve as training sets for machine learning and deep learning methods. Table 4 lists a part of the databases that deposit RRI data, which can be used to construct benchmark datasets for training machine-learning-based methods.
Table 4.
Some databases and their websites for depositing RRI data. All the websites were accessed before 1 February 2021.
This topic could be formulated as a binary class classification problem. Although there are many positive samples (interactions) derived from experimental data, there are much fewer experimentally verified negative samples (no interactions). Obviously, it would be unreasonable to treat all unknown pairs as negative samples. On the one hand, some pairs can form interactions, but current experiments have not yet uncovered these. On the other hand, from statistical and computational points of view, the dataset will be extremely imbalanced due to the huge number of negative samples generated in this way, which will mean that the model prefers to reflect the pattern of negative samples. Thus, generating negative samples in this binary class classification task is still a challenging problem.
In lncRNA–miRNA interaction prediction, some methods [33,54,60] use traditional prediction tools to predict the possibility of a random lncRNA and a random miRNA in the positive dataset and treat lncRNA–miRNA pairs with a low interaction possibility as negative samples. Other methods [55,56] divide lncRNAs into two groups according to existing experimental observations as to whether these lncRNAs interact with miRNAs. Then, a random lncRNA that does not interact with miRNAs is paired with a random miRNA, and this lncRNA–miRNA pair is added to the negative dataset. In miRNA–mRNA interaction prediction, a common approach is to randomly generate mock miRNAs which have different seed region sequences from observed miRNAs in databases, which then screen out mRNAs with a high interaction possibility by traditional prediction tools to pair with mock miRNAs [51,52,53,57]. Generating representative negative samples for training the model is still a challenging task worth investigating in future studies.
3. Deep-Learning-Based Methods for RRI Prediction
Deep-learning-based methods, also known as deep neural networks, are the extension of artificial neural networks. Data-driven deep learning models have been widely applied in analyzing the RNA sequences, including convolutional neural network (CNN), recurrent neural network (RNN), auto-encoder (AE), and graph convolution network (GCN) [32,33,51,52,53,54,55,56,57,59,60]. The basic idea can be divided into two steps: training a neural network and making prediction using the trained neural network. In the first step, the labeled RNA sequences are mapped to digital matrices by expert features, one-hot encoding, or embedding representation as the input of the neural networks. Then, the network is trained by continuously updating the parameters to minimize the predefined loss function. In the second step, the unlabeled RNA sequences can be labeled by the trained neural network.
Some CNN-based methods for RRI prediction include MiRTDL [53], miRAW [52], CIRNN [55], PmliPred [56], miTAR [57], LncMirNet [33], and lncIBTP [59], etc. CNN is a type of neural network that is invariant to shift and has two special layers: the convolutional layer and the subsampling layer (also called the pooling layer). The convolutional layer can extract feature maps from the input data, and the subsampling layer can remove redundant features from the feature maps [70,71]. CNN can extract the spatial features of the RNA sequences well.
Some RNN-based methods for RRI prediction include deepTarget [51], CIRNN [55], PmliPred [56], and miTAR [57], etc. RNN is a type of neural network widely used to process sequence data. It establishes connections between neurons in the same layer [72,73]. In sequence data, there exists connections between adjacent units, such as the connection between adjacent words in a sentence, and the connection between adjacent nucleotides in an RNA sequence. In an RRI prediction task, every two adjacent nucleotides in the RNA sequence would have some correlations and the order information can be captured well by RNN.
Some AE-based methods for RRI prediction include deepTarget [51], DeepMirTar [53], and GCLMI [54], etc. Auto-encoder is an unsupervised deep learning algorithm, which consists of two modules: an encoder and a decoder. The encoder module encodes the input data X to a hidden layer representation of H, and then the decoder module decodes H to the output data of Y [74]. The training objective of AE is to minimize the reconstruction loss between the output data, Y, and the input data, X, for denoising the AE [75,76]. Noise can be added to the AE to strengthen the generalization ability of the AE model by randomly forcing some neurons of the input layer to be zeros, which is called denoising the AE. The AE can be used to extract abstract features from the raw RNA sequences with the representations of the hidden layer.
To the best of our knowledge, there are only a few reported GCN-based methods proposed for RRI prediction, such as GCLMI [54]. GCN is an extension method of CNN in processing non-Euclidean data such as a graph, which is a powerful technique to deal with the network data, and is expected to be more developed for RRI network prediction tasks.
3.1. miRNA–mRNA Interaction Prediction
Many early studies [7,40,77,78,79] for miRNA–mRNA interactions made predictions on some specific binding modes, such as “Seed match”. The region from the 2nd nt to the 8th nt of the miRNA sequence is generally called “Seed”, and finding the complementary match in the 3′ UTR of mRNA is accordingly called “Seed match” [80,81]. With the development of more experiments, more interesting patterns have been revealed. For instance, some interacting miRNA–mRNA pairs would be beyond the predefined interaction mode [82,83], and many noninteracting miRNA–mRNA pairs could also be complementary. Thus, recent studies have gradually introduced more complicated knowledge features, including seed match type [79], conservation score [39], binding energy [28,29,38], and site accessibility [40,84] to enhance the model’s capability of catching different types of interaction patterns.
Deep-learning-based methods can automatically extract abstract features from the data, which will reduce the requirements of predefined expert features [85,86,87]. In 2016, the deep-learning-based method for miRNA–mRNA interaction prediction miRTDL [32] is proposed, which is based on expert features and CNNs. It first calculates three kinds of scores for each binding site: evolutionary conservation score, complementation score, and site accessibility score. Then, these features are concatenated as the input of CNN for classification. The CNN architecture in miRTDL consists of six layers, including an input layer, two convolutional layers, two subsampling layers, and a fully connected layer. Overall, miRTDL achieves promising high-precision scores, illustrating the power of deep learning models.
DeepMirTar [53] uses 750 features to describe miRNA–mRNA pairs, these features not only include expert features (seed match type and free energy), but also include one-hot-encoded features of the raw sequence, as illustrated in Table 5. DeepMirTar uses a stacked denoising auto-encoder (SdAE) model, which consists of multiple layers of denoising auto-encoder. This approach gradually performs unsupervised pretraining on denoising the autoencoder (DA) by minimizing the reconstruction loss. When all DAs are trained, the entire network is trained to minimize the negative log-likelihood loss. The model architecture of DeepMirTar is shown in Figure 3.
Table 5.
One-hot representation and an initial embedding representation of RNA nucleotides.
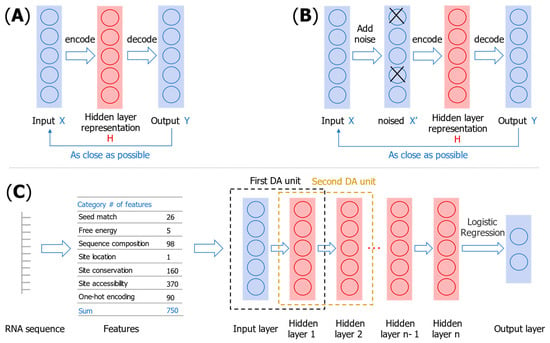
Figure 3.
Illustration of an auto-encoder (A), denoising the auto-encoder (B), and the structure of the DeepMirTar model (C).
The above two methods use deep neural networks to classify miRNA–mRNA pairs with expert features as input. DeepTarget [51] is an end-to-end miRNA–mRNA prediction method based on RNN and AE. It first pretrains two AEs as the encoders to obtain the hidden representations of miRNAs and mRNAs: and . Then, the learned and are concatenated to be the input of the next RNN layer. It uses learned embeddings for encoding sequences instead of the one-hot-encoded representation. One-hot encoding has the inevitable sparsity problem and could not reflect the internal correlation in the raw sequence. Specifically, for miRNA–mRNA interaction prediction task, there exists an internal correlation among the four nucleotides “AUCG”. “A” can pair with “U” to form a dihydrogen bond, and “C” can pair with “G” to form a triple hydrogen bond. Embedding representations are dense vectors, as shown in Table 5, correlation can be evaluated by the cosine similarity between two embedding vectors.
Another model of miRAW [52] applies AE and a deep ANN to identify miRNA–mRNA interactions. It first uses a 30 nts sliding window with a step of 5 nts to process the 3’ UTR of the mRNA sequence to achieve site-level prediction. Then, two traditional feature filters are used to select miRNA–mRNA pairs with a high binding potential. The Vienna RNACofold software [28] checks the stability, and the candidate site selection method (CSSM) checks whether the seed region meets the predefined criteria. The concatenated one-hot encoding of miRNA–mRNA pairs after two filters is fed into an eight-layer-deep ANN classifier. The first five layers are pretrained AEs, and the last the three layers are trained to make a classification from the learned features of the AEs.
A recent computational study on miRNA–mRNA interaction prediction based on deep learning, miTAR [57], combines the advantages of CNN and RNN. CNN learns the spatial features of miRNA–mRNA interaction, and RNN learns the sequential features of RNA sequences. The embedding representations of miRNAs and mRNAs pass through a convolutional layer, a maximum pooling layer, a bidirectional RNN layer, and two fully connection layers.
3.2. lncRNA–miRNA Interaction (LMI) Prediction
Different from the interactions between miRNA–mRNA, the interactions between lncRNAs and miRNAs are more complicated and could be roughly divided into three groups. (1) lncRNAs can act as precursors of miRNAs [11]. (2) lncRNAs can competitively bind to miRNAs and act as “miRNA sponges” [12] to regulate the interactions between miRNA and mRNA, as proposed in the ceRNA hypothesis [14]. (3) miRNA can directly target lncRNA for certain biological functions, such as promoting the degradation of lncRNA [17,18].
Recently, deep-learning-based methods have been applied to predict LMI, bringing a promising improvement compared with traditional methods. CIRNN [55] and PmliPred [56] are two deep-learning-based methods for plant LMI. Both methods use a hybrid CNN and RNN. A batch normalization (BN) layer is added in the PmliPred model. In deep neural networks, as the number of layers increases, and the data distribution of the subsequent layers will change, resulting in a decline in the generalization ability. Adding BN layers can help make the data distribution of the subsequent layer more stable, speed up the training process, and prevent model overfitting [88].
In addition to the above methods based on learned representations from RNA sequences, there is also a type of graph-representation-learning-based methods that can be used to predict LMI. Graph representation learning is a powerful method for learning and processing graph data. In the biological field, there are numerous graph data, such as protein interaction networks, drug molecular, and gene–disease networks. Graph neural network (GNN)-based methods are attracting widespread attention in the bioinformatics field [50,89,90,91]. For LMI tasks, it first constructs a heterogeneous graph, where the nodes are miRNAs and lncRNAs, and the edges are known LMI. Then, the LMI prediction will be transformed into finding the potential edges.
GCLMI [54] is an LMI prediction method based on graph convolution network (GCN) and AE. It consists of a graph convolution encoder and a graph convolution decoder. The adjacency matrix of the lncRNA–miRNA interaction network (a sparse network) and the feature matrix of the node (the feature information of lncRNAs and miRNAs) are fed into the encoder to learn a graph embedding representation. The decoder reconstructs the graph and produces a new adjacency matrix M’. In M’, there is a weight for each lncRNA–miRNA pair representing the probability of the interaction between them. The model architecture of GCLMI is shown in Figure 4.
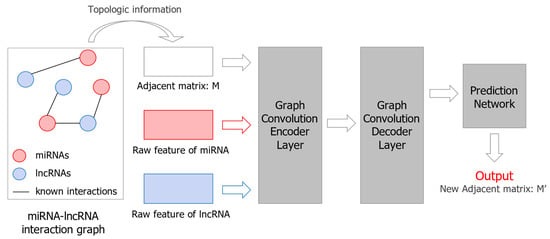
Figure 4.
Illustration of GCLMI model structure for LMI prediction.
Another GNN-based method, GEEL-FI [60], is based on graph embeddings and deep attention neural networks. It first learns the graph embeddings from the lncRNA–miRNA interaction network by an ensemble method, which consists of five different graph embedding methods. Then, the graph embeddings are fed into a deep attention network for making classifications. The attention mechanism is introduced to assign different weights to these five different graph embeddings, avoiding redundant information caused by simply concatenating them.
The LncMirNet [33] method combines graph embeddings with CNN. First, it extracts two expert features k-mer compositions and the composition/transition/distribution (CTD) feature, and learned embeddings of RNA sequences by doc2vec [92]. In addition, LncMirNet extracts graph embedding representations of lncRNAs and miRNAs by role2vec [93], which is a node-level graph embedding method that learns a representation of each node rather than the whole graph. This graph is derived from the lncRNA–lncRNA similarity network and the miRNA–miRNA similarity network instead of the lncRNA–miRNA interaction network. The lncRNA–lncRNA similarity network is constructed by forming an edge between a lncRNA and its top 15 similar lncRNAs, k-mer, CTD, and doc2vec features are used as the feature matrix of the nodes. Then, the above four features are reshaped to be input to a CNN model for prediction. All these methods have indicated a promising application of the graph representation and learning for the challenging LMI prediction task.
4. Discussion
Most deep-learning-based methods [51,52,55,56,57] use different RNA sequence encoding (one-hot encoding or embeddings) as the model input. Some methods [32,33,52,53,56] combine domain features such as seed match and site accessibility. Other methods [33,54,60] construct RNA similarity network to extract features for each node, or directly use the RRI network for learning graph embeddings. These current deep-learning-based methods not only have their own characteristics, but also combine the advantages of traditional methods, so that they outperform the traditional methods overall. Although the deep-learning-based methods are proven to achieve remarkable success in RRI prediction, their limitations should be considered. The deep-learning-based methods have high data dependency, which requires RRI data with high quality. Besides, the complexity and numerous parameters of the deep neural networks make them hard to interpret the prediction results.
In the future, more efforts can be devoted. In terms of the training dataset, generating reasonable negative samples with diversity for training is a difficult problem that remains for further study. In organisms, there are far fewer positive samples (interactions) than negative samples (no interactions): that is, it would form an imbalanced dataset. Thus, collecting representative negative samples which cover the real situation as much as possible, while being balanced with the positive samples to enable the statistical model is not biased from the huge negative data, is a future direction.
In terms of the discriminative features, classical thermodynamic free-energy features can be more explored in the deep-learning-based methods. A recent study [94] proposes that SHAPE data [95] can be applied to RRI prediction; SHAPE data is a type of RNA structure data at the single nucleotide level, and it can accurately obtain the contribution of each nucleotide to the overall free energy. It is expected to achieve better effectiveness if the free-energy information of the single nucleotide level is integrated into deep learning for RRI predictions.
Besides performing the predictions, mining more interpretable binding sequence motifs is another future direction. Binding motifs can provide biological interpretability as well as extra information in RRI prediction, and detecting potential binding motifs can be a significant aspect to be considered in future RRI prediction methods [49].
Some recent studies investigate biological mechanisms using RRI data, which also highlight a future application of the data-driven machine learning models. For instance, MVMTMDA [96] infers microRNA-disease associations (MDA) from lncRNA–microRNA interactions by multi-view, multi-task learning. DeepLGP [97] applies LMI data to infer the target genes of lncRNAs. It encodes lncRNAs and genes as vectors through their interactions with miRNAs, and infer the potential correlation between lncRNA and genes by measuring the cosine distance of the vectors.
5. Conclusions
Deep-learning-based methods have been applied in predicting RNA–RNA interactions with remarkable performance. This paper introduces an overview of the application of deep learning methods in predicting miRNA–mRNA and lncRNA–miRNA interactions. We expect that this paper will provide a useful resource and guide for future RRI prediction studies.
Author Contributions
Conceptualization, X.P. and H.-B.S.; methodology, Y.F.; writing—original draft preparation, Y.F.; writing—review and editing, X.P. and H.-B.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2020AAA0107600), the National Natural Science Foundation of China (No. 61725302, 62073219, 61903248, 61972251), and the Science and Technology Commission of Shanghai Municipality (20S11902100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used to support this study are included within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.J.; Tay, Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Greco, S.; Gorospe, M.; Martelli, F. Noncoding RNA in age-related cardiovascular diseases. J. Mol. Cell. Cardiol. 2015, 83, 142–155. [Google Scholar] [CrossRef]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microRNA targets. Nat. Struct. Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Pan, X.; Wenzel, A.; Jensen, L.J.; Gorodkin, J. Genome-wide identification of clusters of predicted microRNA binding sites as microRNA sponge candidates. PLoS ONE 2018, 13, e0202369. [Google Scholar] [CrossRef]
- Liang, M.; Hu, K.; He, C.; Zhou, J.; Liao, Y. Upregulated lncRNA Gm2044 inhibits male germ cell development by acting as miR-202 host gene. Anim. Cells Syst. 2019, 23, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic targeting of long non-coding RNAs in cancer. Trends Mol. Med. 2018, 24, 257–277. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Zahid, S.; Ashraf, M.A.B.; Qazi, M.H.; Asif, M.; Zaheer, A.; Arshad, M.; Raza, A.; Jamal, M.S. Non-coding RNAs in cancer diagnosis and therapy. Non-Coding RNA Res. 2016, 1, 69–76. [Google Scholar] [CrossRef]
- Leucci, E.; Patella, F.; Waage, J.; Holmstrøm, K.; Lindow, M.; Porse, B.; Kauppinen, S.; Lund, A.H. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013, 3, 2535. [Google Scholar] [CrossRef]
- Guo, G.; Liu, X.; Sun, F.; Cao, J.; Huo, N.; Wuda, B.; Xin, M.; Hu, Z.; Du, J.; Xia, R.; et al. Wheat miR9678 Affects Seed Germination by Generating Phased siRNAs and Modulating Abscisic Acid/Gibberellin Signaling. Plant Cell 2018, 30, 796–814. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell Mol Life Sci 2018, 75, 467–484. [Google Scholar] [CrossRef]
- Heinemann, U.; Roske, Y. Symmetry in Nucleic-Acid Double Helices. Symmetry 2020, 12, 737. [Google Scholar] [CrossRef]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Ascano, M.; Hafner, M.; Cekan, P.; Gerstberger, S.; Tuschl, T. Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 2012, 3, 159–177. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef]
- Xie, F.L.; Huang, S.Q.; Guo, K.; Xiang, A.L.; Zhu, Y.Y.; Nie, L.; Yang, Z.M. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007, 581, 1464–1474. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Mon. Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mitra, R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics 2009, 25, 2625–2631. [Google Scholar] [CrossRef]
- Kim, S.K.; Nam, J.W.; Rhee, J.K.; Lee, W.J.; Zhang, B.T. miTarget: microRNA target gene prediction using a support vector machine. BMC Bioinform. 2006, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Shuang, C.; Maozu, G.; Chunyu, W.; Xiaoyan, L.; Yang, L.; Xuejian, W. MiRTDL: A Deep Learning Approach for miRNA Target Prediction. IEEE ACM Trans. Comput. Biol. Bioinform. 2016, 13, 1161–1169. [Google Scholar]
- Yang, S.; Wang, Y.; Lin, Y.; Shao, D.; He, K.; Huang, L. LncMirNet: Predicting LncRNA–miRNA Interaction Based on Deep Learning of Ribonucleic Acid Sequences. Molecules 2020, 25, 4372. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhu, F.; Tian, G.; Wang, H. LCBNI: Link completion bipartite network inference for predicting new lncRNA-miRNA interactions. In Proceedings of the 2018 IEEE International Conference of Safety Produce Informatization (IICSPI), Miami, FL, USA, 10–12 April 2018; pp. 873–877. [Google Scholar]
- Huang, Y.-A.; Chan, K.C.; You, Z.-H. Constructing prediction models from expression profiles for large scale lncRNA–miRNA interaction profiling. Bioinformatics 2018, 34, 812–819. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef]
- Alkan, C.; Karakoç, E.; Nadeau, J.H.; Sahinalp, S.C.; Zhang, K. RNA-RNA interaction prediction and antisense RNA target search. J. Comput. Biol. 2006, 13, 267–282. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yada, T. miRNA-target prediction based on transcriptional regulation. BMC Genom. 2013, 14 (Suppl. 2), S3. [Google Scholar] [CrossRef]
- Marín, R.M.; Vanícek, J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011, 39, 19–29. [Google Scholar] [CrossRef]
- Jha, A.; Shankar, R. Employing machine learning for reliable miRNA target identification in plants. BMC Genom. 2011, 12, 636. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, J.; Ding, P. Inferring MicroRNA Targets Based on Restricted Boltzmann Machines. IEEE J. Biomed. Health Inform. 2019, 23, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guan, X.; Khan, M.T.; Xiong, Y.; Wei, D.Q. LMI-DForest: A deep forest model towards the prediction of lncRNA-miRNA interactions. Comput. Biol. Chem. 2020, 89, 107406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Deng, Y.; Liu, Q.; Ye, B.; Dai, Z.; Chen, Y.; Dai, X. Identifying Circular RNA and Predicting Its Regulatory Interactions by Machine Learning. Front. Genet. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Kalchbrenner, N.; Grefenstette, E.; Blunsom, P. A Convolutional Neural Network for Modelling Sentences. In Proceedings of the 52nd Annual Meeting of the Association for Computational Linguistics, Baltimore, MD, USA, 23–25 June 2014; Toutanova, K., Wu, H., Eds.; Assoc Computational Linguistics-Acl: Stroudsburg, PA, USA, 2014; Volume 1, pp. 655–665. [Google Scholar]
- Taigman, Y.; Yang, M.; Ranzato, M.; Wolf, L. DeepFace: Closing the Gap to Human-Level Performance in Face Verification. In Proceedings of the 2014 IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 23–28 June 2014; IEEE: New York, NY, USA; pp. 1701–1708. [Google Scholar]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.L.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Shen, H.-B. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 2018, 34, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2018; pp. 6410–6421. [Google Scholar]
- Lee, B.; Baek, J.; Park, S.; Yoon, S. deepTarget: End-to-end learning framework for microRNA target prediction using deep recurrent neural networks. In Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Seattle, WA, USA, 2–5 October 2016. [Google Scholar]
- Pla, A.; Zhong, X.; Rayner, S. miRAW: A deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. PLoS Comput. Biol. 2018, 14, e1006185. [Google Scholar] [CrossRef]
- Wen, M.; Cong, P.; Zhang, Z.; Lu, H.; Li, T. DeepMirTar: A deep-learning approach for predicting human miRNA targets. Bioinformatics 2018, 34, 3781–3787. [Google Scholar] [CrossRef]
- Huang, Y.A.; Huang, Z.A.; You, Z.H.; Zhu, Z.; Huang, W.Z.; Guo, J.X.; Yu, C.Q. Predicting lncRNA-miRNA Interaction via Graph Convolution Auto-Encoder. Front. Genet. 2019, 10, 758. [Google Scholar] [CrossRef]
- Zhang, P.; Meng, J.; Luan, Y.; Liu, C. Plant miRNA-lncRNA Interaction Prediction with the Ensemble of CNN and IndRNN. Interdiscip. Sci. 2020, 12, 82–89. [Google Scholar] [CrossRef]
- Kang, Q.; Meng, J.; Cui, J.; Luan, Y.; Chen, M. PmliPred: A method based on hybrid model and fuzzy decision for plant miRNA-lncRNA interaction prediction. Bioinformatics 2020, 36, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhao, X.; Barbazuk, W.B.; Lee, J.-H. miTAR: A hybrid deep learning-based approach for predicting miRNA targets. BMC Bioinform. 2021, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, B. Deep Learning-Based microRNA Target Prediction Using Experimental Negative Data. IEEE Access. 2020, 8, 197908–197916. [Google Scholar] [CrossRef]
- Zhang, Y.; Jia, C.; Kwoh, C.K. Predicting the interaction biomolecule types for lncRNA: An ensemble deep learning approach. Brief. Bioinform. 2020, 22, bbaa228. [Google Scholar] [CrossRef]
- Zhao, C.; Qiu, Y.; Zhou, S.; Liu, S.; Zhang, W.; Niu, Y. Graph embedding ensemble methods based on the heterogeneous network for lncRNA-miRNA interaction prediction. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Rabin, D.; Crothers, D. Analysis of RNA secondary structure by photochemical reversal of psoralen crosslinks. Nucleic Acids Res. 1979, 7, 689–703. [Google Scholar] [CrossRef][Green Version]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA–target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Liu, C.-J.; Gao, C.; Ma, Z.; Cong, R.; Zhang, Q.; Guo, A.-Y. lncRInter: A database of experimentally validated long non-coding RNA interaction. J. Genet. Genom. 2017, 44, 265–268. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. lncRNASNP2: An updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Shrestha, S.; Yang, C.-D.; Chang, N.-W.; Lin, Y.-L.; Liao, K.-W.; Huang, W.-C.; Sun, T.-H.; Tu, S.-J.; Lee, W.-H. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2. 0: A comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Oerlemans, A.; Lao, S.; Wu, S.; Lew, M.S. Deep learning for visual understanding: A review. Neurocomputing 2016, 187, 27–48. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Zheng, P.; Xu, S.-t.; Wu, X. Object detection with deep learning: A review. IEEE Trans. Neural Netw. Learn. Syst. 2019, 30, 3212–3232. [Google Scholar] [CrossRef]
- Mikolov, T.; Kombrink, S.; Burget, L.; Černocký, J.; Khudanpur, S. Extensions of recurrent neural network language model. In Proceedings of the 2011 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Prague, Czech Republic, 22–27 May 2011; pp. 5528–5531. [Google Scholar]
- Mikolov, T.; Karafiát, M.; Burget, L.; Černocký, J.; Khudanpur, S. Recurrent neural network based language model. In Proceedings of the Eleventh annual conference of the international speech communication association, Chiba, Japan, 26–30 September 2010. [Google Scholar]
- Wang, Y.; Yao, H.; Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing 2016, 184, 232–242. [Google Scholar] [CrossRef]
- Bengio, Y.; Yao, L.; Alain, G.; Vincent, P. Generalized denoising auto-encoders as generative models. arXiv 2013, arXiv:1305.6663. [Google Scholar]
- Du, B.; Xiong, W.; Wu, J.; Zhang, L.; Zhang, L.; Tao, D. Stacked convolutional denoising auto-encoders for feature representation. IEEE Trans. Cybern. 2016, 47, 1017–1027. [Google Scholar] [CrossRef]
- Lai, E.C. Predicting and validating microRNA targets. Genome Biol. 2004, 5, 1–6. [Google Scholar] [CrossRef][Green Version]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA–target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.h.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Wainberg, M.; Merico, D.; Delong, A.; Frey, B.J. Deep learning in biomedicine. Nat. Biotechnol. 2018, 36, 829–838. [Google Scholar] [CrossRef]
- Zheng, X.; Fu, X.; Wang, K.; Wang, M. Deep neural networks for human microRNA precursor detection. BMC Bioinform. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Pan, X.; Rijnbeek, P.; Yan, J.; Shen, H.-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genom. 2018, 19, 511. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. arXiv 2015, arXiv:1502.03167. [Google Scholar]
- Xuan, P.; Pan, S.; Zhang, T.; Liu, Y.; Sun, H. Graph convolutional network and convolutional neural network based method for predicting lncRNA-disease associations. Cells 2019, 8, 1012. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Wang, Z.; Huang, J.; Parthasarathy, S.; Moosavinasab, S.; Huang, Y.; Lin, S.M.; Zhang, W.; Zhang, P.; Sun, H. Graph embedding on biomedical networks: Methods, applications and evaluations. Bioinformatics 2020, 36, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Y.; Shen, H.B. Inferring Disease-Associated MicroRNAs Using Semi-supervised Multi-Label Graph Convolutional Networks. Iscience 2019, 20, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.H.; Baldwin, T. An empirical evaluation of doc2vec with practical insights into document embedding generation. arXiv 2016, arXiv:1607.05368. [Google Scholar]
- Ahmed, N.; Rossi, R.A.; Lee, J.; Willke, T.; Zhou, R.; Kong, X.; Eldardiry, H. Role-based Graph Embeddings. IEEE Trans. Knowl. Data Eng. 2020, 34, 2401–2415. [Google Scholar] [CrossRef]
- Miladi, M.; Montaseri, S.; Backofen, R.; Raden, M. Integration of accessibility data from structure probing into RNA-RNA interaction prediction. Bioinformatics 2019, 35, 2862–2864. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Huang, Y.-A.; Chan, K.C.; You, Z.-H.; Hu, P.; Wang, L.; Huang, Z.-A. Predicting microRNA–disease associations from lncRNA–microRNA interactions via Multiview Multitask Learning. Brief. Bioinform. 2021, 22, bbaa133. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, Y.; Peng, J.; Cheng, L. DeepLGP: A novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 2020, 36, 4466–4472. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).