3.1. The E⊗e Potential Energy Surfaces in the Linear Coupling Model
We consider the simplest case, namely the prototype of the
E⊗
e effect under linear coupling spanning the so-called “Mexican hat” potential energy surface. The coupled vibrational–electronic problem is, of course, an issue of wave-mechanics, but we ignore some of the details, taking directly the equations of the related surfaces [
2,
3] and proceeding to their analysis in terms of geodesics and curvature. Thus, we start concisely with the effective form of the equations defining the simplest
E⊗
e problem, the energy of the mentioned
E states being eigenvalues of the following Hamiltonian matrix:
In the physical sense,
K has the meaning of a force constant when the system is stressed along the
ρ coordinate, while
V, the first-order vibronic parameter, measures the strength of the coupling between the initially degenerate (equivalent) states. The
e-type coordinates can be expressed in the polar format by
ρ and
φ, the above matrix being independent of the angular variable. The eigenvalues are:
being expressible in the unified form
e(
ρ) ≡
e−(
ρ) or
e(
ρ) ≡
e+(
ρ) if we conventionally allow the radial coordinate to take negative values. In this system, we have axial symmetry, which allows for the use of the idea of a rotational surface. The points of the surface can be described as the following set of coordinates:
For the concepts and formulae used in the further expansion, we refer the reader to the do Carmo monograph [
35]. The metric is defined by the scalar products in the following manner:
Denoting by
G the matrix with the above elements, one prepares its inverse in order to obtain the
= (
G−1)
ij elements. It follows that:
The Christoffel symbols in the given case are:
where
and
,
. By straightforward calculations, one obtains:
The equations of geodesics on this surface are:
where the dot and double-dot upper-scripts denote the first and second derivatives, respectively, with respect to an evolution parameter (say,
t):
Rewritten with the above defined parameters, Equation (9a) turns into:
On the other hand, geodesic Equation (9b) is related to the axial symmetry of the problem. Let us introduce
, having the meaning of angular velocity, and subsequently perform certain rearrangements:
The last equality is nothing other than the conservation of angular momentum. By rescaling conventions, we eliminated the explicit factor of mass that appears in the physical definition of this quantity. In our circumstance, we retain the conservation of the product:
This quantity is settled by the initial conditions of the differential equations.
Replacing the last equality in Equation (10), one obtains:
The problem is formally reduced to a one-variable second-order differential equation. Except for the L = 0 case, it becomes invalid at ρ = 0, such a singularity being physically forbidden for a particle in circular motion because it would imply an infinite angular velocity. In other words, a general geodesic cannot touch the radial origin.
To the best of our knowledge, general analytic solutions are not available, confining the discussion to numeric illustrations suggesting the patterns of geodesics placed in different areas of the coupled surfaces. In this view, we will consider conventional parameters and inspect the different types of geodesic curves.
3.2. Illustrating Geodesic Patterns on the “Mexican Hat” and Conical Intersection Surfaces
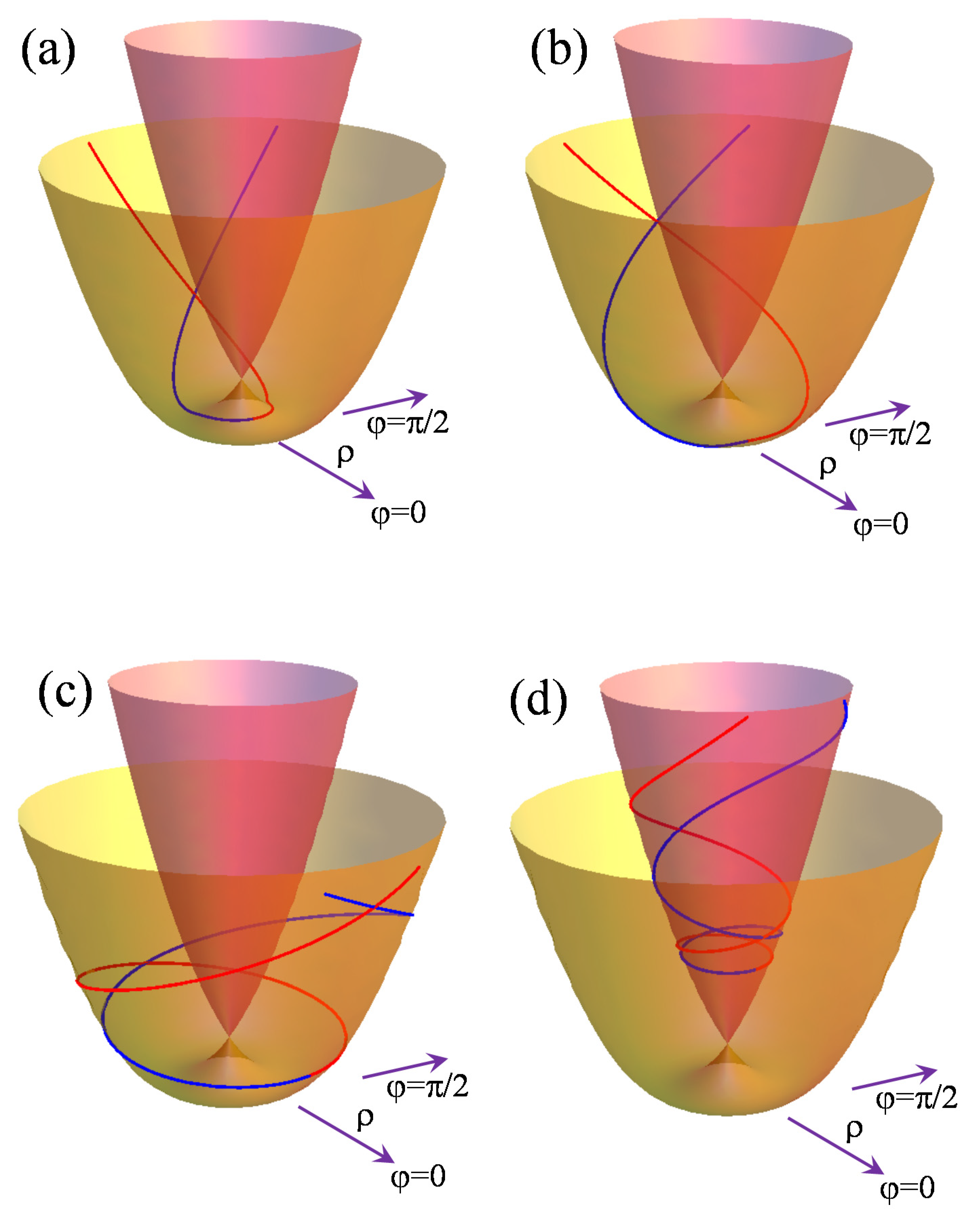
Figure 1 shows the general shape of the
E⊗
e problem. One may observe a composed surface, with the lower sheet having the minimum and the upper one having a steep slope everywhere. The two surfaces meet at the
ρ = 0 degeneracy point. In this vicinity, the first-order term predominates, indicating another name given to such a topological pattern, namely a conical intersection [
36,
37]. This sort of crossing may also occur in other instances than the nominal Jahn–Teller effect, but it also has vibronic origins. The upper surface corresponds to the
e+(
ρ) solution in Equation (2), being depicted with darker (violet) coloring in
Figure 1, while the lowest sheet
e−(
ρ) is drawn in a lighter color (yellow). The
e−(
ρ) surface has a circular trough at the the
ρmin =
V/
K radius. With a certain scaling and cropping in the graphical representation of the surfaces, a certain resemblance to a Mexican hat can be observed, the
E⊗
e profile also being called this name. In our vertically elongated scaling, the “Mexican hat” name, as figure of speech, is not so obvious, but this is an irrelevant aspect.
The frames of
Figure 1 illustrate qualitatively distinct placements of point
P crossed by geodesics obeying the
initial condition in the corresponding differential equations. The initial polar coordinate is set to
φP = 0, other arbitrary values being obtainable by the rotation with
φP of the presented figures around the vertical axis.
The panels (a–c) explore the curves on the lower e−(ρ) surface, produced with P at the respective smaller, equal, and larger radial coordinates, in comparison to the radius ρmin of the minimum trough. Strictly speaking, the non-null initial condition due to the first-order derivative of the angular coordinate determines only one half of the geodesic line evolving in the clockwise or anti-clockwise direction with respect to P’s origin depending on the or < 0 case. We represented these halves in different colors, their meeting marking the starting point. In the discussed situation, the geodesic halves modulated by the sign of are symmetrical with respect to a mirror plane passing through the point P and the vertical axis at ρ = 0. The two branches of the geodesic spiral on the surface, crossing each other at polar coordinates equal to those of the initial point φ = φP and, on the diametrically opposed side, φ = φP + π. For the starting points on the upper surface (see panel d), e+(ρ), and for the ones placed on the lower surface, e−(ρ), after the minimum radius (see panels b and c), ρP ≥ ρmin, the curves evolve upwards at elevations higher than the initial point e±(ρ) > e±(ρP).
For a point on the lower sheet, e−(ρ), comprised of the origin and radius of the minimum trough, 0< ρP < ρmin (see panel a), the geodesic goes first toward the minimum and then starts spiraling upward after crossing the valley. The rotation of the geodesics on the surfaces around the ρ = 0 vertical axis can be assigned to the positive curvature of the e±(ρ) surfaces with respect to the ρ coordinate. This situation tells us that, in the long term, the geodesics cannot be interpreted with the intuition of the shortest path between two points because the cycling imposes lengthier routes than when drawing a curve with a monotonous variation in the angular parameter between the φP and φQ values of the generally distant P and Q references placed on the same geodesic. In turn, the minimum path interpretation holds in the infinitesimal sense of Q’s arrival occurring in the vicinity of P’s starting point.
Another special geodesic equation is the case, i.e., a movement without an angular impetus, that actually keeps the angular coordinate constant along the curve φ = φP. This is the trivial case of the geodesic and is defined as the intersection of the e±(ρ) surfaces with the vertical plane containing the ρ = 0 axis and the initial P point. These are the analogs of meridians, which are geodesics on the sphere. In turn the circles defined at a constant radial parameter ρ = ρP, are not allowed to be geodesics in similar manner to a globe in which parallels other than the equator do not fulfill the underlying equations.
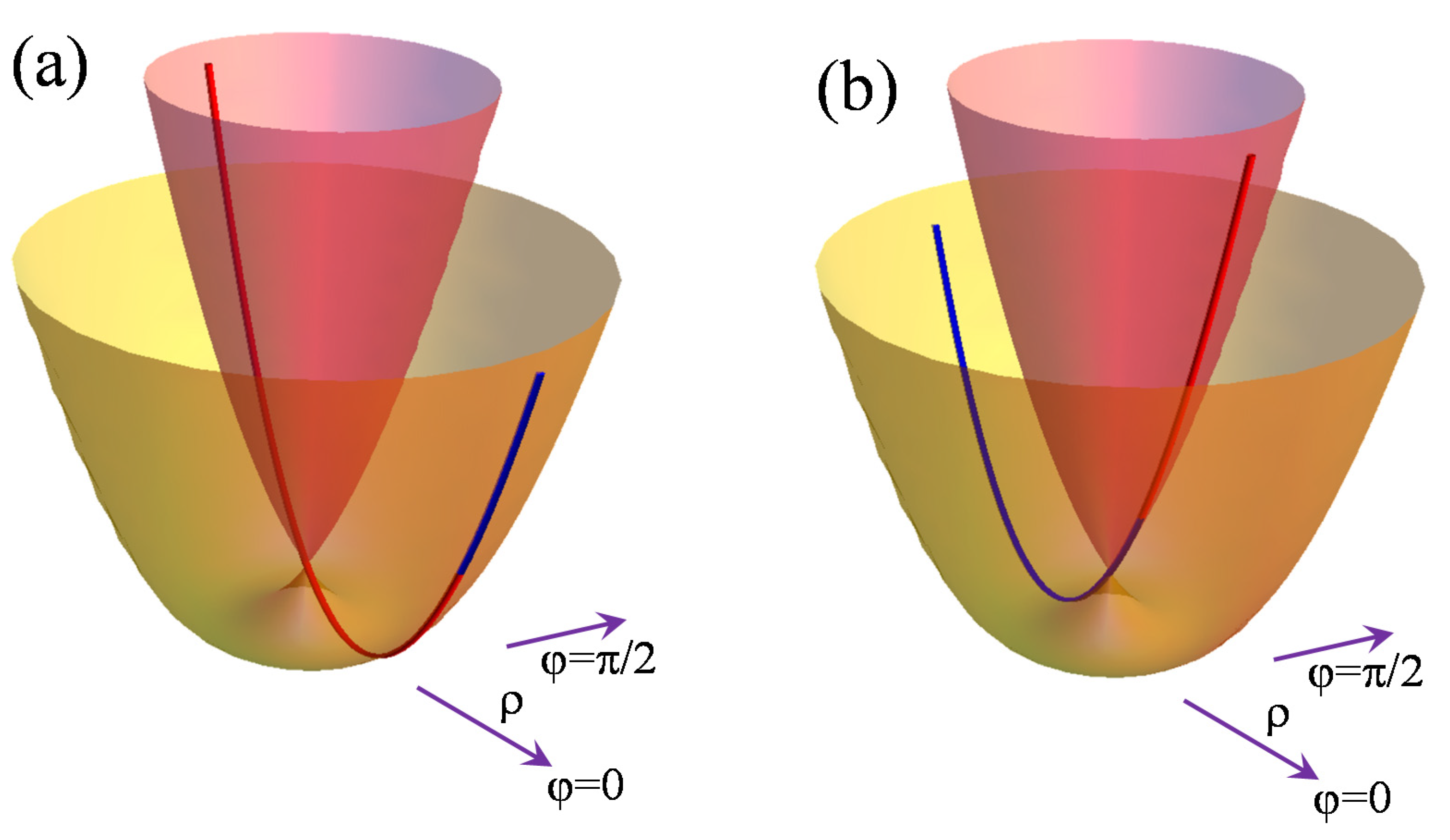
Figure 2 presents examples of solutions based on the
initial condition. One can observe that the curve continues on the other surface. If the starting point
P is on the
e_(
ρ) sheet, as in panel (a), it crosses the degeneracy at
ρ = 0 and goes onto the
e+(
ρ) surface. Vice versa, as shown in panel (b), it can pass from
e+(
ρ) to
e−(
ρ), the different colors of the curve branches helping us to visually locate the initial point at their confluence. Actually, in this situation, the geodesics are parabolas with the minimum located at the
ρmin contained in the plane defined by the vertical
ρ = 0 axis and the point
P.
The degeneracy crossing indicates the above-suggested perspective of a unified description of the reunion of the surfaces as a single object. Formally, this is realized by choosing only one of the equalities shown in Equation (2), say e(ρ) = e−(ρ), the lower surface resulting then for ρ > 0 and the upper one being accounted for with an enforced negative ρ having e+(ρ) = e(−ρ) = e_(−ρ). Accepting such a non-standard convention is not essential to the discussion, but it helps us to deal with the inter-surface evolution of the particular meridian-like geodesics. In turn, in the above-mentioned case, as well as in the cases discussed below, with a non-null initial angular derivative the geodesics are not allowed to either touch the ρ conical intersection or to pass through onto the companion surface. This is determined by the avoidance of a singularity in the last term of Equation (13), or, in other words, by the physical denial of infinite angular momentum in the conservation law from (12).
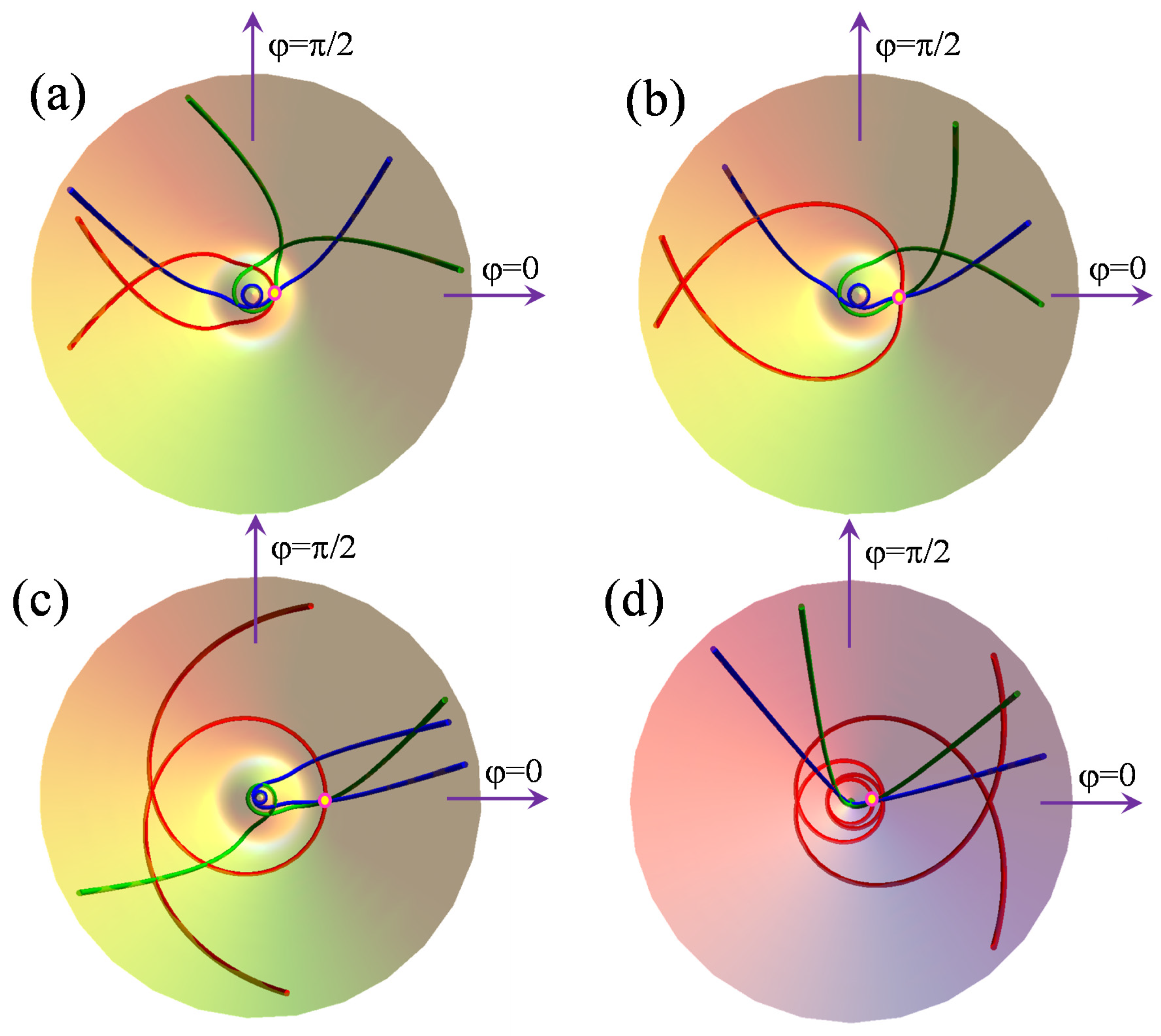
Figure 3 illustrates the situation of geodesics with non-null initial derivatives with respect to both the radial and angular variables (green and blue lines, respectively). However, for the sake of comparison, the particular situation of a null initial radial derivative was added (red geodesics). The blue line has a larger starting radial derivative than the green one (at the same angular value).
One may notice that the geodesics with the initial condition are not symmetric with respect to the initial point in the sense that the halves going in opposite directions from P are not identical, as is the case for . However, ignoring the starting point, each geodesic admits a symmetry plane where the condition is reached at the point Q. A geodesic has one unique point obeying the condition. At the same time, any arbitrary point P is passed by an infinite family of geodesics obtainable by tuning the local angle of the curves with respect to the particular reference with . This angle is determined by the sign and magnitude of the ratio of the and initial conditions. The factor was included in order to convert the angular derivative to the dimension of the radial one, akin to the physical meaning of the linear impulse.
On the lower surface,
e−(
ρ), the geodesic has a part that approaches an orbit near the conical intersection. The larger the initial radial derivative, the closer the loop comes to the conical intersection, as seen in frames (a) and (b) in
Figure 3, where the blue line climbs to a smaller radius and performs a complete cycle around the conical intersection, while the green one escapes before completing a cycle. Both branches evolve, in the long term, towards the upper part of
e_(
ρ) at
ρ > ρmin. In the frames (c) and (d), both the blue and green lines perform a tour of the conical intersection, although, in the (d) panel, corresponding to the point
P on the
e+(
ρ) sheet, this is not easily visible because of the small radius near the conical point.
We refrain from adopting a classical mechanics explanation since the Jahn–Teller surface is a quantum object, but one may suggest that the cases (c) and (d) can be compared with the free fall from higher potential energies. Then, the line behaves analogously to one having a larger amount of momentum that allows for one or more circles at smaller radii. As discussed in the cases from
Figure 1, the more general instances from
Figure 3 do not allow a geodesic to escape the surface of the starting point,
e+(
ρ) or
e−(
ρ), this fact being possible only in the particular condition shown in
Figure 2.
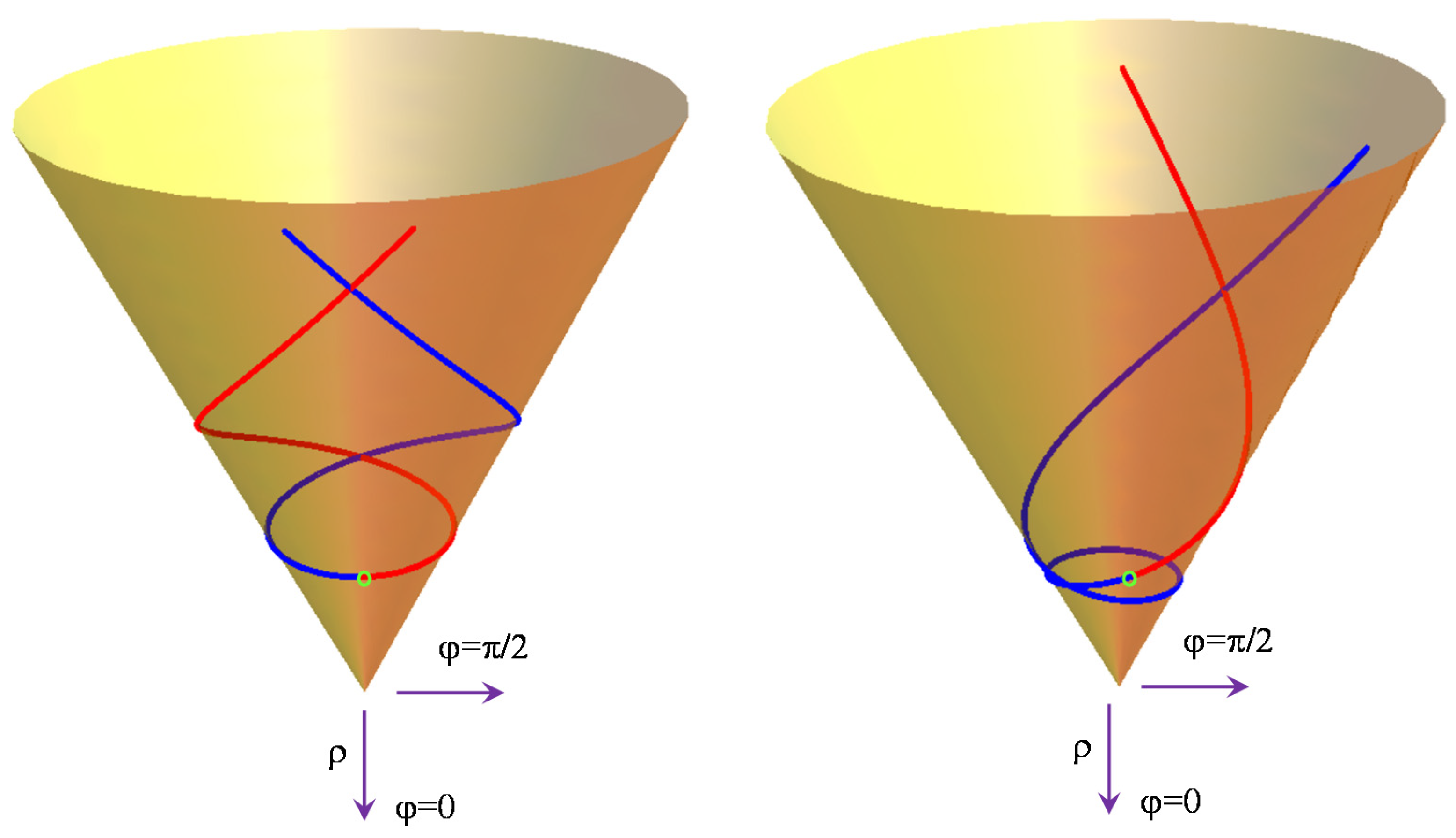
In the vicinity of the crossing point, both surfaces are approximated by cones with shared vertices. As pointed out previously, for this reason, in computational chemistry, this pattern is known as a conical intersection [
34,
35]. The equations for the cone and their geodesics can be derived if we impose
K = 0 in the above formulas, i.e.,
e±(
ρ) = ±
V ρ. Note that a cone is obtained if we take the half-difference of the eigenvalues:
V ρ = (
e+(
ρ) −
e−(
ρ))/2.
In this simplified hypothesis, one may find analytical expressions for geodesics based on the fact that a cone can be presented as an isometric transformation of the plane (both surfaces have zero Gauss curvature); thus, any straight line in the initial plane is mapped onto a geodesic on the cone surface (see pages 246–247 in reference [
35] and page 250 in [
38]). In a parametric dependence on
t, the cone geodesics starting from the point
ρ0 = r0,
φ0 = 0, with the initial gradients
and
, can be ascribed as follows:
where
One may observe that we used the conventional angular value u0 to define a ratio between the linear tangential and radial velocities if the evolution parameter t is formally regarded as time.
The in-plane line related by isometry to the cone geodesics is:
We checked that the numeric solutions of the generic differential Equations (9) and (10) with the above initial conditions render the same curves as the analytical ones from Equations (14) and (15). The
t > 0 and
t < 0 branches are related by a sign swap in the initial derivatives (represented in red and blue, respectively, in
Figure 4).
3.3. Quantum Chemical Calculation of the Jahn–Teller Effect in Triangular Molecular Systems
In this section, we aim to make concrete the
E⊗
e effect by state-of-the art calculations on an illustrative example. The simplest molecule imaginable for such a goal is the tri-hydrogen H
3. However, although it presents a conical intersection, the potential energy surface of H
3 does not show bonded minima [
39,
40], the system being prone to rapid dissociation in the molecular and atomic hydrogen H
2 + H, or to ionization, the H
3+ cation being a well-known molecule in the interstellar space [
41]. Then, the next molecule on the simplicity scale would be the cyclo-propenyl radical C
3H
3. This molecule can be described as a triangle composed of carbon atoms bordered by a triangle composed of hydrogens bonded to carbons. According to chemical intuition, the Jahn–Teller effect is due to the so-called π electrons on the C
3 unit, the H
3 moiety just following the distortion of the carbon frame.
This radical is an intermediate in combustion processes [
42]. Early electron spin resonance measurements proved the distortions to be due to the Jahn–Teller effect [
43,
44], and this finding was corroborated by similar conclusions from nuclear magnetic resonance spectra [
45]. The first approaches to distortion effects in the cyclo-propenyl radical were put in the heuristic key of the so-called antiaromaticity [
46] and were limited to the rather rudimentary calculation procedures available at the dawn of computational chemistry [
47,
48]. The studies were continuously re-enacted as progress was made in the calculation methods, retrieving the same qualitative and semi-quantitative conclusions [
49,
50]. The most recent and deep study of the C
3H
3 radical [
51] used highly rated computational methods, the so-called coupled cluster routines [
52], which, although not suited for multi-configurational and degenerate wave functions, are good at tackling the optimal distorted molecular geometries at the energy minima of the Jahn–Teller problem at hand.
Here, we performed Complete Active Space Self-Consistent Field (CASSCF) [
53,
54] calculations consisting of three electrons in three orbitals corresponding to the π subsystem of the planar molecule. A comprehensive modern study [
55], carried out with accurate multi-configurational computations, treated in detail the Jahn–Teller and pseudo Jahn–Teller effects in the cyclo-propenyl radical and anion, outlining potential energy curves. However, full potential energy surfaces representing the ab initio complete mapping of the
E⊗
e problem in C
3H
3 have not been presented previously.
The upper part of
Figure 5 shows the geometry of the unstable equilateral triangle, optimized in a conical intersection CASSCF procedure, along with the isosceles geometry stabilized as energy minimum. The change in bond lengths is not very large, but clearly illustrates that the stabilization can be interpreted as the formation of a simple C-C bond in concert with the delocalization of a double bond over two equal carbon–carbon lines.
The degenerate ground-state named previously,
E, in a generic manner is, more precisely, a
2E″ term in the case of the π-type electronic system of the cyclo-propenyl in the
D3h symmetry point group. The molecular term has the same symmetry as those of the singly occupied degenerate orbital ascribed conventionally the non-capitalized label
e″ (see the scheme in the left-bottom part of
Figure 5). The double degeneracy of the
2E″ state comes from the two equivalent possibilities to place the unpaired electron in the two lodges of the
e″ orbital set. One may see in the qualitative scheme shown in the lower part of
Figure 5 that the distortion leads to the removal of orbital degeneracy (energy equivalence) in the
e″ orbitals. The stable molecular geometry is an isosceles triangle with one elongated edge, the frontier orbital having the
a2 representation that gives rise to the corresponding
2A2 ground state in the
C2v point group.
We conducted the calculations to generate the ab initio potential energy surfaces of the Jahn–Teller effect in the C3H3 system. In this view, we did a relaxed geometry scan, where the structure of the C3 moiety is generated by tuning the distortion coordinates having the e′ symmetry, while the hydrogen atoms, i.e., the C-H bond lengths and CCH bond angles, were the subject of gradient refinement to optimized values.
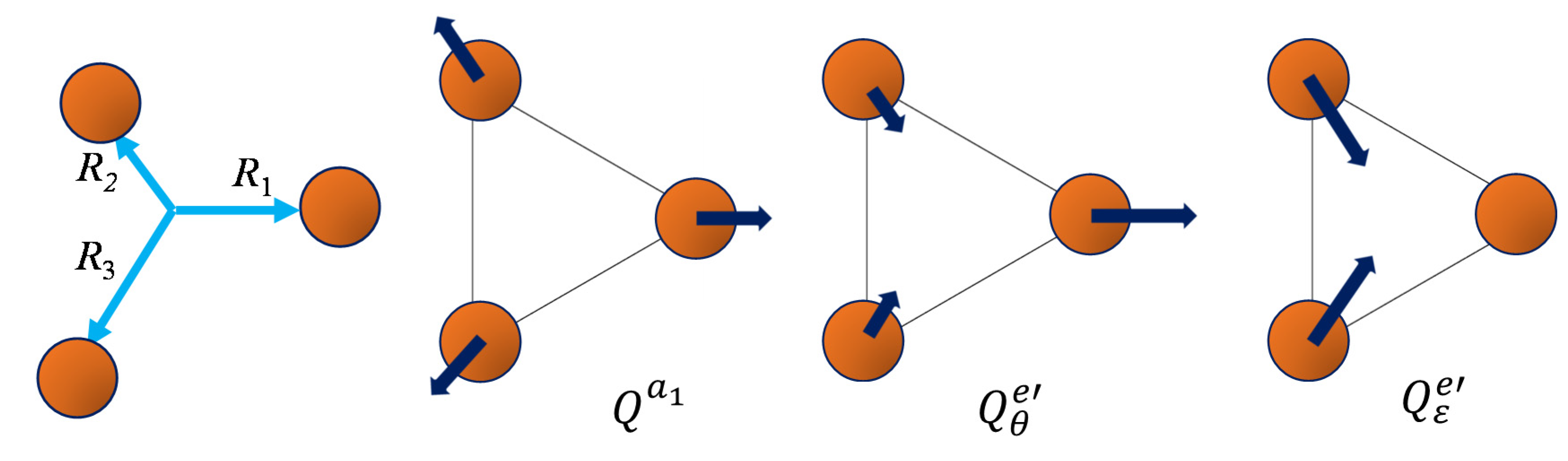
The coordinates tuning the geometry of the triangle are chosen as symmetrized combinations of the edge lengths [
47]. Here, we propose a different way, starting from the idea of defining a general triangle by three radii,
R1,
R2, and
R3, along axes fixed at mutual 120° angles. In spite of the constrained directions, the degrees of freedom are sufficient for defining any triangular configuration. In principle, even a negative
Ri can be admitted, but such an extreme evolution is not needed in our problem since relatively small displacements from the equilateral reference are expected when
as a function of the carbon–carbon bond-length in the system at the conical intersection. Then, we chose the symmetrized coordinates as a linear transformation over the above-defined radii:
Figure 6 illustrates the above-defined coordinates.
The total symmetric coordinate is not of direct interest in the discussed potential energy surface, being fixed at the geometry of the optimized conical intersection:
. The coordinates generically labeled
e in the introductory discussion are, more specifically,
e′ in the case of the triangle. The components with the
θ and
ε subscripts in Equations (17b) and (17c) correspond to the
ρ·cos(
φ) and
ρ·sin(
φ) definitions, respectively, with the polar coordinates discussed in
Section 3.1.
As a technical detail, we performed an ab initio scan on a grid in polar coordinates and backward converted these to the geometry of the carbon-based triangle. The radial points run between
ρ = 0 and
ρ = 0.2 Å, with a δ
ρ = 0.02 Å step, the polar coordinate going around the circle with a δ
φ = 15° increment. The points collected for the lowest two CASSCF states are represented as surfaces in
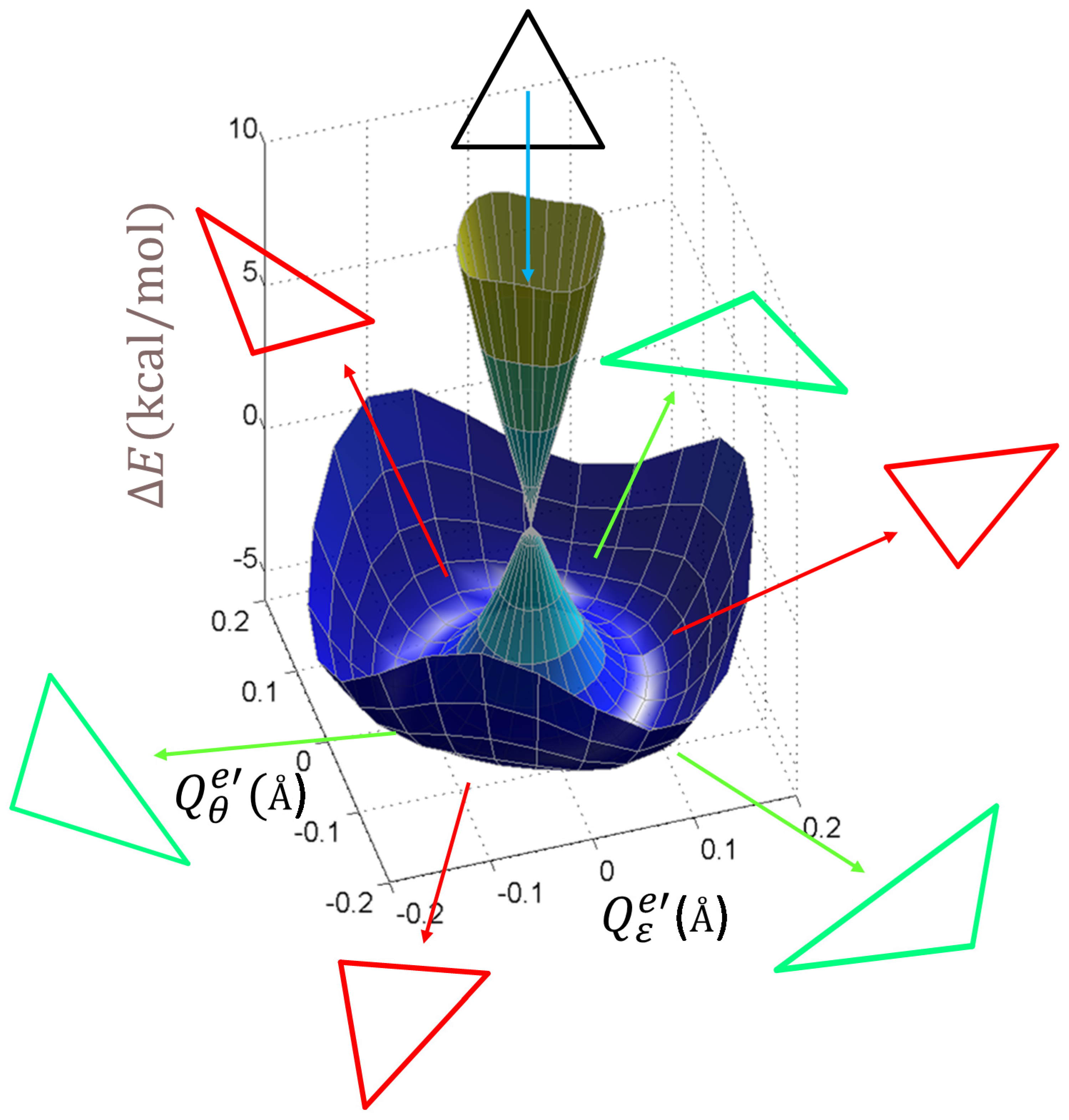
Figure 7. To distinguish it from the idealized model in
Section 3.1, which, confined to first-order vibronic coupling terms, had a continuous circular minimum at the bottom of the
e−(
ρ) surface, the realistic system is warped in a trigonal pattern, showing three minima and three saddle points in the quasi-circular valley. To account for this, the model should be updated with second-order vibronic coupling terms [
2,
3], namely the
W parameter in the new eigenvalue equations:
This model acquires a dependence on the polar angle, the 3φ term in Equation (18) accounting for the three-fold symmetry implied by the C3 axis of the molecule. The angular dependence modulates three minima and three maxima around the former circular trough of the minimum from the previous simplified level of the model (with W = 0). Since on the bottom of the valley the curvature with respect to the radial variable remains positive (corresponding to the minimum), the maxima with respect to the angular parameter (negative curvature) are actually saddle points. The absolute minima (the three equivalent positions at φ = 0 and ±120°) show a positive curvature (the second derivative of the above-defined eigenvalues) with respect to both the ρ and φ variables.
The computed points represented as surfaces in
Figure 3 were reasonably fitted with Equation (18) by the following optimized parameters:
K = 942.256 kcal·mol
−1Å
−2,
V = 102.996 kcal·mol
−1Å
−1, and
W = 34.349 kcal
2·mol
−2Å
−4. The goodness of the fit can be measured by the linearity in the representation of the
ecalc ab initio calculated energies vs. the
efit values fitted by Equation (18). The least-square line
efit =
a·
ecalc +
b has a slope
a = 1.001 and an
R2 = 0.994, close to the ideal unity values, while the intercept is small (
b = 0.202 kcal/mol).
Given the novelty of the proposed geodesic analysis of the potential energy surfaces, we opted to proceed systematically from the simplest level of the prototypical
E⊗
e model, represented by Equations (1) and (2), while the real cases are better accounted for with the more complicated form (Equation (18)). As a compromise, we consider in the following the parameters fitted from the application to the cyclo-propenyl radical, dropping, however, the term corresponding the second-order coupling (i.e., imposing
W = 0). This numeric experiment is presented in
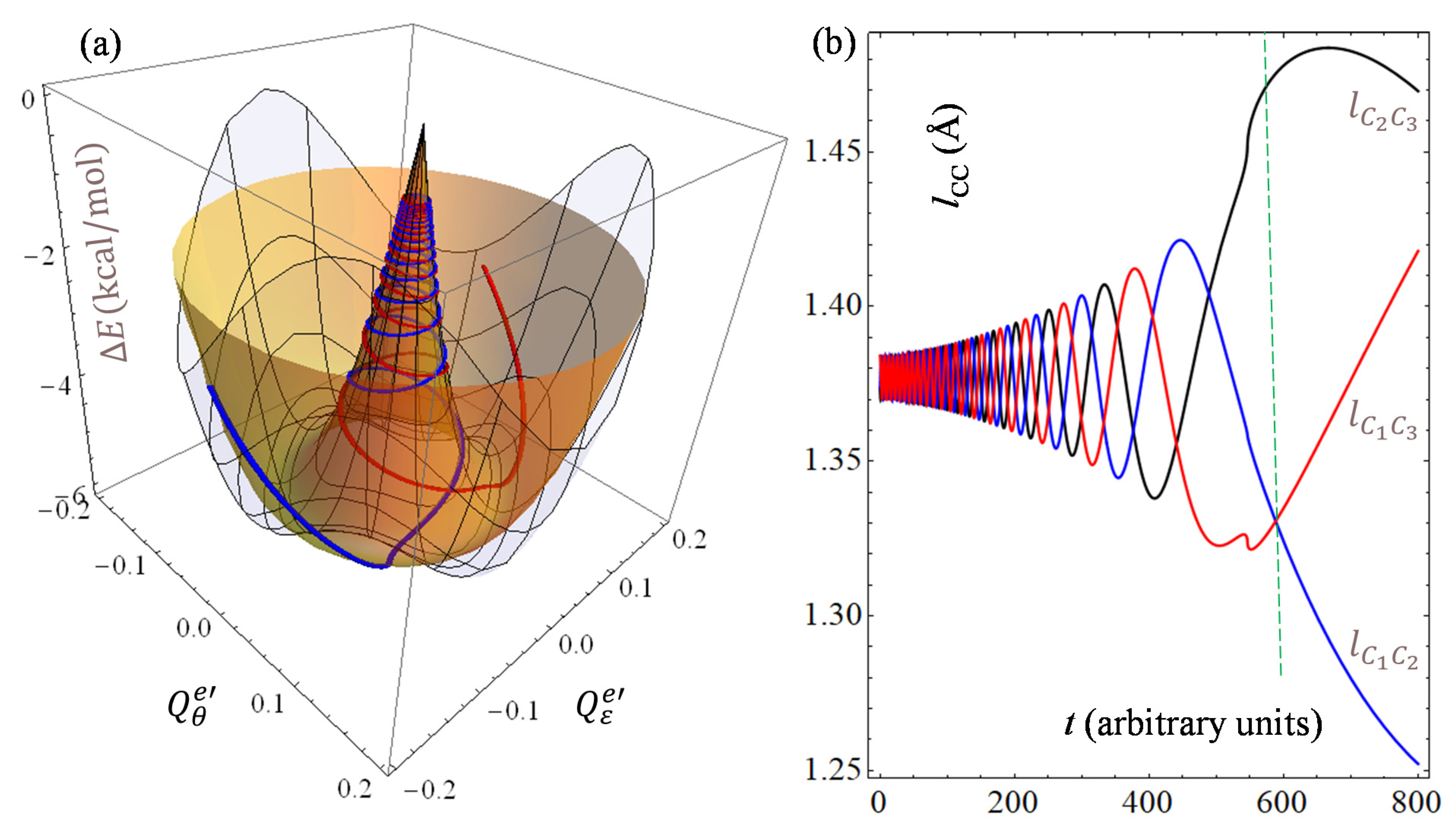
Figure 8.
Although the evolution parameter,
t, can be intuitively thought of as time, we are considering it here in arbitrary units. In qualitative terms, the situation presented in
Figure 8 is similar to panel (a) from
Figure 1, while the aspect of the curve is sensibly different because of another interplay of parameters. In the
Figure 8 case, we placed the initial point close to the conical intersection in order to simulate the evolution toward distortion, imposing a conventional value for the angular derivative. The larger the initial derivative, the steeper the descent on the conical part, with fewer coils until the minimum valley. A smaller initial derivative yields more loops, with successive spirals closer to each other. A mechanical interpretation of geodesics is the trajectory in the absence of a continuous source of acceleration [
35]. Here, we do not consider this meaning, first of all because a mechanical trajectory is not literally accepted in a quantum model. At the same time, the geodesics appear as infinite curves, while the model itself is not conceivable over a very long range, being conceptually limited in the area containing the minimum of the lowest state and the conical intersection. On the other hand, the spiraling pattern of general geodesics can be tentatively put in connection with the intrinsic oscillatory nature of the problem, as the
E⊗
e model can be thought of as a sort of coupling between harmonic oscillators. The geodesic example from
Figure 8 may suggest that, in the hypothetical case of instantaneous high symmetry, the system will probably evolve toward the distorted minimum in oscillatory progressions. The geodesics also continue after passing through the minimum valley. In a rough interpretation, this can be considered as being due to the impetus gained during the descent, but the continuation at a larger radius has no physical meaning. Therefore, there is a dissociation between the geometric geodesic and the mechanical or Born–Oppenheimer trajectory, which presumably encounters a barrier in terms of the reachable energy height. We let the connection between geodesics and the Born–Oppenheimer molecular dynamics trajectory be a matter of further investigations and debate. We hypothesize that a certain connection with the nodes of vibrational wave functions can be established, but we cannot exhaust here all the possible lemmas emerging from the problem.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}