
Orientation of Chiral Molecules by External Electric Fields: Focus on Photodissociation Dynamics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Background
2.1. Molecular Alignment and Oientation
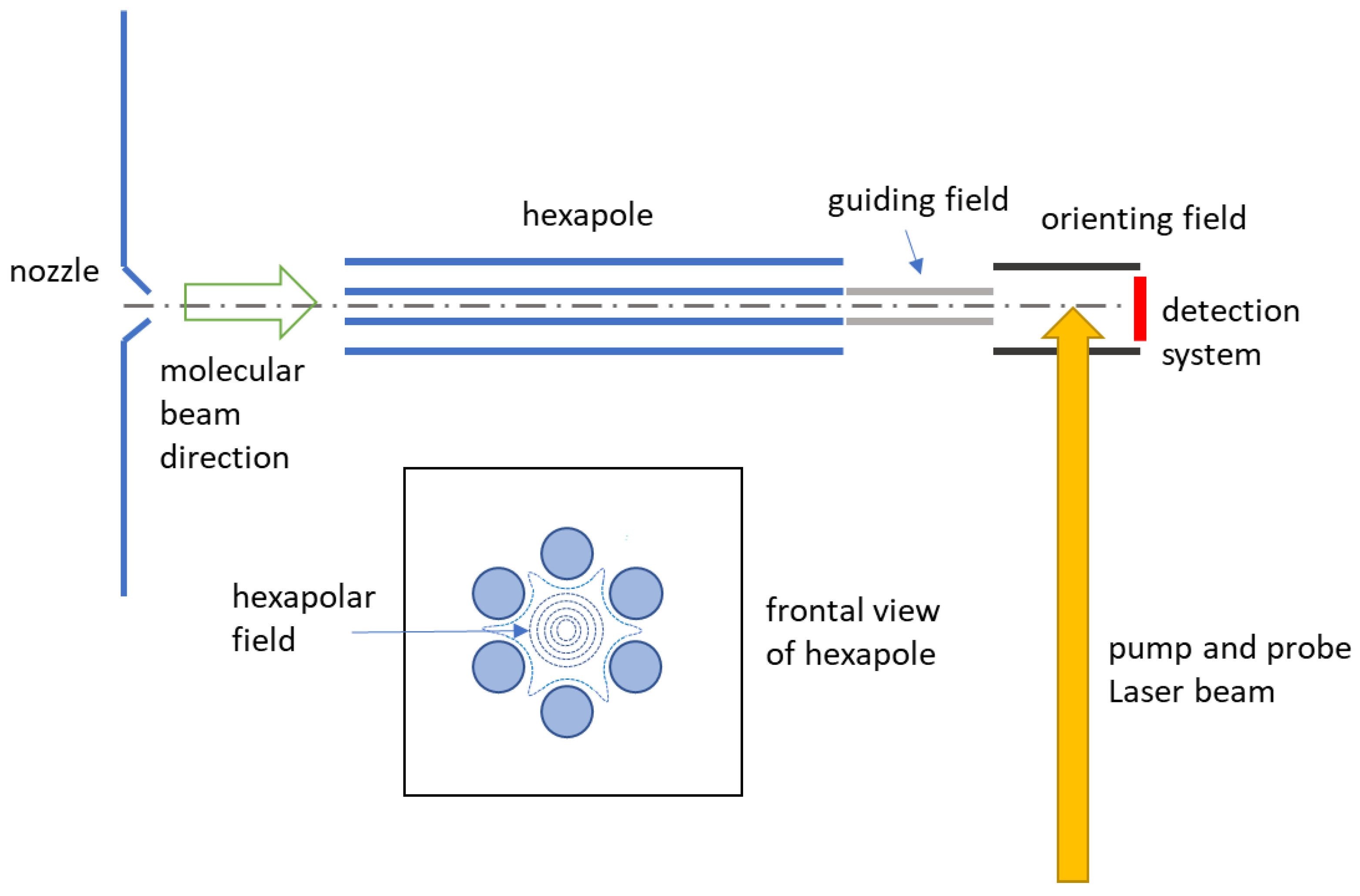
2.2. Experimental Set-Up
2.3. Alignment and Orientation Induced by Electric Fields
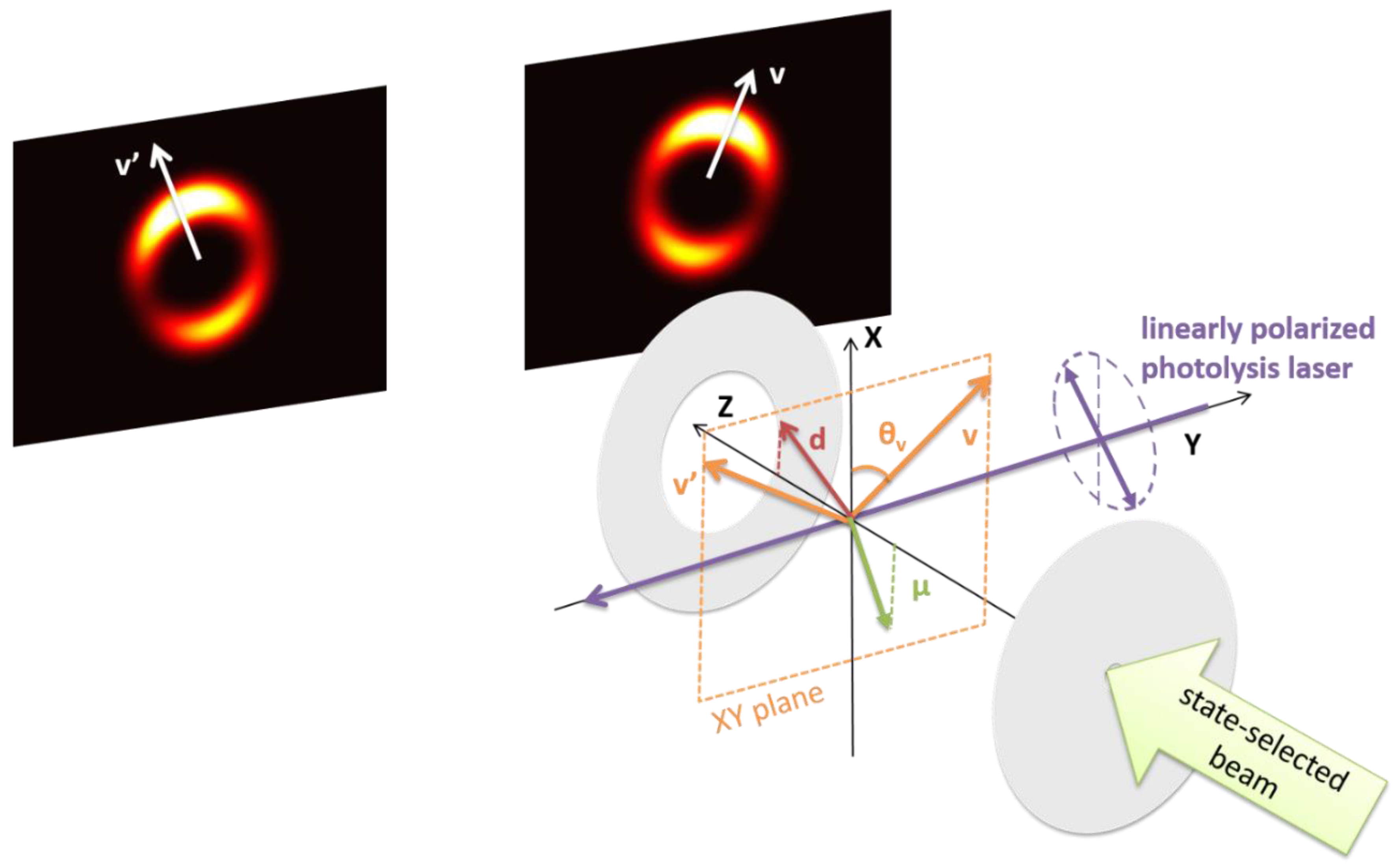
2.4. Photofragment Ion-Imaging
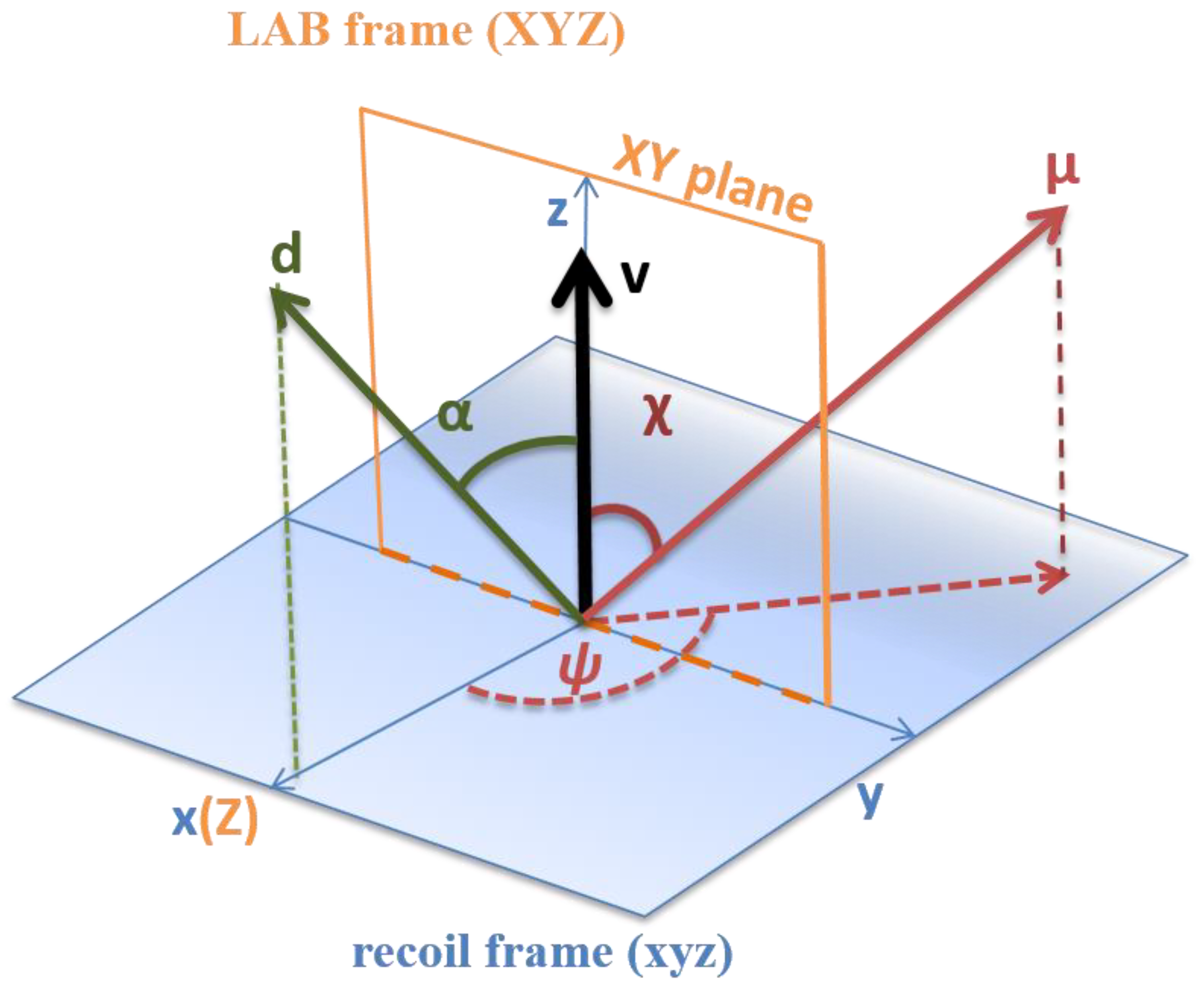
2.5. The Semiclassical Vector Model
3. Review of Photodissociation Experiments on Oriented Chiral Molecules
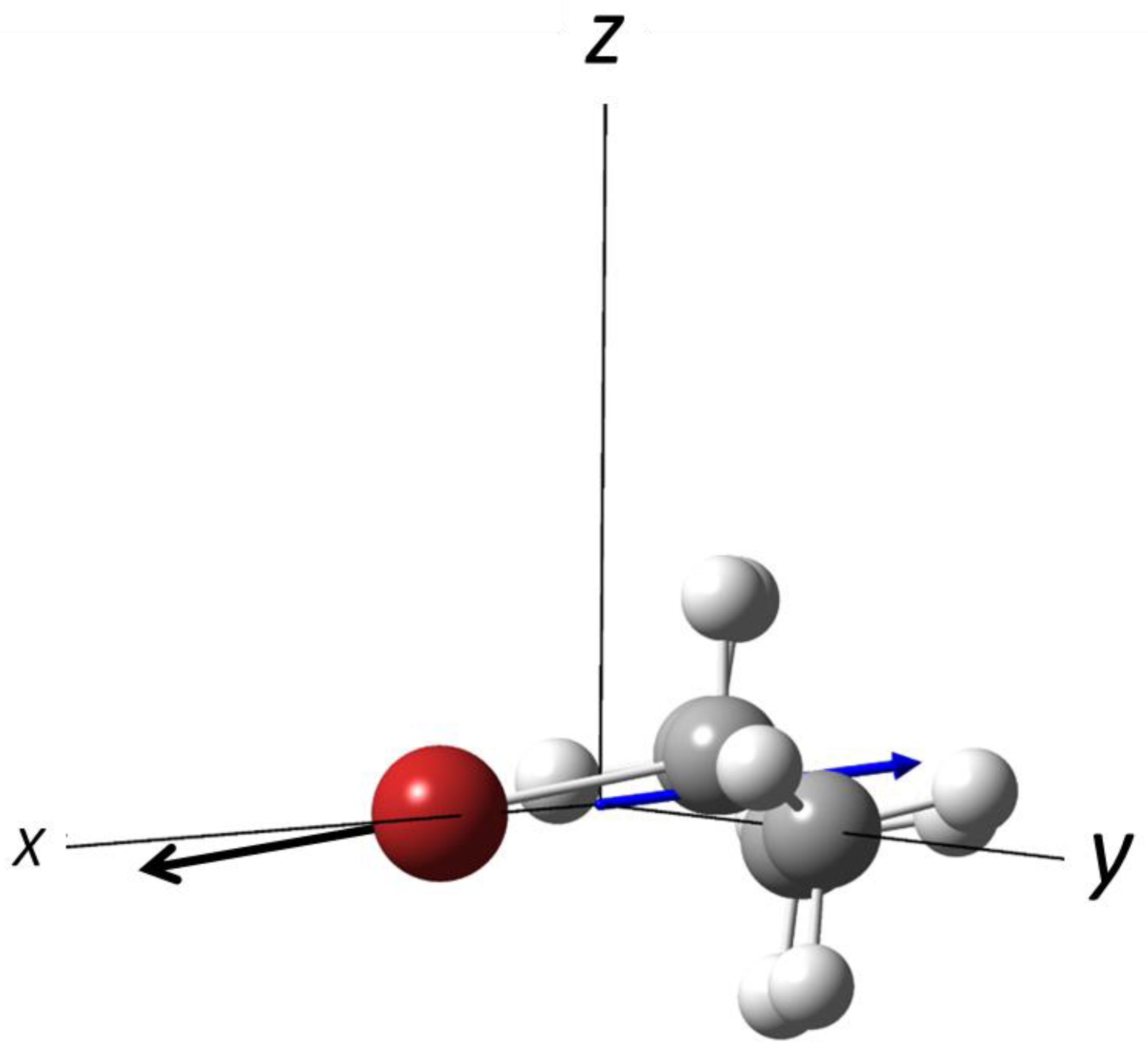
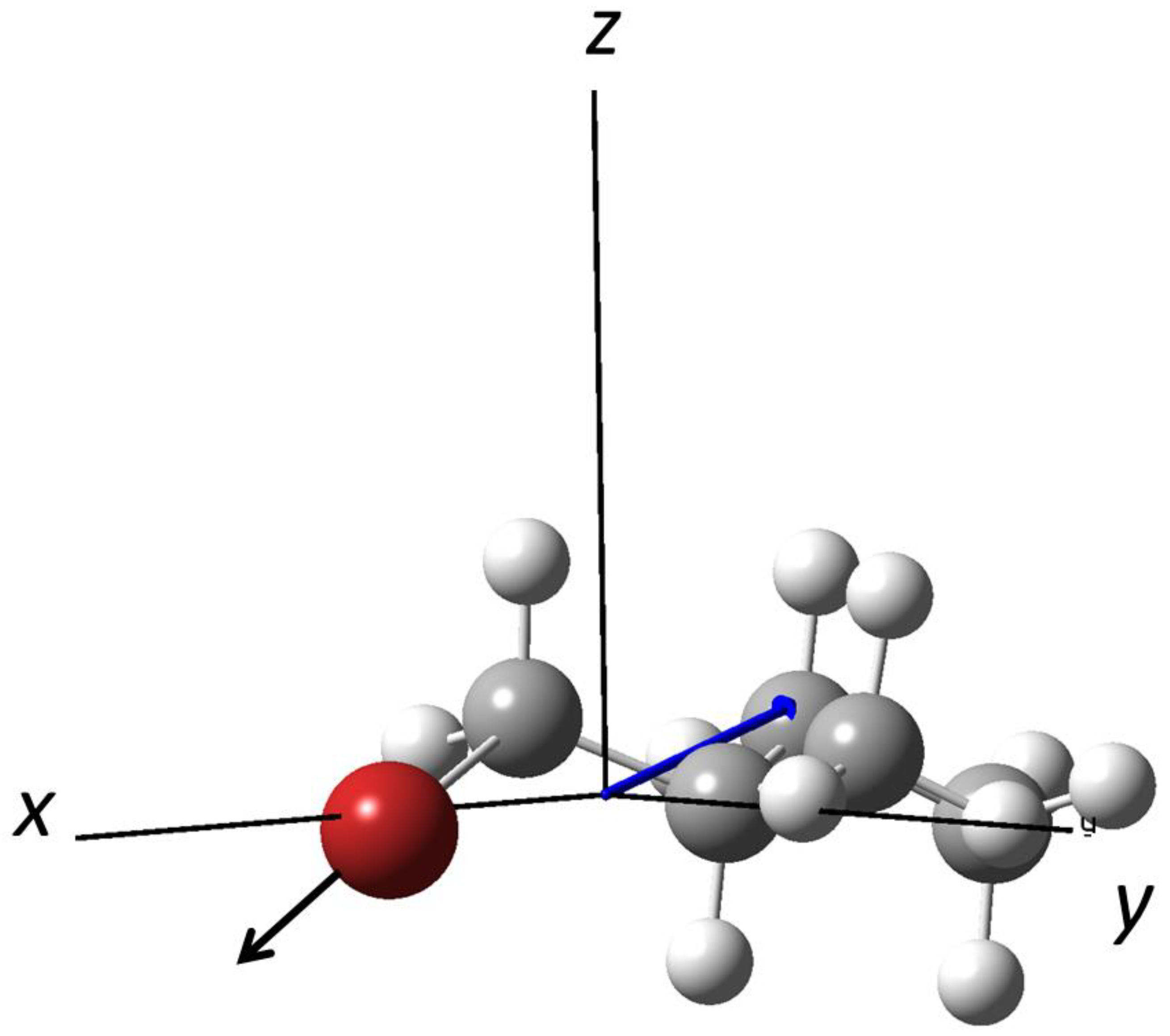
3.1. Photodissociation of Oriented 2-Bromobutane
3.2. Photodissociation of Oriented 1-Bromo-2-Methylbutane
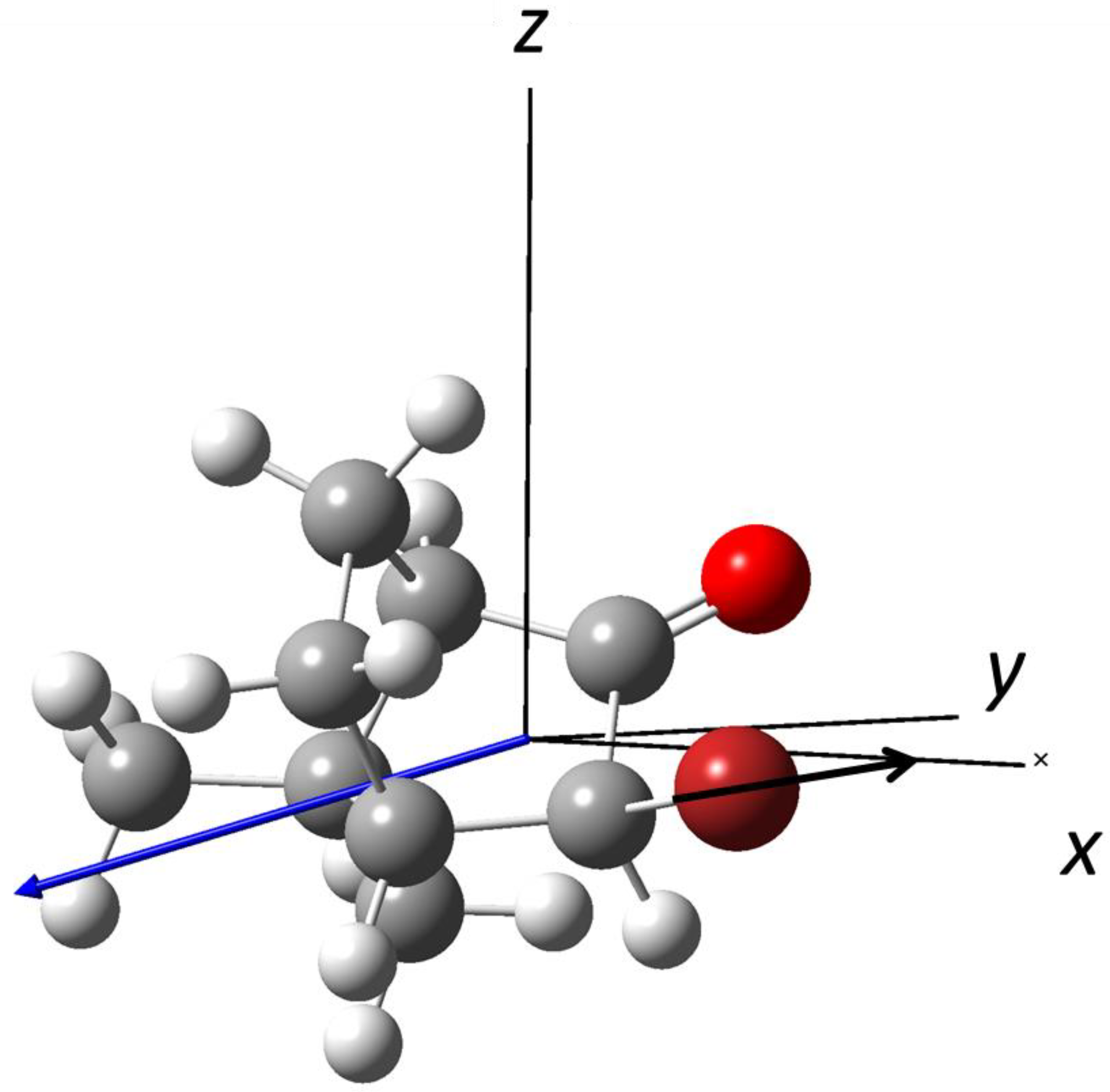
3.3. Photodissociation of (R)-3-Bromocamphor at λ = 234 nm
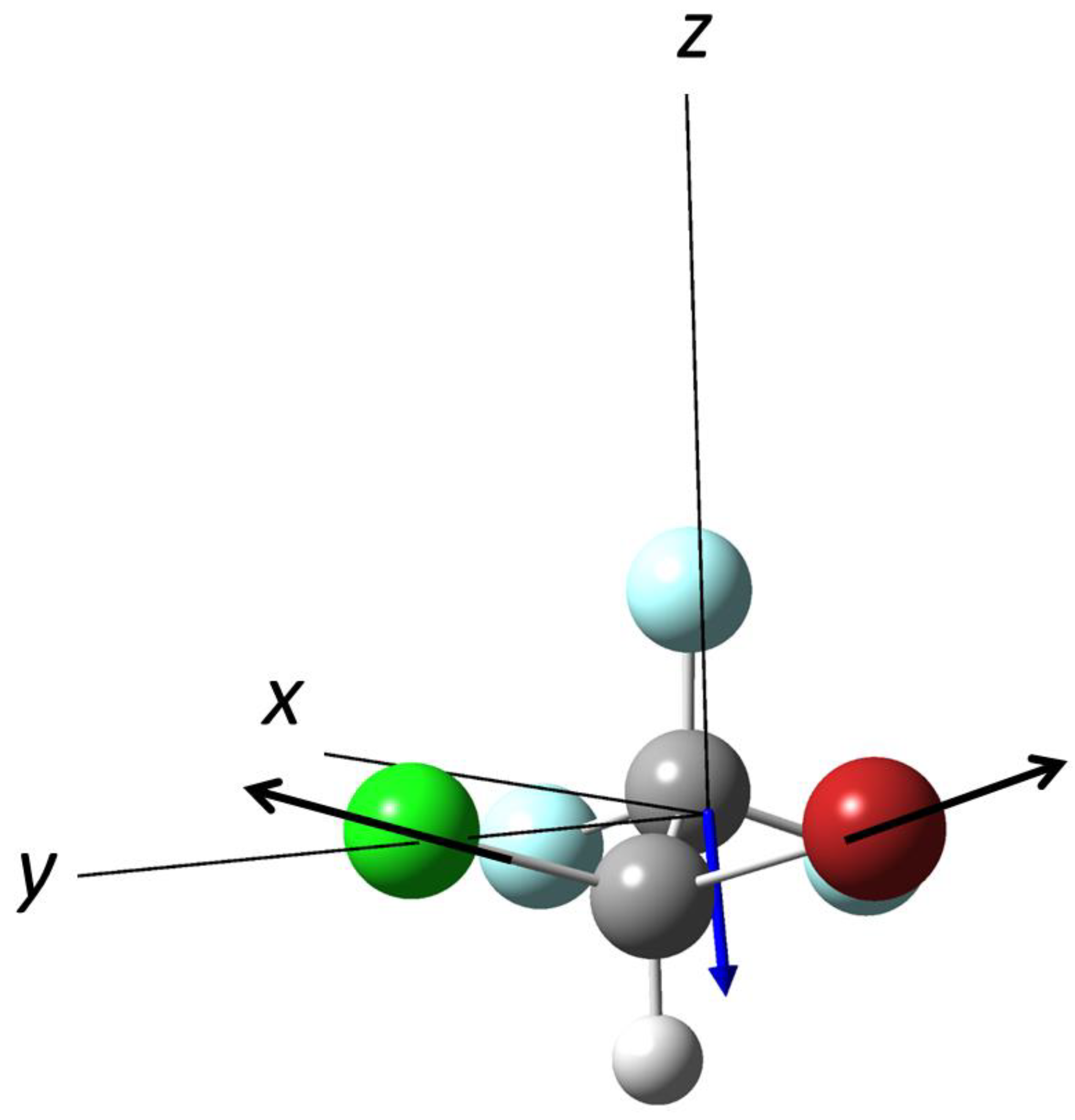
3.4. Photodissociation of Halothane, CF3CH2Cl at λ = 254.1 nm
4. Final Remarks and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scoles, G. Atomic and Molecular Beam Methods; Oxford University Press: New York, NY, USA, 1988; ISBN 0195042808. [Google Scholar]
- Boesl, U. Time-of-flight mass spectrometry: Introduction to the basics. Mass Spectr. Rev. 2017, 36, 86–109. [Google Scholar] [CrossRef]
- Pirani, F.; Cappelletti, D.; Vecchiocattivi, F.; Vattuone, L.; Gerbi, A.; Rocca, M.; Valbusa, U. A simple and compact mechanical velocity selector of use to analyze/select molecular alignment in supersonic seeded beams. Rev. Sci. Instr. 2004, 75, 349. [Google Scholar] [CrossRef]
- Gilijamse, J.J.; Hoekstra, S.; van de Meerakker, S.Y.T.; Groenenboom, G.C.; Meijer, G. Near-Threshold Inelastic Collisions Using Molecular Beams with a Tunable Velocity. Science 2006, 313, 1617–1620. [Google Scholar] [CrossRef][Green Version]
- Van de Meerakker, S.Y.T.; Bethlem, H.L.; Meijer, G. Taming molecular beams. Nat. Phys. 2008, 4, 595–602. [Google Scholar] [CrossRef]
- Suits, A.G. Invited Review Article: Photofragment imaging. Rev. Sci. Instr. 2018, 89, 111101. [Google Scholar] [CrossRef]
- Friedrich, B.; Herschbach, D.R. Spatial orientation of molecules in strong electric fields and evidence for pendular states. Nature 1991, 353, 412–414. [Google Scholar] [CrossRef]
- Rakitzis, T.P.; van den Brom, A.J.; Janssen, M.H.M. Molecular and Laboratory Frame Photofragment Angular Distributions from Oriented and Aligned Molecules. Chem. Phys. Lett. 2003, 372, 187–194. [Google Scholar] [CrossRef]
- Lipciuc, M.L.; van den Brom, A.J.; Dinu, L.; Janssen, M.H.M. Slice Imaging of Photodissociation of Spatially Oriented Molecules. Rev. Sci. Instrum. 2005, 76, 123103. [Google Scholar] [CrossRef]
- Thoman, J.W., Jr.; Chandler, D.W.; Parker, D.H.; Janssen, M.H.M. Two-dimensional Imaging of Photofragments. Laser Chem. 1988, 9, 27–46. [Google Scholar] [CrossRef]
- Rakitzis, T.P.; van den Brom, A.J.; Janssen, M.H.M. Directional Dynamics in the Photodissociation of Oriented Molecules. Science 2004, 303, 1852–1854. [Google Scholar] [CrossRef]
- Van den Brom, A.J.; Rakitzis, T.P.; Janssen, M.H.M. Molecular frame properties from photodissociation of laboratory-oriented symmetric top and chiral molecules. Phys. Scr. 2006, 73, C83–C88. [Google Scholar] [CrossRef]
- Blum, K.; Musigmann, M.; Thompson, D. Elastic Electron Collision with Chiral and Oriented Molecules. In Supercomputing, Collision Processes, and Applications. Physics of Atoms and Molecules; Bell, K.L., Berrington, K.A., Crothers, D.S.F., Hibbert, A., Taylor, K.T., Eds.; Springer: Boston, MA, USA, 2002. [Google Scholar] [CrossRef]
- Lombardi, A.; Palazzetti, F.; Maciel, G.S.; Aquilanti, V.; Sevryuk, M.B. Simulation of oriented collision dynamics of simple chiral molecules. Int. J. Quant. Chem. 2011, 111, 1651–1658. [Google Scholar] [CrossRef]
- Rezende, M.V.C.S.; Coutinho, N.D.; Palazzetti, F.; Lombardi, A.; Carvalho-Silva, V.A. Nucleophilic substitution vs elimination reaction of bisulfide ions with substituted methanes: Exploration of chiral selectivity by stereodirectional first-principles dynamics and transition state theory. J. Mol. Model. 2019, 25, 227. [Google Scholar] [CrossRef] [PubMed]
- Hashinokuchi, M.; Che, D.-C.; Watanabe, D.; Fukuyama, T.; Koyano, I.; Shimizu, Y.; Woelke, A.; Kasai, T. Single |J Ω MJ> state-selection of OH radicals using an electrostatic hexapole field. Phys. Chem. Chem. Phys. 2003, 5, 3911–3915. [Google Scholar] [CrossRef]
- He, L.; Bulthuis, J.; Luo, S.; Wang, J.; Lu, C.; Stolte, S.; Ding, D.; Roeterdink, W.G. Laser induced alignment of state-selected CH3I. Phys. Chem. Chem. Phys. 2015, 17, 24121–24128. [Google Scholar] [CrossRef]
- Cho, V.A.; Bernstein, R.B. Tight focusing of beams of polar polyatomic molecules via the electrostatic hexapole lens. J. Phys. Chem. 1991, 95, 8129–8136. [Google Scholar] [CrossRef]
- Hain, T.D.; Moision, R.M.; Curtiss, T.J. Hexapole state-selection and orientation of asymmetric top molecules: CH2F2. J. Chem. Phys. 1999, 111, 6797. [Google Scholar] [CrossRef]
- Che, D.-C.; Palazzetti, F.; Okuno, Y.; Aquilanti, V.; Kasai, T. Electrostatic Hexapole State-Selection of the Asymmetric-Top Molecule Propylene Oxide. J. Phys. Chem. A 2010, 114, 3280–3286. [Google Scholar] [CrossRef]
- Palazzetti, F.; Maciel, G.S.; Kanda, K.; Nakamura, M.; Che, D.-C.; Kasai, T.; Aquilanti, V. Control of conformers combining cooling by supersonic expansion of seeded molecular beams with hexapole selection and alignment: Experiment and theory on 2-butanol. Phys. Chem. Chem. Phys. 2014, 16, 9866–9875. [Google Scholar] [CrossRef]
- Caglioti, C.; Nakamura, M.; Che, D.-C.; Tsai, P.-Y.; Palazzetti, F. Conformer Selection by Electrostatic Hexapoles: A Theoretical Study on 1-Chloroethanol and 2-Chloroethanol. Symmetry 2022, 14, 317. [Google Scholar] [CrossRef]
- Che, D.-C.; Kanda, K.; Palazzetti, F.; Aquilanti, V.; Kasai, T. Electrostatic hexapole state-selection of the asymmetric-top molecule propylene oxide: Rotational and orientational distributions. Chem. Phys. 2012, 399, 180–192. [Google Scholar] [CrossRef]
- Nakamura, M.; Yang, S., Jr.; Tsai, P.Y.; Kasai, T.; Che, D.-C.; Lombardi, A.; Palazzetti, F.; Aquilanti, V. Hexapole-Oriented Asymmetric-Top Molecules and Their Stereodirectional Photodissociation Dynamics. J. Phys. Chem. A 2016, 120, 5389–5398. [Google Scholar] [CrossRef]
- Nakamura, M.; Yang, S.-J.; Lin, K.-C.; Kasai, T.; Che, D.-C.; Lombardi, A.; Palazzetti, F.; Aquilanti, V. Stereodirectional images of molecules oriented by a variable-voltage hexapolar field: Fragmentation channels of 2-bromobutane electronically excited at two photolysis wavelengths. J. Chem. Phys. 2017, 147, 013917. [Google Scholar] [CrossRef]
- Nakamura, M.; Palazzetti, F.; Tsai, P.-Y.; Yang, S., Jr.; Lin, K.-C.; Kasai, T.; Che, D.-C.; Lombardi, A.; Aquilanti, V. Vectorial imaging of the photodissociation of 2-bromobutane oriented via hexapolar state selection. Phys. Chem. Chem. Phys. 2019, 21, 14164–14172. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Chang, H.-P.; Lin, K.-C.; Kasai, T.; Che, D.-C.; Palazzetti, F.; Aquilanti, V. Stereodynamic Imaging of Bromine Atomic Photofragments Eliminated from 1-Bromo-2-methylbutane Oriented via Hexapole State Selector. J. Phys. Chem. A 2019, 123, 6635–6644. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-P.; Nakamura, M.; Kasai, T.; Lin, K.-C. Photodissociation study of spatially oriented (R)-3-bromocamphor by the hexapole state selector. Mol. Phys. 2021, 120, e1985643. [Google Scholar] [CrossRef]
- Che, D.-C.; Nakamura, M.; Chang, H.-P.; Lin, K.-C.; Kasai, T.; Aquilanti, V.; Palazzetti, F. UV Photodissociation of Halothane in a Focused Molecular Beam: Space-Speed Slice Imaging of Competitive Bond Breaking into Spin–Orbit-Selected Chlorine and Bromine Atoms. J. Phys. Chem. A 2020, 124, 5288–5296. [Google Scholar] [CrossRef] [PubMed]
- Efrati, A.; Irvine, W.T.M. Orientation-Dependent Handedness and Chiral Design. Phys. Rev. X 2014, 4, 011003. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, Z.; Yang, D.; Wang, D.; Ma, C.; Tao, M.; Sun, K.; Yang, J.; Wang, J. Orientation Ordering and Chiral Superstructures in Fullerene Monolayer on Cd (0001). Nanomaterials 2020, 10, 1305. [Google Scholar] [CrossRef]
- Gershnabel, E.; Averbukh, I.S. Orienting Asymmetric Molecules by Laser Fields with Twisted Polarization. Phys. Rev. Lett. 2018, 120, 083204. [Google Scholar] [CrossRef] [PubMed]
- Milner, A.A.; Fordyce, J.A.M.; MacPhail-Bartley, I.; Wasserman, W.; Milner, V.; Tutunnikov, I.; Averbukh, S. Controlled Enantioselective Orientation of Chiral Molecules with an Optical Centrifuge. Phys. Rev. Lett. 2019, 122, 223201. [Google Scholar] [CrossRef] [PubMed]
- Yachmanev, A.; Onvlee, J.; Zak, E.; Owens, A.; Küpper, J. Field-Induced Diastereomers for Chiral Separation. Phys. Rev. Lett. 2019, 123, 243202. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-N.; Chang, L.-C.; Su, T.-M. Optical rotamers of substituted simple alkanes induced by macroscopic translation-rotational motions. Chem. Phys. Lett. 2011, 507, 63–68. [Google Scholar] [CrossRef]
- Su, T.M.; Palazzetti, F.; Lombardi, A.; Grossi, G. Molecular alignment and chirality in gaseous streams and vortices. Rend. Fis. Acc. Lincei 2013, 24, 291–297. [Google Scholar] [CrossRef]
- Zare, R.N.; Harter, W.G. Angular Momentum, Understanding Spatial Aspects in Chemistry and Physics; Wiley-Interscience: New York, NY, USA, 1988. [Google Scholar]
- Orr-Ewing, A.J.; Zare, R.N. Orientation and alignment of reaction products. Annu. Rev. Phys. Chem. 1994, 45, 315. [Google Scholar] [CrossRef]
- Eppink, A.T.J.B.; Parker, D.H. Velocity map imaging of ions and electrons using electrostatic lenses: Application in photoelectron and photofragment ion imaging of molecular oxygen. Rev. Sci. Instr. 1997, 68, 3477. [Google Scholar] [CrossRef]
- Gebhardt, C.R.; Rakitzis, T.P.; Samartzis, P.C.; Ladopoulos, V.; Kitsopoulos, T.N. Slice imaging: A new approach to ion imaging and velocity mapping. Rev. Sci. Inst. 2001, 72, 3848. [Google Scholar] [CrossRef]
- Townsend, D.; Minitti, M.P.; Suits, A.G. Direct current slice imaging. Rev. Sci. Instr. 2003, 74, 2530. [Google Scholar] [CrossRef]
- Che, D.-C.; Kawamata, H.; Nakamura, M.; Kasai, T.; Lin, K.-C. A vector correlation study using a hexapole-oriented molecular beam: Photodissociation dynamics of oriented isohaloethane. Phys. Chem. Chem. Phys. 2022, 24, 5914–5920. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, P.-Y.; Palazzetti, F. Orientation of Chiral Molecules by External Electric Fields: Focus on Photodissociation Dynamics. Symmetry 2022, 14, 2152. https://doi.org/10.3390/sym14102152
Tsai P-Y, Palazzetti F. Orientation of Chiral Molecules by External Electric Fields: Focus on Photodissociation Dynamics. Symmetry. 2022; 14(10):2152. https://doi.org/10.3390/sym14102152
Chicago/Turabian StyleTsai, Po-Yu, and Federico Palazzetti. 2022. "Orientation of Chiral Molecules by External Electric Fields: Focus on Photodissociation Dynamics" Symmetry 14, no. 10: 2152. https://doi.org/10.3390/sym14102152
APA StyleTsai, P.-Y., & Palazzetti, F. (2022). Orientation of Chiral Molecules by External Electric Fields: Focus on Photodissociation Dynamics. Symmetry, 14(10), 2152. https://doi.org/10.3390/sym14102152