
Structure Determination Feasibility of Three-Dimensional Electron Diffraction in Case of Limited Data

 ,
,  , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
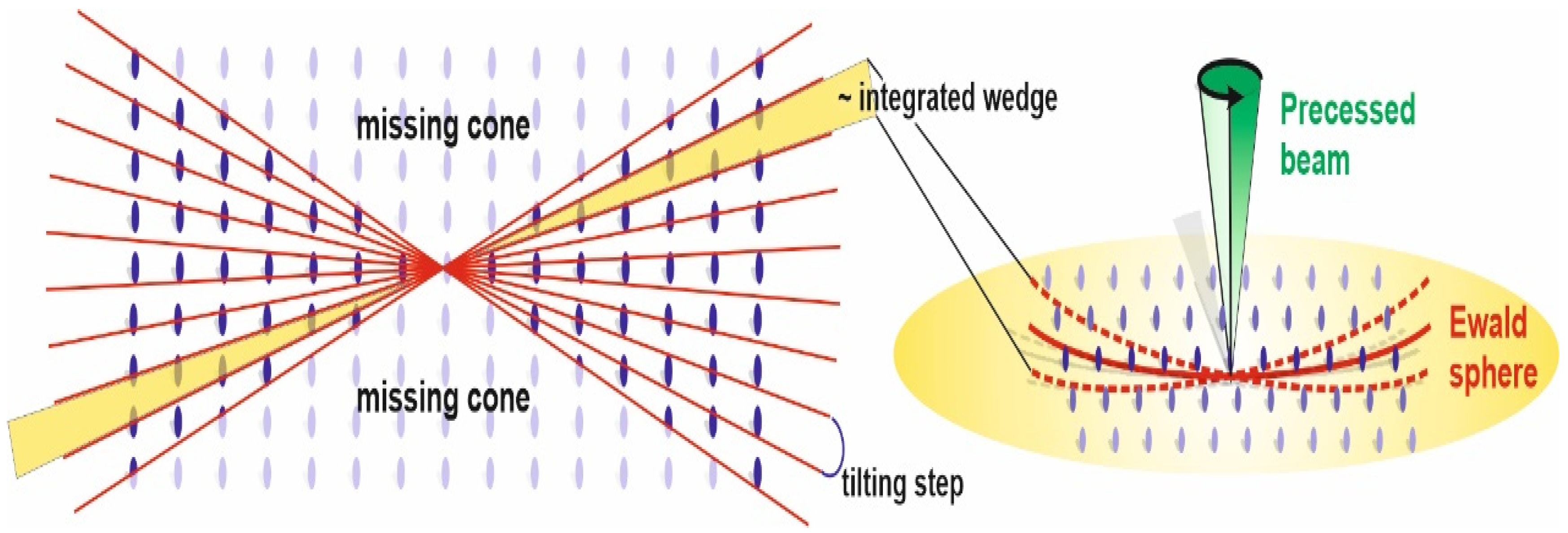
2.2. 3D ED Acquisitions
2.3. Structure Determination Tools
Direct Methods & Charge-Flipping
2.4. Global Optimization Method
3. Results and Discussion
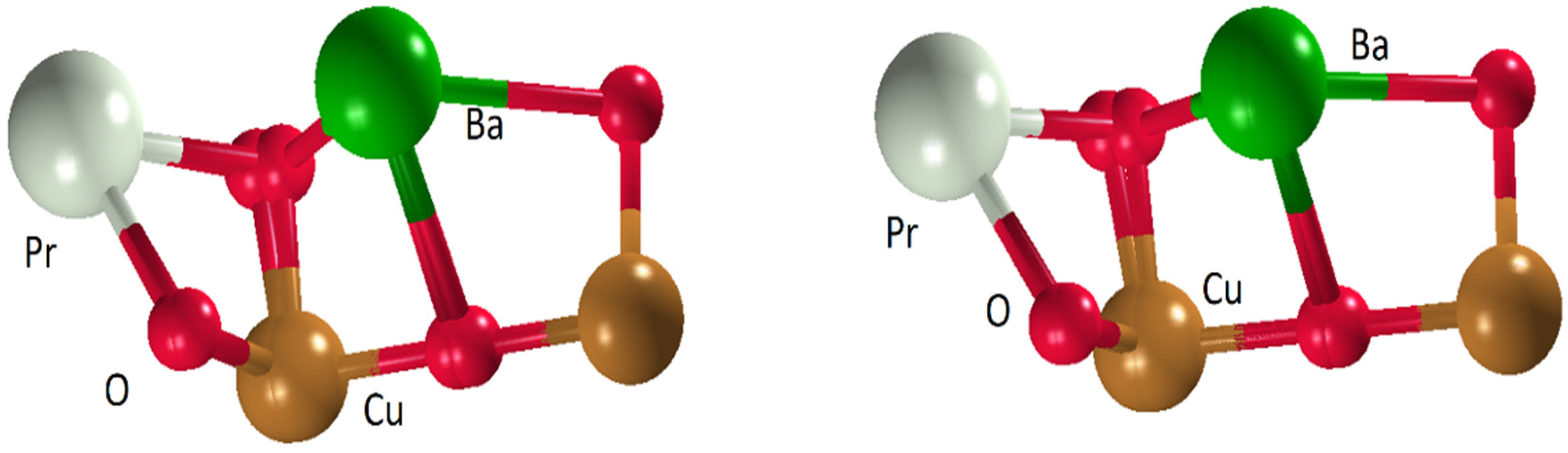
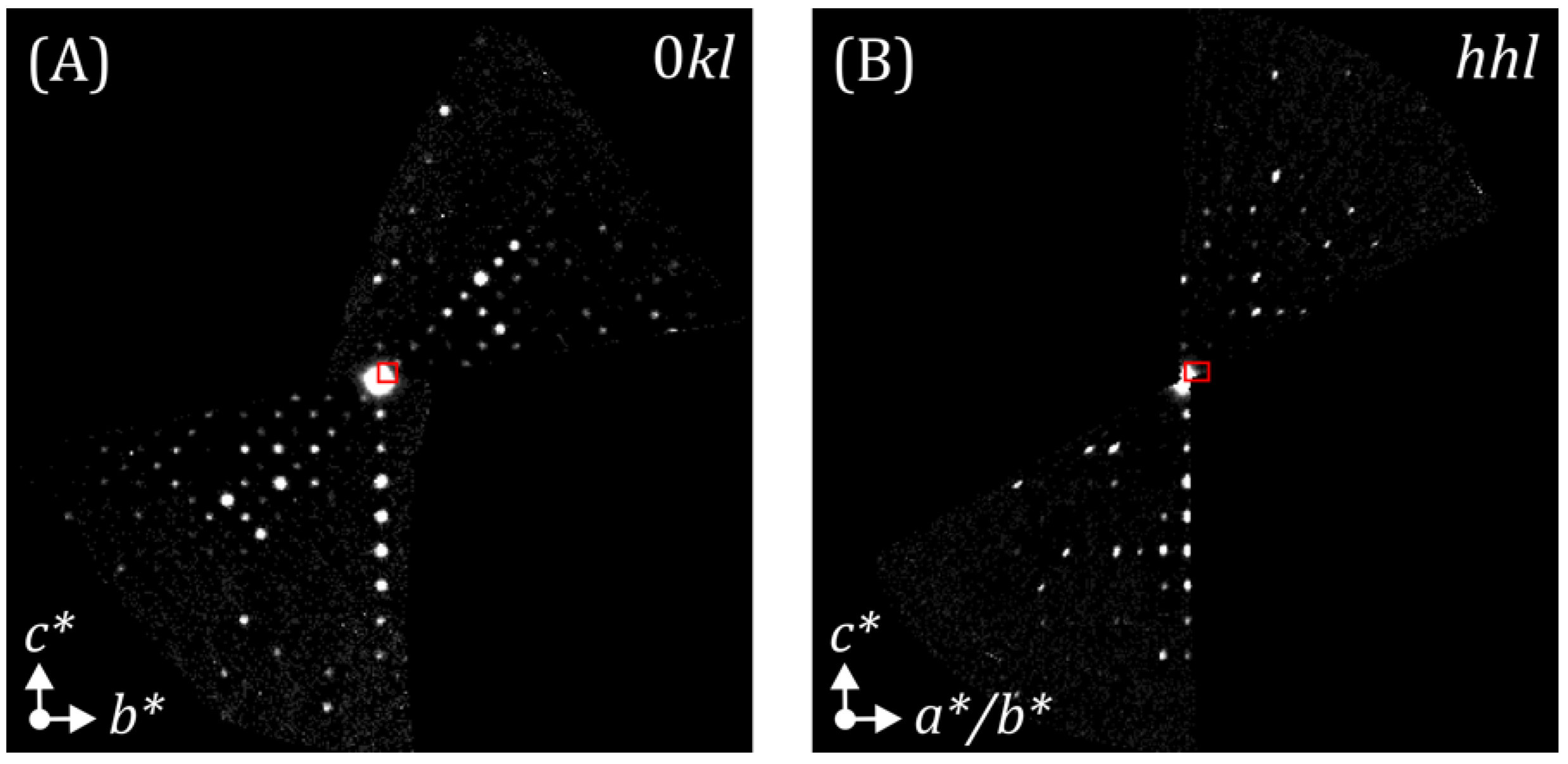
3.1. PrBa2Cu3O7
3.1.1. Direct Methods
3.1.2. Charge-Flipping
3.1.3. Global Optimization Method
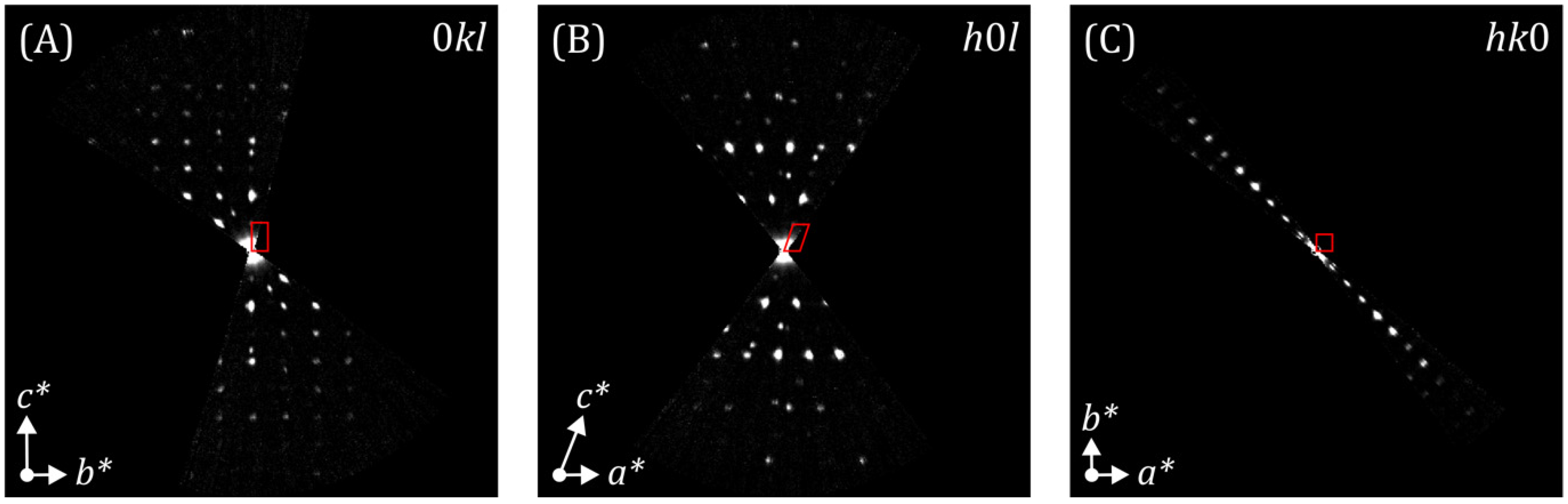
3.2. BaCuO2
3.2.1. Direct Methods
3.2.2. Charge-Flipping
3.2.3. Global Optimization Method
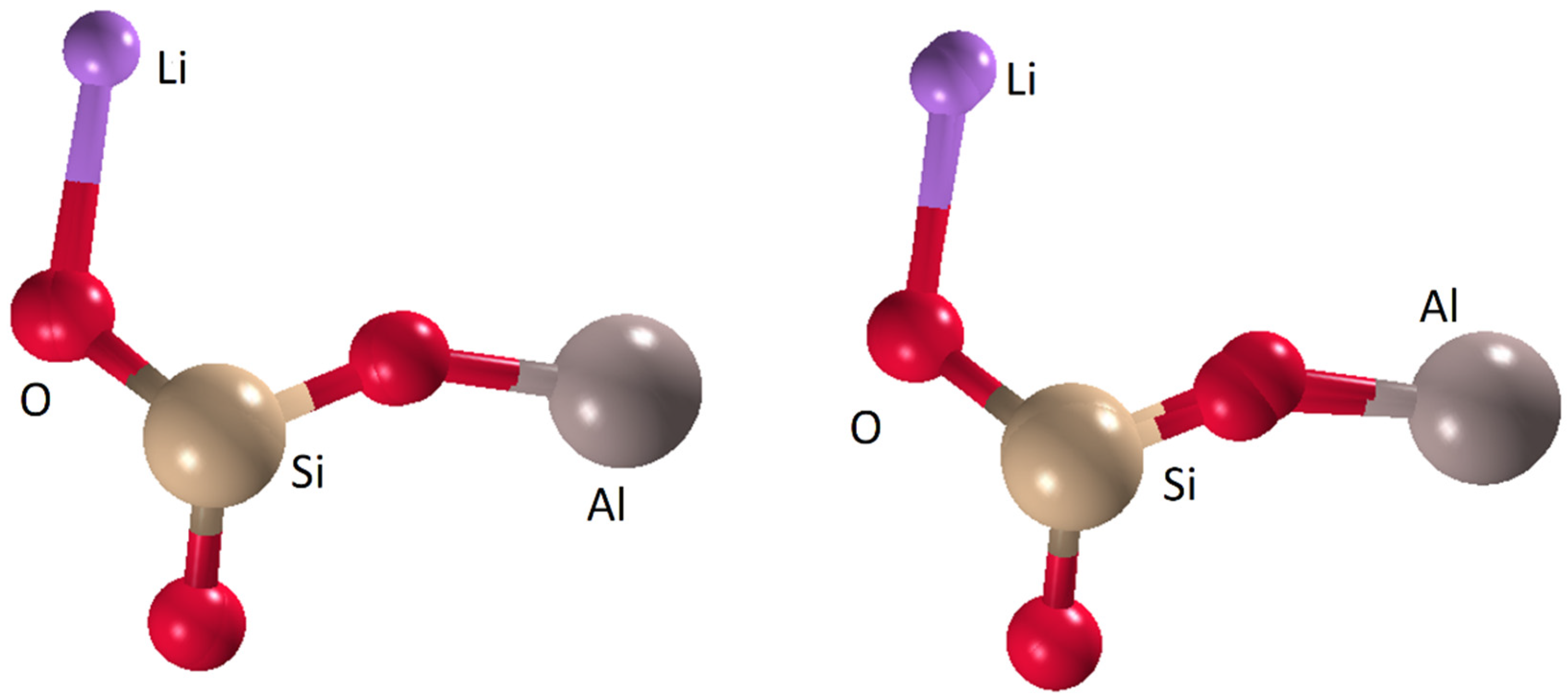
3.3. LiAl0.8Fe0.2(SiO3)2
3.3.1. Direct Methods
3.3.2. Charge-Flipping
3.3.3. Global Optimization Method
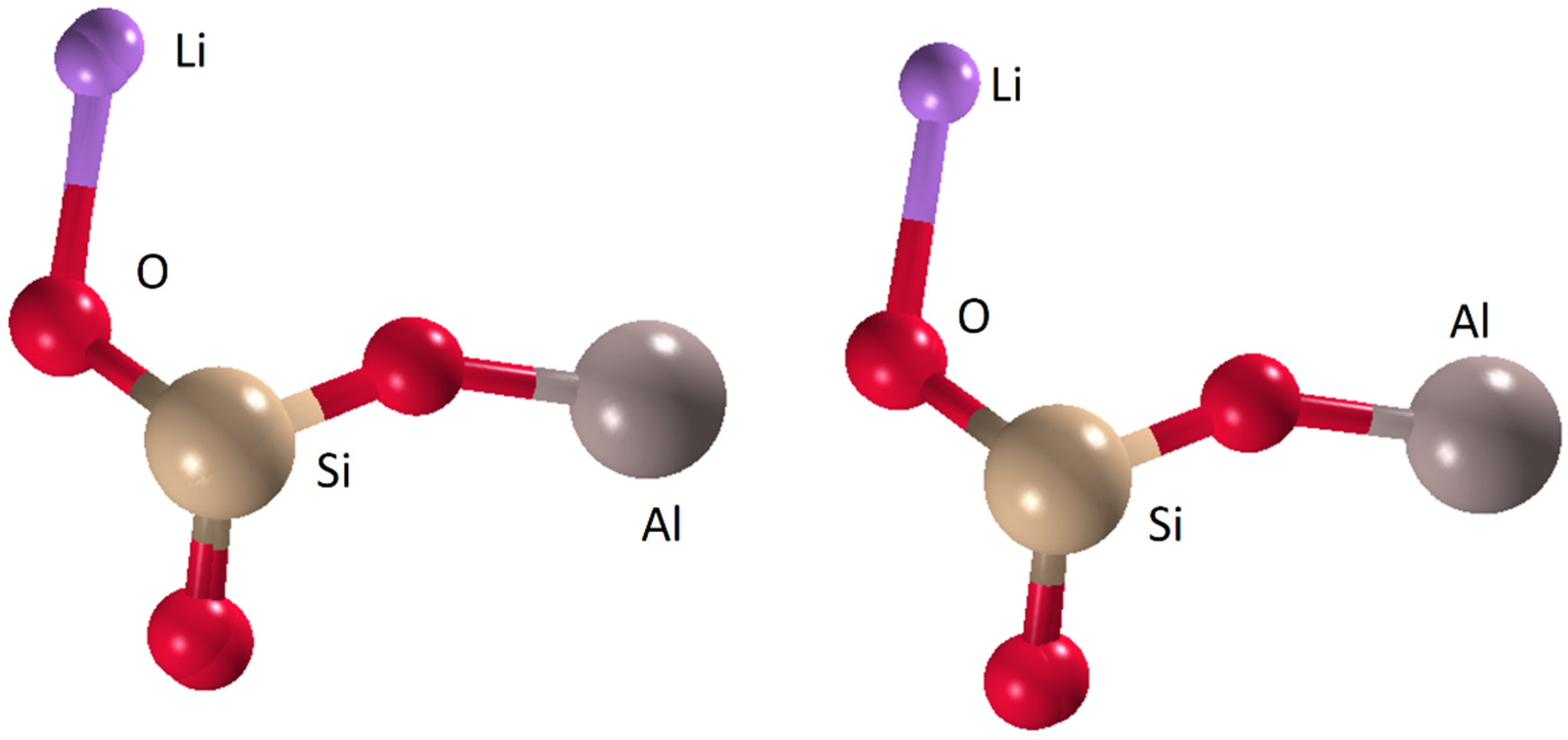
3.4. LiAl(SiO3)2
3.4.1. Direct Methods
3.4.2. Charge-Flipping
3.4.3. Global Optimization Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowley, J.M. Electron Diffraction Techniques; Oxford University Press: Oxford, UK, 1992; Volumes 1 and 2. [Google Scholar]
- Dorset, D.L. Structural Electron Crystallography; Plenum Press: New York, NY, USA, 1995. [Google Scholar]
- Dorset, D.L.; Hauptman, H.A. Direct phase determination for quasi-kinematical electron diffraction intensity data from organic microcrystals. Ultramicroscopy 1976, 1, 195–201. [Google Scholar] [CrossRef]
- Dorset, D.L. Electron crystallography. Acta Crystallogr. Sect. B Struct. Sci. 1996, 52, 753–769. [Google Scholar] [CrossRef]
- Zhou, X.; Hovmöller, S. Electron crystallography: Imaging and single-crystal diffraction from powders. Acta Crystallogr. Sect. A 2008, 64, 149–160. [Google Scholar] [CrossRef]
- Sinkler, W.; Own, C.S.; Marks, L.D. Application of a 2-beam model for improving the structure factors from precession electron diffraction intensities. Ultramicroscopy 2007, 107, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Ciston, J.; Deng, B.; Marks, L.D.; Own, C.S.; Sinkler, W. A quantitative analysis of the cone-angle dependence in precession electron diffraction. Ultramicroscopy 2008, 108, 514–522. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Eggeman, A.S.; Midgley, P.A. Is precession electron diffraction kinematical? Part I:: “Phase-scrambling” multislice simulations. Ultramicroscopy 2010, 110, 763–770. [Google Scholar] [CrossRef]
- Palatinus, L.; Jacob, D.; Cuvillier, P.; Klementová, M.; Sinkler, W.; Marks, L.D. Structure refinement from precession electron diffraction data. Acta Crystallogr. Sect. A 2013, 69, 171–188. [Google Scholar] [CrossRef]
- Vincent, R.; Midgley, P.A. Double conical beam-rocking system for measurement of integrated electron diffraction intensities. Ultramicroscopy 1994, 53, 271–282. [Google Scholar] [CrossRef]
- Own, C.S. System Design and Verification of the Precession Electron Diffraction Technique. Ph.D. Thesis, Northwestern University, Evanston, IL, USA, 2005. [Google Scholar]
- Nicolopoulos, S.; Weirich, T.E. ELCRYST 2005 proceedings of the electron crystallography school 2005: New frontiers in electron crystallography. Ultramicroscopy 2007, 107, 431–558. [Google Scholar]
- Available online: www.nanomegas.com (accessed on 10 June 2022).
- Portillo, J.; Rauch, E.F.; Nicolopoulos, S.; Gemmi, M.; Bultreys, D. Precession Electron Diffraction Assisted Orientation Mapping in the Transmission Electron Microscope. Mater. Sci. Forum 2010, 644, 1–7. [Google Scholar] [CrossRef]
- Darbal, A.D.; Narayan, R.D.; Vartuli, C.; Lian, G.; Graham, R.; Shaapur, F.; Nicolopoulos, S.; Weiss, J.K. Automated High Precision Strain Measurement Using Nanobeam Diffraction Coupled with Precession. Microsc. Microanal. 2013, 19, 702–703. [Google Scholar] [CrossRef][Green Version]
- Hoque, M.M.; Vergara, S.; Das, P.P.; Ugarte, D.; Santiago, U.; Kumara, C.; Whetten, R.L.; Dass, A.; Ponce, A. Structural Analysis of Ligand-Protected Smaller Metallic Nanocrystals by Atomic Pair Distribution Function under Precession Electron Diffraction. J. Phys. Chem. C 2019, 123, 19894–19902. [Google Scholar] [CrossRef]
- Kolb, U.; Gorelik, T.; Kübel, C.; Otten, M.T.; Hubert, D. Towards automated diffraction tomography: Part I—Data acquisition. Ultramicroscopy 2007, 107, 507–513. [Google Scholar] [CrossRef]
- Kolb, U.; Gorelik, T.; Otten, M.T. Towards automated diffraction tomography. Part II—Cell parameter determination. Ultramicroscopy 2008, 108, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Mugnaioli, E.; Gorelik, T.; Kolb, U. “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy 2009, 109, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Kolb, U.; Mugnaioli, E.; Gorelik, T.E. Automated electron diffraction tomography—A new tool for nano crystal structure analysis. Cryst. Res. Technol. 2011, 46, 542–554. [Google Scholar] [CrossRef]
- Kolb, U.; Gorelik, T.; Mugnaioli, E. Automated diffraction tomography combined with electron precession: A new tool for ab initio nanostructure analysis. MRS Online Proc. Libr. 2009, 1184, 38–50. [Google Scholar] [CrossRef]
- Zhang, D.; Oleynikov, P.; Hovmoller, S.; Zou, X. Collecting 3D electron diffraction data by the rotation method. Z. Krist. Cryst. Mater. 2010, 225, 94–102. [Google Scholar] [CrossRef]
- Wan, W.; Sun, J.; Su, J.; Hovmöller, S.; Zou, X. Three-dimensional rotation electron diffraction: Software RED for automated data collection and data processing. Appl. Crystallogr. 2013, 46, 1863–1873. [Google Scholar] [CrossRef]
- Palatinus, L.; Brázda, P.; Jelínek, M.; Hrdá, J.; Steciuk, G.; Klementová, M. Specifics of the data processing of precession electron diffraction tomography data and their implementation in the program PETS2.0. Acta Crystallogr. Sect. B 2019, 75, 512–522. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Altomare, A.; Corriero, N.; Cuocci, C.; Moliterni, A.; Rizzi, R. The hybrid big bang-big crunch method for solving crystal structure from powder diffraction data. J. Appl. Crystallogr. 2013, 46, 779–787. [Google Scholar] [CrossRef]
- Weirich, T.E.; Zou, X.D.; Ramlau, R.; Simon, A.; Cascarano, G.L.; Giacovazzo, C.; Hovmöller, S. Structures of nanometre-size crystals determined from selected-area electron diffraction data. Acta Crystallogr. Sect. A 2000, 56, 29–35. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Palatinus, L. The charge-flipping algorithm in crystallography. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 1–16. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef]
- Nederlof, I.; van Genderen, E.; Li, Y.-W.; Abrahams, J.P. A Medipix quantum area detector allows rotation electron diffraction data collection from submicrometre three-dimensional protein crystals. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, E.; Clabbers, M.T.B.; Das, P.P.; Stewart, A.; Nederlof, I.; Barentsen, K.C.; Portillo, Q.; Pannu, N.S.; Nicolopoulos, S.; Gruene, T.; et al. Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr. Sect. A 2016, 72, 236–242. [Google Scholar]
- Gemmi, M.; la Placa, M.G.I.; Galanis, A.S.; Rauch, E.F.; Nicolopoulos, S. Fast electron diffraction tomography. J. Appl. Crystallogr. 2015, 48, 718–727. [Google Scholar] [CrossRef]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. eLife 2013, 2, e01345. [Google Scholar] [CrossRef]
- Clabbers, M.T.B.; van Genderen, E.; Wan, W.; Wiegers, E.L.; Gruene, T.; Abrahams, J.P. Protein structure determination by electron diffraction using a single three-dimensional nanocrystal. Acta Crystallogr. Sect. D 2017, 73, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Clabbers, M.T.B.; Gruene, T.; Parkhurst, J.M.; Abrahams, J.P.; Waterman, D.G. Electron diffraction data processing with DIALS. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Giacovazzo, C.; Monaco, H.L.; Artioli, G.; Viterbo, D.; Milanesio, M.; Gilli, G.; Gilli, P.; Zanotti, G.; Ferraris, G.; Catti, M. Fundamentals of Crystallography; Oxford University Press: Oxford, UK, 2011; ISBN 9780199573653. [Google Scholar]
- Dorset, D.L. Direct methods and refinement in electron and X-ray crystallography—Diketopiperazine revisited. Z. Krist. 2010, 225, 86–93. [Google Scholar] [CrossRef]
- Palatinus, L. Structure Solution by Charge Flipping. In Unitin Electron Crystallography and Powder Diffraction; NATO Science for Peace and Security Series B: Physics and Biophysics; Kolb, U., Shankland, K., Meshi, L., David, W.I.F., Eds.; Springer: Dordrecht, The Netherlands, 2012; ISBN 9789400755796. [Google Scholar] [CrossRef]
- Palatinus, L.; Van Der Lee, A. Symmetry determination following structure solution in P1. J. Appl. Crystallogr. 2008, 41, 975–984. [Google Scholar]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; Shankland, K.; Shankland, N. Routine determination of molecular crystal structures from powder diffraction data. Chem. Commun. 1998, 8, 931–932. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P.W.; Bohle, D.S.; Kosar, A.D.; Madsen, S.K. The structure of malaria pigment β-haematin. Nature 2000, 404, 307–310. [Google Scholar]
- Favre-Nicolin, V.; Cerny, R. FOX, ‘free objects for crystallography’: A modular approach to ab initio structure determination from powder diffraction. J. Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- Černý, R.; Favre-Nicolin, V. Direct space methods of structure determination from powder diffraction: Principles, guidelines and perspectives. Z. Krist.-Cryst. Mater. 2007, 222, 105–113. [Google Scholar] [CrossRef]
- Andreev, Y.G.; MacGlashan, G.S.; Bruce, P.G. Ab initio solution of a complex crystal structure from powder-diffraction data using simulated-annealing method and a high degree of molecular flexibility. Phys. Rev. B 1997, 55, 12011–12017. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Johnston, R.L.; Kariuki, B.M. The Genetic Algorithm: Foundations and Applications in Structure Solution from Powder Diffraction Data. Acta Crystallogr. A 1998, 54, 632–645. [Google Scholar] [CrossRef]
- Shankland, K.; David, W.I.F.; Csoka, T. Crystal structure determination from powder diffraction data by the application of a genetic algorithm. Z. Krist.-Cryst. Mater. 1997, 212, 550–552. [Google Scholar] [CrossRef]
- Deem, M.W.; Newsam, J.M. Framework crystal structure solution by simulated annealing: Test application to known zeolite structures. J. Am. Chem. Soc. 1992, 114, 7189–7198. [Google Scholar] [CrossRef]
- Putz, H.; Schön, J.C.; Jansen, M. Combined method for ab initio structure solution from powder diffraction data. J. Appl. Crystallogr. 1999, 32, 864–870. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Endeavour—Structure Solution from Powder Diffraction. Crystal Impact. Available online: https://www.crystalimpact.de/endeavour (accessed on 19 February 2021).
- Hadermann, J.; Abakumov, A.M.; Tsirlin, A.A.; Filonenko, V.P.; Gonnissen, J.; Tan, H.; Verbeeck, J.; Gemmi, M.; Antipov, E.V.; Rosne, H. Direct space structure solution from precession electron diffraction data: Resolving heavy and light scatterers in Pb13Mn9O25. Ultramicroscopy 2010, 110, 881–890. [Google Scholar] [CrossRef]
- Gorelik, T.; Matveeva, G.; Kolb, U.; Schleuß, T.; Kilbinger, A.F.M.; van de Streek, J.; Bohle, A.; Brunklaus, G. H-bonding schemes of di- and tri-p-benzamides assessed by a combination of electron diffraction, X-ray powder diffraction and solid-state NMR. CrystEngComm 2010, 12, 1824–1832. [Google Scholar] [CrossRef]
- Das, P.P.; Mugnaioli, E.; Nicolopoulos, S.; Tossi, C.; Gemmi, M.; Galanis, A.; Borodi, G.; Pop, M.M. Crystal Structures of Two Important Pharmaceuticals Solved by 3D Precession Electron Diffraction Tomography. Org. Process Res. Dev. 2018, 22, 1365–1372. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Faber, J. A New Relational Database Format for Powder Diffraction, Data Mining and Materials Characterization. Suppl. J. Ceram. Soc. Jpn. 2004, S1434–S1438. [Google Scholar] [CrossRef]
- Grazulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.T.; Quiros, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database—An open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Dill, K.A.; Phillips, A.T.; Rosen, J.B. Molecular Structure Prediction by Global Optimization. In Developments in Global Optimization; Bomze, I.M., Csendes, T., Horst, R., Pardalos, P.M., Eds.; Kluwer: Dordrecht, The Netherlands, 1997; pp. 217–234. [Google Scholar]
- Calestani, G.; Rizzoli, C. Crystal structure of the YBa2Cu3O7 superconductor by single-crystal X-ray diffraction. Nature 1987, 328, 606–607. [Google Scholar] [CrossRef]
- Calamiotou, M.; Gantis, A.; Margiolaki, I.; Palles, D.; Siranidi, E.; Liarokapis, E. Phase separation, microstructure and superconductivity in the Y1−xPrxBa2Cu3Oy compounds. J. Phys. Condens. Matter 2008, 20, 395224. [Google Scholar] [CrossRef]
- Paulus, E.F.; Miehe, G.; Fuess, H.; Yehia, I.; Löchner, U. The crystal structure of BaCuO2. J. Solid State Chem. 1991, 90, 17–26. [Google Scholar] [CrossRef]
- Kuntzinger, S.; Ghermani, N.E. Electron density distribution and Madelung potential in α-spodumene, LiAl(SiO3)2, from two-wavelength high-resolution X-ray diffraction data. Acta Crystallogr. Sect. B 1999, 55, 273–284. [Google Scholar] [CrossRef]
- Redhammer, G.J.; Roth, G. Structural variation and crystal chemistry of LiMe3+Si2O6 clinopyroxenes Me3+ = Al, Ga, Cr, V, Fe, Sc and In. Z. Krist.-Cryst. Mater. 2004, 219, 278–294. [Google Scholar] [CrossRef]
- Redhammer, G.J.; Roth, G. Structural changes upon the temperature dependent C2/c → P21/c phase transition in LiMe3+Si2O6 clinopyroxenes, Me = Cr, Ga, Fe, V, Sc and In. Z. Krist.-Cryst. Mater. 2004, 219, 585–605. [Google Scholar] [CrossRef]
- Iezzi, G.; Bromiley, G.D.; Cavallo, A.; Das, P.P.; Karavassili, F.; Margiolaki, I.; Stewart, A.A.; Tribaudino, M.; Wright, J.P. Solid solution along the synthetic LiAlSi2O6-LiFeSi2O6 (spodumene-ferri-spodumene) join: A general picture of solid solutions, bond lengths, lattice strains, steric effects, symmetries, and chemical compositions of Li clinopyroxenes. Am. Mineral. 2016, 101, 2498–2513. [Google Scholar] [CrossRef]
- Bertrand, C.; Galez, P.; Jorda, J.L.; Gladyshevskii, R.E. The Pr (Ba1−xPrx)2 Cu3O7 + d solid solution. A crystal structure and phase diagram study. Phys. C Supercond. 1999, 321, 151–161. [Google Scholar] [CrossRef]
- Kipka, R.; Müller-Buschbaum, H. Über Oxocuprate, XX Ein Erdalkalioxocuprat(II) mit geschlossenen Baugruppen: BaCuO2/about Oxocuprates, XX Alkaline-earth Oxocuprate(II) with Closed Structural Groups: BaCuO2. Z. Nat. B 1977, 32, 121–123. [Google Scholar] [CrossRef]
- Plana-Ruiz, S. Development & Implementation of an Electron Diffraction Approach for Crystal Structure Analysis—TUprints (TU-Darmstadt.De). Available online: https://www.tesisenred.net/handle/10803/670887#page=1 (accessed on 12 January 2022).
- Plana-Ruiz, S.; Krysiak, Y.; Portillo, J.; Alig, E.; Estradé, S.; Peiró, F.; Kolb, U. Fast-ADT: A fast and automated electron diffraction tomography setup for structure determination and refinement. Ultramicroscopy 2020, 211, 112951. [Google Scholar] [CrossRef]
- Plana-Ruiz, S.; Portillo, J.; Estradé, S.; Peiró, F.; Kolb, U.; Nicolopoulos, S. Quasi-parallel precession diffraction: Alignment method for scanning transmission electron microscopes. Ultramicroscopy 2018, 193, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Smeets, S.; Wang, B.; Cichocka, M.O.; Ångström, J.; Wan, W. Continuous rotation electron diffraction data for zeolite SSZ-27. Instamatic Zenodo 2018. [Google Scholar] [CrossRef]
- Gorelik, T.E.; Stewart, A.A.; Kolb, U. Structure solution with automated electron diffraction tomography data: Different instrumental approaches. J. Microsc. 2011, 244, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kolb, U.; Krysiak, Y.; Plana-Ruiz, S. Automated electron diffraction tomography—Development and applications. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 463–474. [Google Scholar] [CrossRef]
- Cascarano, G.L.; Giacovazzo, C.; Carrozzini, B. Crystal structure solution via precession electron diffraction data: The BEA algorithm. Ultramicroscopy 2010, 111, 56–61. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Krist.-Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M. Crystal Structure Determination from Powder Diffraction Data. Chem. Mater. 1996, 8, 2554–2570. [Google Scholar] [CrossRef]
- Louër, D. Advances in Powder Diffraction Analysis. Acta Cryst. Sect. A 1998, 54, 922–933. [Google Scholar] [CrossRef]
- Kaplow, R.; Rowe, T.A.; Averbach, B.L. Atomic Arrangement in Vitreous Selenium. Phys. Rev. 1968, 168, 1068–1079. [Google Scholar] [CrossRef]
- McGreevy, R.L. 6—Reverse Monte Carlo Methods for Structural Modelling. In Computer Modeling in Inorganic Crystallography; Catlow, C.R.A., Ed.; Academic Press: London, UK, 1997; pp. 151–184. [Google Scholar] [CrossRef]
- Pareto, V. Cours d’Économie Politique; F. Rouge: Laussane, Switzerland, 1896. [Google Scholar]
- De Leeuw, S.W.; Perram, J.W.; Smith, E.R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. A 1980, 373, 27–57. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, A.; Hundt, R.; Schoen, J.C.; Jansen, M. A New Algorithm for Space-Group Determination. J. Appl. Cryst. 1998, 31, 922–928. [Google Scholar] [CrossRef]
- Hundt, R.; Schoen, J.C.; Hannemann, A.; Jansen, M. Determination of Symmetries and Idealized Cell Parameters for Simulated Structures. J. Appl. Cryst. 1999, 32, 413–416. [Google Scholar] [CrossRef]
- Prince, E. (Ed.) International Tables for Crystallography. Vol. C: Mathematical, Physical and Chemical Tables, 3rd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Moeck, P.; (Portland State University, Portland, OR, USA). Personal communication, 2008.
- Ge, M.; Yang, T.; Wang, Y.; Carraro, F.; Liang, W.; Doonan, C.; Falcaro, P.; Zheng, H.; Zou, X.; Huang, Z. On the completeness of three-dimensional electron diffraction data for structural analysis of metal-organic frameworks. Faraday Discuss. 2021, 231, 66–80. [Google Scholar] [CrossRef]
- Wennmacher, J.T.C.; Zaubitzer, C.; Li, T.; Bahk, Y.K.; Wang, J.; van Bokhoven, J.A.; Gruene, T. 3D-structured supports create complete data sets for electron crystallography. Nat. Commun. 2019, 10, 3316. [Google Scholar] [CrossRef]
- Palatinus, L.; Petříček, V.; Corrêa, C.A. Structure refinement using precession electron diffraction tomography and dynamical diffraction: Theory and implementation. Acta Crystallogr. Sect. A 2015, 71, 235–244. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Brown, I.D. 14—The Bond-Valence Method: An Empirical Approach to Chemical Structure and Bonding. Ind. Chem. Libr. 1981, 2, 1–30. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B 1985, 41, 244–247. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-ray: Powder [66] | 3D ED with PED | ||||||
|---|---|---|---|---|---|---|---|
| Direct Methods | Charge-Flipping | Global Optimization | |||||
| ADT3D | PETS2 | ADT3D | PETS2 | ADT3D | PETS2 | ||
| Used Resolution (Å) | - | 0.8 | 0.8 | 0.8 | 0.8 | - | - |
| Total/Used 1 Refl. (#) | - | 425/95 | 426/96 | 425/96 | 426/97 | 425 | 426 |
| Completeness (%) | - | 40.4 | 39.8 | 41.3 | 40.2 | - | - |
| Rint (%) 2 | - | 11.4 | 13.5 | 11.4 | 13.5 | 11.4 | 13.5 |
| R(F)/R1(obs) (%) 3 | - | 10.7 | 11.0 | 37.5 | 56.6 | 59.35 5 | 60.89 5 |
| RMSD 4 | - | 0.123 | 0.079 | 0.013 | 0.104 | 0.130 | 0.120 |
| Atomic fractional coordinates (x, y, z) | |||||||
| Pr1 | 0.5 0.5 0.5 | 0.5 0.5 0.5 | 0.5 0.5 0.5 | 0.5 0.5 0.5 | 0.5 0.5 0.5 | 0.5 0.5 0.5 | 0.5 0.5 0.5 |
| Ba1 | 0.5 0.5 0.1804 | 0.5 0.5 0.165 | 0.0 0.0 0.159 | 0.5 0.5 0.177 | 0.5 0.5 0.164 | 0.5 0.5 0.185 | 0.5 0.5 0.185 |
| Cu1 | 0.0 0.0 0.0 | 0.0 0.0 0.0 | 0.0 0.0 0.0 | 0.0 0.0 0.0 | 0.0 0.0 0.0 | 0.0 0.0 0.0 | 0.0 0.0 0.0 |
| Cu2 | 0.0 0.0 0.3515 | 0.0 0.0 0.293 | 0.0 0.0 0.336 | 0.0 0.0 0.349 | 0.0 0.0 0.320 | 0.0 0.0 0.359 | 0.0 0.0 0.359 |
| O1 | 0.0 0.5 0.0 | 0.0 0.5 0.0 | Not found | Not found | Not found | 0.0 0.5 0.0 | 0.0 0.5 0.0 |
| O2 | 0.0 0.0 0.168 | Not found | Not found | Not found | Not found | 0.0 0.0 0.181 | 0.0 0.0 0.178 |
| O3 | 0 0.5 0.365 | Not found | Not found | Not found | Not found | 0.0 0.5 0.391 | 0 0.5 0.390 |
| O4 | 0.5 0 0.383 | 0.5 0.0 0.3655 | Not found | Not found | Not found | 0.5 0.0 0.391 | 0.5 0 0.389 |
| X-ray: Single-Crystal [67] | 3D ED without PED | |||
|---|---|---|---|---|
| Direct Methods | Charge-Flipping | Global Optimization | ||
| Used Resolution (Å) | - | 0.87 | 0.87 | - |
| Total/Used 1 Refl. (#) | - | 14719/514 | 14719/514 | 14719 |
| Completeness (%) | - | 99.4 | 100 | - |
| Rint (%) 2 | - | 60.2 | 60.2 | |
| R(F)/R1(obs) (%) 3 | - | 15.6 | 116.1 | 112.50 5 |
| RMSD 4 | - | 0.069 | 0.382 | 0.698 |
| Atomic fractional coordinates (x, y, z, a) | ||||
| Ba1 | 0.0 0.151 0.31 | 0.0 0.1455 0.3051 | 0.0 0.2916 0.1831 | 0 0.1396 0.3283 |
| Ba2 | 0.0 0.364 0.364 | 0.0 0.3646 0.3646 | 0.0 0.3475 0.3475 | 0.137 0.137 0.5 |
| Ba3 | 0.177 0.177 0.177 | 0.1785 0.1785 0.1785 | 0.1335 0.1335 0.1335 | 0.1710 0.1710 0.1710 |
| Ba4 | 0.0 0.0 0.0 | Not found | Not found | 0.0 0.0 0.0 |
| Cu1 | 0.25 0.15 0.35 | 0.25 0.1519 0.3481 | 0.2659 0.1014 0.3827 | 0.25 0.1445 0.3555 |
| Cu2 | 0.0 0.125 0.125 | 0.0 0.1177 0.1177 | Not found | 0.0 0.1281 0.1281 |
| Cu3 | 0.206 0.0 0.0 | 0.2259 0.0 0.0 | 0.1502 0.0 0.0 | - |
| Cu4 | 0.43 0.0 0.0 (0.5) | 0.4302 0.0 0.0 (1) | 0.3701 0.0 0.0 (1) | - |
| O1 | 0.072 0.072 0.186 | Not found | 0.0911 0.0911 0.2380 | - |
| O2 | 0.144 0.144 0.343 | 0.1488 0.1488 0.3151 | Not found | 0.1497 0.1497 0.3283 |
| O3 | 0.267 0.267 0.085 | 0.2637 0.2637 0.0897 | Not found | - |
| O4 | 0.25 0.0 0.5 | 0.25 0.0 0.5 | Not found | 0.25 0.0 0.5 |
| O5 | 0.338 0.0 0.0 | Not found | Not found | - |
| O6 | 0.0 0.112 0.44 (0.25) | Not found | Not found | - |
| X-ray: Single-Crystal [65] | 3D ED with PED | ||||||
|---|---|---|---|---|---|---|---|
| Direct Methods | Charge-Flipping | Global Optimization | |||||
| ADT3D | PETS2 | ADT3D | PETS2 | ADT3D | PETS2 | ||
| Used Resolution (Å) | - | 0.7 | 0.7 | 0.7 | 0.7 | - | - |
| Total/Used 1 Refl. (#) | - | 7007/312 | 1670/322 | 7007/313 | 1670/323 | 7007 | 1670 |
| Completeness (%) | - | 52.7 | 54.4 | 53.3 | 54.8 | - | - |
| Rint (%) 2 | - | 10.0 | 19.3 | 10.0 | 19.3 | 10.0 | 19.3 |
| R(F)/R1(obs) (%) 3 | - | 16.5 | 15.3 | 71.8 | 65.8 | 94.27 5 | 76.32 5 |
| RMSD 4 | - | 0.026 | 0.015 | 0.063 | 0.020 | 0.056 | 0.105 |
| Atomic fractional coordinates (x, y, z) | |||||||
| Li1 | 0.0 0.2746 0.25 | Not found | Not found | Not found | Not found | 0.0 0.2734 0.25 | 0.0 0.2609 0.25 |
| Al1/Fe1 | 0.0 0.9067 0.25 | 0.0 0.0973 0.75 | 0.0 0.0981 0.75 | 0.0 0.0989 0.75 | 0.0 0.0980 0.75 | 0.500 0.404 0.25 | 0.500 0.408 0.25 |
| Si1 | 0.2941 0.0935 0.2559 | 0.2926 0.0904 0.2509 | 0.2948 0.0913 0.2544 | 0.2922 0.0983 0.2491 | 0.2901 0.0937 0.2528 | 0.2956 0.0905 0.2572 | 0.3003 0.0888 0.2648 |
| O1 | 0.1096 0.0825 0.1404 | 0.1012 0.0851 0.1385 | 0.1064 0.0809 0.1414 | 0.1157 0.0726 0.1488 | 0.1091 0.0803 0.1412 | 0.1172 0.0896 0.1483 | 0.1141 0.0836 0.1499 |
| O2 | 0.3647 0.2669 0.3004 | 0.3737 0.2638 0.3040 | 0.3682 0.2635 0.3032 | 0.3800 0.2613 0.2826 | 0.3732 0.2644 0.3040 | 0.3589 0.2620 0.2890 | 0.3619 0.2560 0.2933 |
| O3 | 0.3565 0.9867 0.0585 | 0.3546 0.9872 0.0549 | 0.3569 0.9876 0.0566 | Not found | 0.3561 0.9870 0.0558 | 0.3537 −0.014 0.0625 | 0.3532 −0.013 0.0576 |
| X-ray: Single-Crystal [63] | 3D ED with PED | ||||||
|---|---|---|---|---|---|---|---|
| Direct Methods | Charge-Flipping | Global Optimization | |||||
| ADT3D | PETS2 | ADT3D | PETS2 | ADT3D | PETS | ||
| Used Resolution (Å) | - | 0.7 | 0.7 | 0.7 | 0.7 | - | - |
| Total/Used 1 Refl. (#) | - | 1597/256 | 749/261 | 1597/257 | 749/262 | 1597 | 749 |
| Completeness (%) | - | 44.8 | 45.7 | 46.7 | 47.4 | - | - |
| Rint (%) 2 | - | 11.8 | 13.1 | 11.8 | 13.1 | 11.8 | 13.1 |
| R(F)/R1(obs) (%) 3 | - | 12.8 | 9.3 | 38.0 | 48.9 | 46.59 5 | 44.82 5 |
| RMSD 4 | - | 0.017 | 0.025 | 0.024 | 0.017 | 0.035 | 0.027 |
| Atomic fractional coordinates (x, y, z) | |||||||
| Li1 | 0.0 0.2746 0.25 | 0.0 0.2688 0.25 | 0.0 0.2791 0.25 | Not found | Not found | 0.0 0.2615 0.25 | 0.0 0.2725 0.25 |
| Al1 | 0.0 0.9067 0.25 | 0.0 0.9029 0.25 | 0.0 0.9066 0.25 | 0.0 0.9065 0.25 | 0.0 0.9044 0.25 | 0.500 0.408 0.246 | 0.500 0.407 0.246 |
| Si1 | 0.2941 0.0935 0.2559 | 0.2980 0.0951 0.2555 | 0.2974 0.0945 0.2517 | 0.2892 0.0951 0.2630 | 0.2917 0.0941 0.2565 | 0.2899 0.0942 0.2690 | 0.2941 0.0944 0.2720 |
| O1 | 0.1096 0.0825 0.1404 | 0.1040 0.0856 0.1378 | 0.1096 0.0830 0.1400 | 0.1019 0.0828 0.1420 | 0.1049 0.0844 0.1430 | 0.1137 0.0840 0.1310 | 0.1129 0.0846 0.1250 |
| O2 | 0.3647 0.2669 0.3004 | 0.3647 0.2691 0.2972 | 0.3565 0.2658 0.3073 | 0.3628 0.2666 0.2999 | 0.3646 0.2663 0.3014 | 0.3586 0.2665 0.2980 | 0.3611 0.2652 0.2960 |
| O3 | 0.3565 0.9867 0.0585 | 0.3555 0.9912 0.0598 | 0.3628 0.9872 0.0469 | 0.3601 0.9920 0.0579 | 0.3618 0.9914 0.0598 | 0.3492 −0.016 0.0697 | 0.3543 −0.022 0.0621 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, P.P.; Plana-Ruiz, S.; Galanis, A.S.; Stewart, A.; Karavasili, F.; Nicolopoulos, S.; Putz, H.; Margiolaki, I.; Calamiotou, M.; Iezzi, G. Structure Determination Feasibility of Three-Dimensional Electron Diffraction in Case of Limited Data. Symmetry 2022, 14, 2355. https://doi.org/10.3390/sym14112355
Das PP, Plana-Ruiz S, Galanis AS, Stewart A, Karavasili F, Nicolopoulos S, Putz H, Margiolaki I, Calamiotou M, Iezzi G. Structure Determination Feasibility of Three-Dimensional Electron Diffraction in Case of Limited Data. Symmetry. 2022; 14(11):2355. https://doi.org/10.3390/sym14112355
Chicago/Turabian StyleDas, Partha Pratim, Sergi Plana-Ruiz, Athanassios S. Galanis, Andrew Stewart, Fotini Karavasili, Stavros Nicolopoulos, Holger Putz, Irene Margiolaki, Maria Calamiotou, and Gianluca Iezzi. 2022. "Structure Determination Feasibility of Three-Dimensional Electron Diffraction in Case of Limited Data" Symmetry 14, no. 11: 2355. https://doi.org/10.3390/sym14112355
APA StyleDas, P. P., Plana-Ruiz, S., Galanis, A. S., Stewart, A., Karavasili, F., Nicolopoulos, S., Putz, H., Margiolaki, I., Calamiotou, M., & Iezzi, G. (2022). Structure Determination Feasibility of Three-Dimensional Electron Diffraction in Case of Limited Data. Symmetry, 14(11), 2355. https://doi.org/10.3390/sym14112355