
Hybrid Feature Selection of Breast Cancer Gene Expression Microarray Data Based on Metaheuristic Methods: A Comprehensive Review

Abstract
1. Introduction
2. Review Methodology
- Articles on hybrid feature selection, bio-inspired meta-heuristic algorithms, and machine learning;
- Full articles on the outcomes of microarray BC and other cancer.
- Commentaries, reviews, and articles with no full text and book chapters;
- Study on cancer or a certain type of human disease.
3. Microarray Gene Expression Profiles
4. Feature Selection, Metaheuristics, and Classification Methods for Microarray BC Data
4.1. Feature Selection (FS)
4.2. Metaheuristic Methods
4.2.1. Genetic Algorithm
4.2.2. Particle Swarm Optimization (PSO)
4.2.3. Harmony Search Algorithm (HSA)
4.2.4. Ant Colony Optimization (ACO)
4.2.5. Artificial Bee Colony Optimization (ABC)
4.2.6. Firefly Algorithm (FA)
4.2.7. Cuckoo Search Algorithm (CS)
4.2.8. Gravitational Search Algorithm (GSA)
4.2.9. Grey Wolf Optimization (GWO)
4.2.10. Whale Optimization Algorithm (WOA)
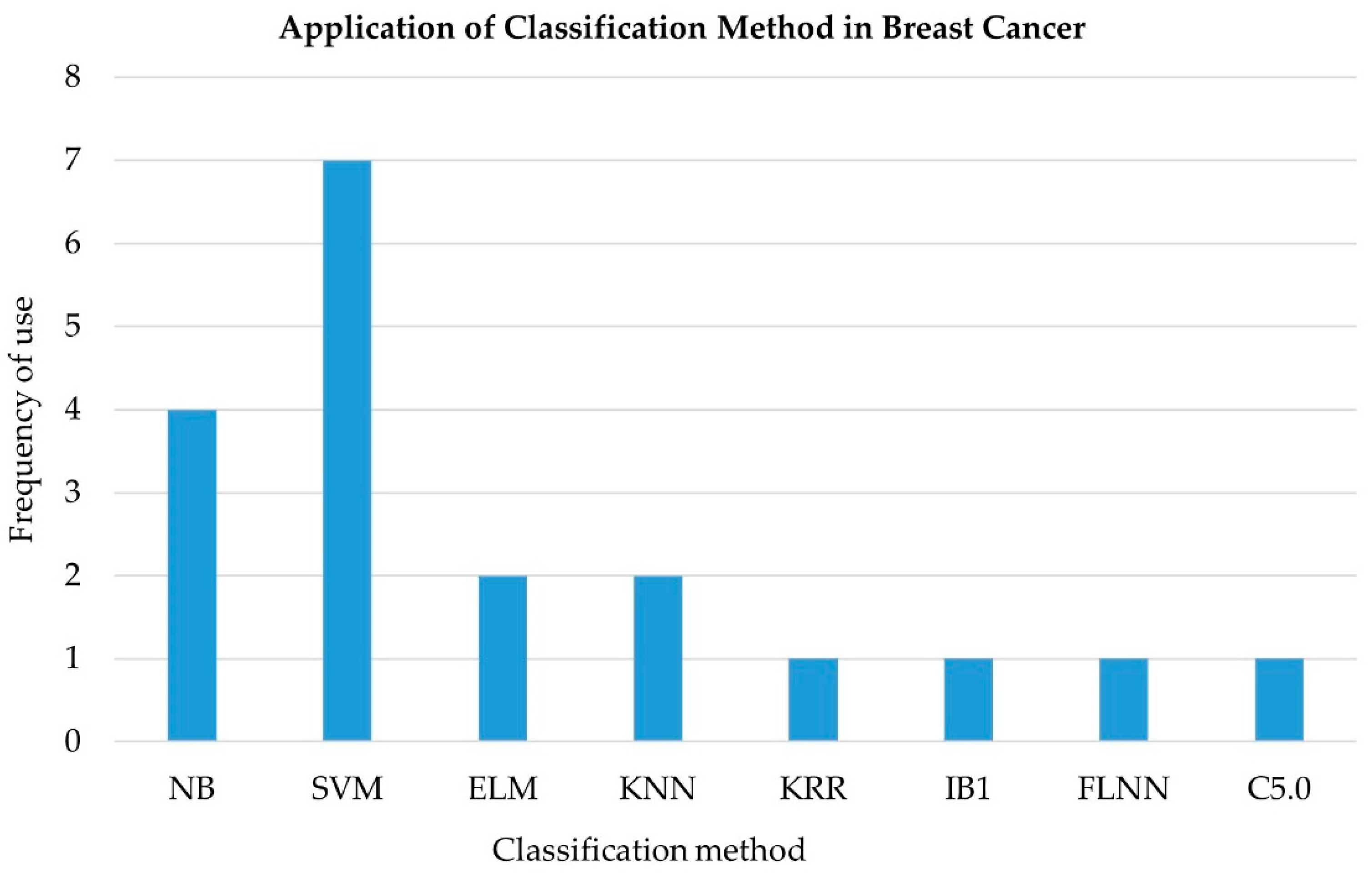
4.3. Classification
4.3.1. Support Vector Machine (SVM)
4.3.2. Random Forest (RF)
4.3.3. K-Nearest Neighbor (KNN)
4.3.4. Naïve Bayes (NB)
4.3.5. Logistic Regression (LR)
4.3.6. Fuzzy Logic
4.3.7. Artificial Neural Network (ANN)
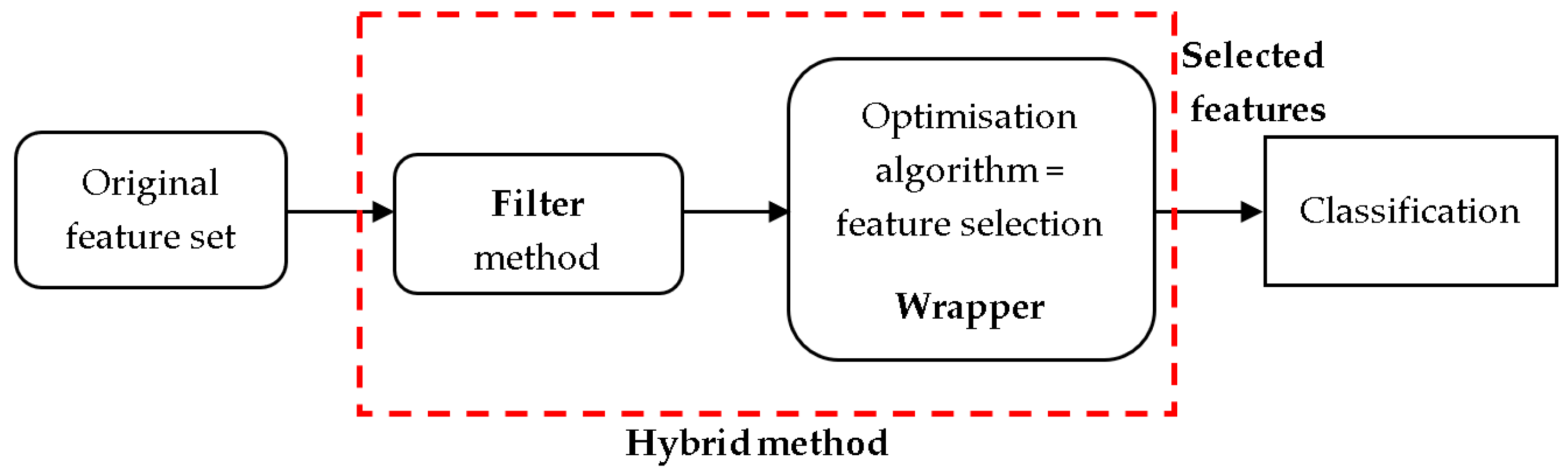
5. Hybrid Feature Selection Based on Metaheuristic Optimization Methods
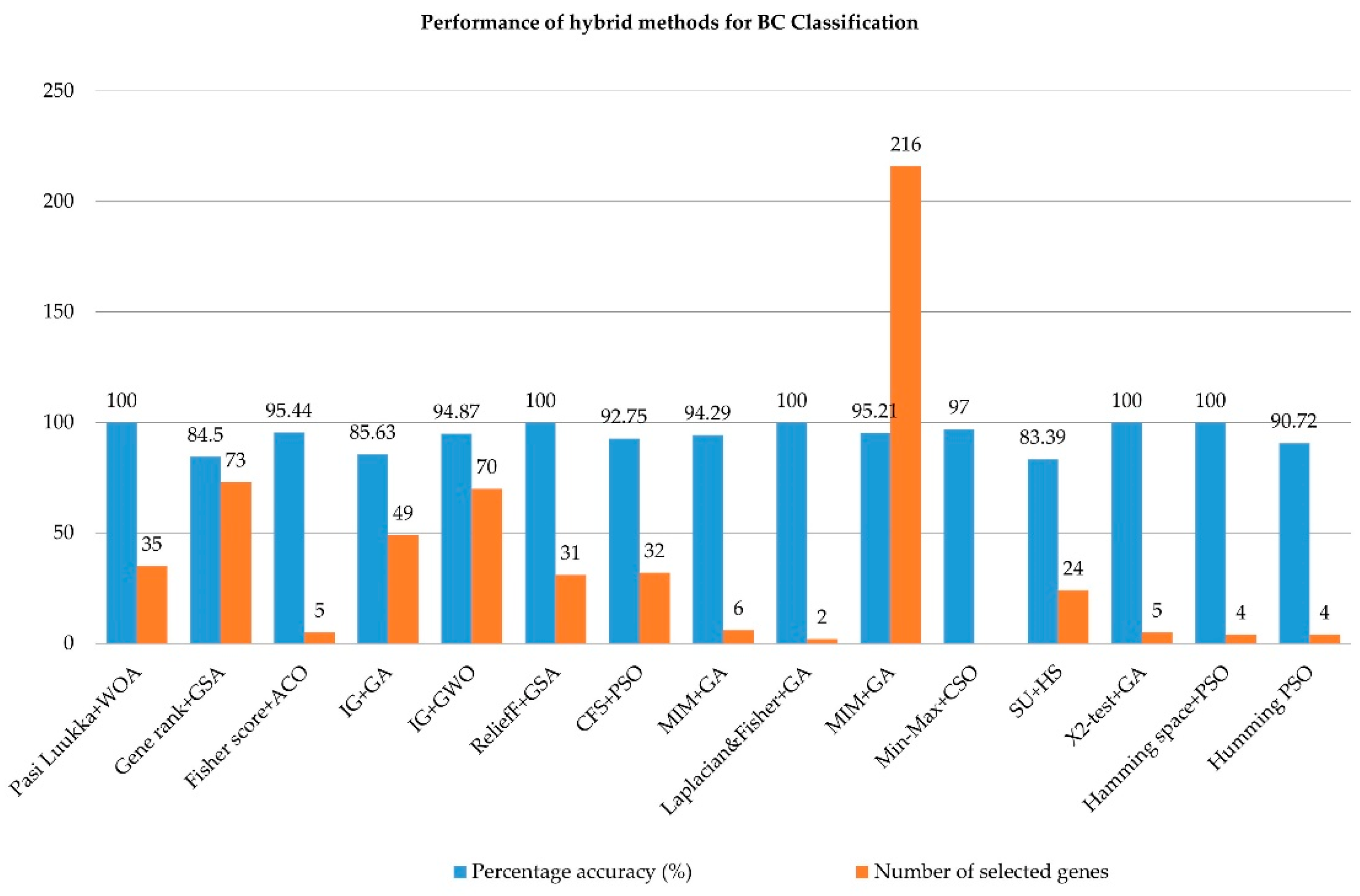
5.1. Hybrid Methods for BC Classification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Studies | Hybrid Based Optimization Method | Classification Method | No. of Selected Genes | No. of Genes | Percentage of Selected Genes (%) | Classification Accuracy | Breast Cancer-Associated | |
|---|---|---|---|---|---|---|---|---|---|
| Filter Method | Wrapper-Based Optimization Method | (%) | /Reference | ||||||
| 1 | [46] | Pasi Luukka | WOA | SVM | 35 | 456 | 7.68 | 100 | [58] |
| 44 | 2905 | 1.51 | 94.12 | [59] | |||||
| 2 | [47] | Gene Rank | GSA | SVM | 73 | 20,545 | 0.355 | 84.5 | [60] |
| 3 | [48] | Fisher Score | ACO | C5.0 | 5 | 24,481 | 0.02 | 95.44 | [61] |
| 4 | [49] | IG | GA | FLNN | 49 | 24,481 | 0.2 | 85.63 | [62] |
| 5 | [50] | IG | GWO | SVM | 70 | 24,481 | 0.29 | 94.87 | [63] |
| 6 | [51] | ReliefF | GSA | NB | 31 | 24,481 | 0.13 | 100 | - |
| 7 | [52] | CFS | PSO | NB | 32 | 24,481 | 0.13 | 92.75 | [64,65] |
| 8 | MIM | GA | ELM | 6 | 24,481 | 0.02 | 94.29 | [61] | |
| [53] | SVM | - | - | 87.06 | |||||
| KNN | - | - | 85.17 | ||||||
| 9 | [26] | Laplacian and Fisher Scores | GA | KNN | - | - | - | 95.5 | [66] |
| SVM | 2 | 3226 | 0.06 | 100 | |||||
| NB | - | - | 100 | ||||||
| 10 | [27] | MIM | GA | ELM | 216 | 24,482 | 0.88 | 95.21 | - |
| 11 | [54] | Max-min scaling/normalization | CSO | KRR | - | 24,481 | - | 97 | [67] |
| 12 | [55] | SU | HS | NB | 14 | 24,481 | 0.06 | 75.97 | [65] |
| IB1 | 24 | 0.09 | 83.39 | ||||||
| 13 | [56] | Statistical | GA | SVM | 5 | 3226 | 0.15 | 100 | [66] |
| X2-test | |||||||||
| 14 | [57] | Hamming space | GA | SVM | 4 | 24,481 | 0.02 | 90.72 | [68] |
| PSO | 4 | 0.02 | 100 | ||||||
5.2. Hybrid Methods for Classification of Other Types of Cancer
| Item | Studies | Hybrid Based Optimization Method | Classification Method | No of Selected Genes | Percentage of Selected Genes (%) | No of Genes | Classification Accuracy | Dataset Associated with Cancer for Possible Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| Filter Method | Wrapper-Based Optimization Method | (%) | |||||||
| 1 | [46] | Pasi Luukka | WOA | SVM | 21 | 1.05 | 2000 | 100 | Colon |
| 25 | 1.08 | 2308 | 100 | SRBCT | |||||
| 30 | 0.421 | 7129 | 100 | Leukemia | |||||
| 37 | 0.295 | 12,534 | 100 | 11_Tumors | |||||
| 22 | 0.402 | 5470 | 100 | DLBCL | |||||
| 2 | [69] | SLI | GA | KNN | 29 | 0.407 | 7129 | 99.97 | CNS |
| 11 | 0.55 | 2000 | 100 | Colon | |||||
| 29 | 0.407 | 7129 | 99.99 | Leukemia | |||||
| 3 | [70] | SNR | PSO | KNN | - | - | 7070 | 99.47 | DLBCL |
| - | - | 8280 | 94.62 | Leukemia | |||||
| - | - | 12,533 | 100 | Prostate | |||||
| - | - | 12,625 | 100 | CML | |||||
| - | - | 2000 | 98.81 | Colon | |||||
| 4 | [71] | Spearman Correlation | PSO | SVM | 18 | 0.9 | 2000 | 92 | Colon |
| 44 | 0.617 | 7129 | 97 | CNS | |||||
| 6 | 0.084 | 7128 | 100 | DLBCL | |||||
| 23 | 0.323 | 7129 | 100 | Leukemia | |||||
| 3 | 0.024 | 12,533 | 100 | Lung | |||||
| 9 | 0.086 | 10,509 | 97 | Prostate | |||||
| 6 | 0.26 | 2308 | 100 | SRBCT | |||||
| 5 | [72] | IG | KH | 17 | 0.85 | 2000 | 96.47 | Colon | |
| 14 | 0.196 | 7129 | 90.34 | CNS | |||||
| 4 | 0.056 | 7129 | 100 | ALL-AML | |||||
| 23 | 0.183 | 12,601 | 96.12 | Lung | |||||
| 3 | 0.0198 | 15,154 | 100 | Ovarian | |||||
| 8 | 0.112 | 7129 | 100 | ALL-MLL3 | |||||
| KNN | 15 | 0.21 | 7129 | 99.44 | ALL-MLL4 | ||||
| 11 | 0.087 | 12,582 | 99.72 | MLL | |||||
| 6 | 0.26 | 2308 | 100 | SRBCT | |||||
| 12 | 0.6 | 2000 | 96.06 | Colon | |||||
| 16 | 0.224 | 7129 | 92.78 | CNS | |||||
| 4 | 0.056 | 7129 | 100 | ALL-AML | |||||
| 20 | 0.159 | 12,601 | 95.98 | Lung | |||||
| 4 | 0.026 | 15,154 | 100 | Ovarian | |||||
| 8 | 0.112 | 7129 | 98.89 | ALL-MLL3 | |||||
| SVM | 15 | 0.21 | 7129 | 97.53 | ALL-MLL4 | ||||
| 13 | 0.103 | 12,582 | 99.31 | MLL | |||||
| 7 | 0.303 | 2308 | 98.65 | SRBCT | |||||
| 12 | 0.6 | 2000 | 90.97 | Colon | |||||
| 12 | 0.168 | 7129 | 82.5 | CNS | |||||
| 14 | 0.196 | 7129 | 98.94 | ALL-AML | |||||
| 3 | 0.024 | 12,601 | 73.58 | Lung | |||||
| NB | 10 | 0.066 | 15,154 | 99.82 | Ovarian | ||||
| 12 | 0.168 | 7129 | 92.31 | ALL-MLL3 | |||||
| 3 | 0.042 | 7129 | 77.33 | ALL-MLL4 | |||||
| 20 | 0.159 | 12,582 | 90.97 | MLL | |||||
| 21 | 0.91 | 2308 | 93.15 | SRBCT | |||||
| 6 | [73] | IG | GA | SVM | 1706 | 13.61 | 12,533 | 100 | Lung |
| 2844 | 18.77 | 15,154 | 95.05 | Ovarian | |||||
| 29 | 0.407 | 7129 | 92.86 | CNS | |||||
| 7 | F-Score | FA | SVM | 15 | 0.75 | 2000 | 94.3 | Colon | |
| [74] | 2 | 0.03 | 7129 | 100 | Lung | ||||
| 5 | 0.07 | 7129 | 100 | Leukemia-1 | |||||
| 8 | 0.35 | 2308 | 100 | SRBCT | |||||
| 10 | 0.14 | 7129 | 97.8 | Leukemia-2 | |||||
| 8 | [75] | Fisher Score | SSA | KELM | 3 | 0.04 | 7129 | 99 | Leukemia |
| 5 | 0.25 | 2000 | 95.5 | Colon | |||||
| 4 | 0.03 | 15,154 | 100 | Ovarian | |||||
| 4 | 0.06 | 7129 | 99.38 | ALL-AML-3 | |||||
| 3 | 0.07 | 4026 | 99.71 | Lymphoma-3 | |||||
| 7 | 0.3 | 2308 | 100 | SRBCT | |||||
| 5 | 0.04 | 12,600 | 98.9 | Lung | |||||
| 9 | ICA | ABC | NB | 16 | 0.8 | 2000 | 98.17 | Colon | |
| [76] | 12 | 0.17 | 7129 | 98.18 | Acute | ||||
| 16 | 0.13 | 12,600 | 98.38 | Prostate | |||||
| 9 | 0.07 | 12,625 | 94.39 | High-grade glioma | |||||
| 24 | 0.19 | 12,533 | 92.76 | Lung Cancer II | |||||
| 15 | 0.21 | 7129 | 97.12 | Leukemia-2 | |||||
| SVM | 15 | 0.75 | 2000 | 97.09 | Colon | ||||
| 13 | 0.18 | 7129 | 96.72 | Acute | |||||
| 13 | 0.103 | 12,600 | 97.2 | Prostate | |||||
| 14 | 0.11 | 12,625 | 93.21 | High-grade glioma | |||||
| 22 | 0.18 | 12,533 | 95.23 | Lung Cancer II | |||||
| 19 | 0.27 | 7129 | 96.43 | Leukemia-2 | |||||
| 10 | ANOVA | FOA | SVM | 4 | 0.06 | 7129 | 98.57 | Leukemia-2 | |
| [77] | 3 | 0.15 | 2000 | 96.9 | Colon | ||||
| 4 | 0.03 | 15,154 | 100 | Ovarian | |||||
| 4 | 0.06 | 7129 | 98.55 | Leukemia-3 | |||||
| 3 | 0.07 | 4026 | 99.87 | Lymphoma-3 | |||||
| 5 | 0.22 | 2308 | 97.77 | SRBCT | |||||
| 4 | 0.03 | 12,600 | 94.56 | Lung cancer-5 | |||||
| 11 | Fisher Score and ReliefF | EPO | SVM | 7 | 0.1 | 7129 | 98.82 | Leukemia | |
| [78] | 8 | 0.4 | 2000 | 96.44 | Colon | ||||
| 5 | 0.03 | 15,154 | 100 | Ovarian | |||||
| 8 | 0.11 | 7129 | 97.66 | ALL-AML-3 | |||||
| 6 | 0.15 | 4026 | 99 | Lymphoma-3 | |||||
| 6 | 0.26 | 2308 | 98.91 | SRBCT | |||||
| 5 | 0.04 | 12,600 | 92.45 | Lung-5 | |||||
| 12 | [51] | ReliefF | GSA | NB | 5 | 0.25 | 2000 | 95.02 | Colon |
| 9 | 0.13 | 7129 | 100 | Central Nervous System | |||||
| 5 | 0.07 | 7129 | 100 | ALL-AML | |||||
| 9 | 0.07 | 12,533 | 100 | Lung | |||||
| 4 | 0.03 | 15,154 | 100 | Ovarian | |||||
| 13 | [53] | MIM | GA | 9 | 0.45 | 2000 | 98.89 | Colon | |
| ELM | 7 | 0.17 | 4026 | 99.07 | DLBCL | ||||
| 8 | 0.11 | 7129 | 99.14 | Leukemia | |||||
| 7 | 0.3 | 2308 | 100 | SRBCT | |||||
| 13 | 0.1 | 12,600 | 96.34 | Lung | |||||
| SVM | - | - | 2000 | 89.31 | Colon | ||||
| - | - | 4026 | 97.07 | DLBCL | |||||
| - | - | 7129 | 92.67 | Leukemia | |||||
| - | - | 2308 | 96.79 | SRBCT | |||||
| - | - | 12,600 | 85.96 | Lung | |||||
| - | - | 2000 | 85.97 | Colon | |||||
| KNN | - | - | 4026 | 95.3 | DLBCL | ||||
| - | - | 7129 | 89.79 | Leukemia | |||||
| - | - | 2308 | 95.23 | SRBCT | |||||
| - | - | 12,600 | 87.78 | Lung | |||||
| 14 | [52] | CFS | PSO | NB | 4 | 0.2 | 2000 | 94.89 | Colon |
| 10 | 0.14 | 7129 | 95.84 | Central nervous system | |||||
| 4 | 0.06 | 7129 | 100 | ALL-AML | |||||
| 10 | 0.08 | 12,533 | 100 | Lung | |||||
| 3 | 0.02 | 15,154 | 100 | Ovarian | |||||
| 6 | 0.08 | 7129 | 100 | ALL-AML-3 | |||||
| 20 | 0.28 | 7129 | 97.63 | ALL-AML-4 | |||||
| 24 | 0.6 | 4026 | 100 | Lymphoma | |||||
| 30 | 0.24 | 12,582 | 100 | MLL | |||||
| 34 | 1.47 | 2308 | 100 | SRBCT | |||||
| 15 | [79] | Mutual Information | AC | Fuzzy | - | - | 2000 | 100 | Colon |
| System | - | - | 7129 | 100 | Leukemia | ||||
| - | - | 12,600 | 90.85 | Prostate | |||||
| 16 | [80] | ABC | 9 | 0.45 | 2000 | 96.77 | Colon | ||
| CFS | SVM | 3 | 0.04 | 7129 | 100 | Leukemia1 | |||
| 2 | 0.03 | 7129 | 100 | Lung | |||||
| 4 | 0.17 | 2308 | 100 | SRBCT | |||||
| 2 | 0.05 | 4026 | 100 | Lymphoma | |||||
| 6 | 0.08 | 7129 | 100 | Leukemia2 | |||||
| GA | 8 | 0.4 | 2000 | 90.32 | Colon | ||||
| 24 | 0.34 | 7129 | 100 | Leukemia1 | |||||
| 20 | 0.28 | 7129 | 100 | Lung | |||||
| 38 | 1.65 | 2308 | 100 | SRBCT | |||||
| 17 | 0.42 | 4026 | 100 | Lymphoma | |||||
| 36 | 0.5 | 7129 | 100 | Leukemia2 | |||||
| PSO | 7 | 0.35 | 2000 | 91.94 | Colon | ||||
| 15 | 0.21 | 7129 | 100 | Leukemia1 | |||||
| 5 | 0.07 | 7129 | 100 | Lung | |||||
| 35 | 1.52 | 2308 | 100 | SRBCT | |||||
| - | - | 4026 | - | Lymphoma | |||||
| - | - | 7129 | - | Leukemia2 | |||||
| 17 | MIM | GA | ELM | 7 | 0.1 | 7130 | 97.62 | Leukemia | |
| [27] | 19 | 0.95 | 2000 | 89.09 | Colon | ||||
| 93 | 0.74 | 12,600 | 97.69 | Prostate | |||||
| 3 | 0.02 | 12,535 | 97.8 | Lung | |||||
| 30 | 1.3 | 2309 | 95.8 | SRBCT | |||||
| 18 | [81] | LA | GA | SVM | 8 | 0.4 | 2000 | 100 | Colon |
| 2 | 0.03 | 7129 | 100 | ALL_AML | |||||
| 6 | 0.26 | 2308 | 99.94 | SRBCT | |||||
| 3 | 0.02 | 12,582 | 95.71 | MLL | |||||
| 10 | 0.17 | 5727 | 89.15 | Tumors_9 | |||||
| 10 | 0.08 | 12,534 | 85.23 | Tumors_11 | |||||
| 19 | [82] | ICA | ABC | NB | 16 | 0.8 | 2000 | 98.14 | Colon |
| 12 | 0.17 | 7129 | 98.68 | Leukemia | |||||
| 16 | 0.13 | 12,600 | 98.88 | Prostate | |||||
| 12 | 0.1 | 12,625 | 94.22 | Glioma | |||||
| 24 | 0.19 | 12,533 | 92.45 | Lung | |||||
| 15 | 0.21 | 7129 | 97.33 | Leukemia-2 | |||||
| PSO | 20 | 1 | 2000 | 91.17 | Colon | ||||
| 19 | 0.27 | 7129 | 95.81 | Leukemia | |||||
| 32 | 0.26 | 12,600 | 93.21 | Prostate | |||||
| 23 | 0.18 | 12,625 | 91.11 | Glioma | |||||
| 41 | 0.33 | 12,533 | 89.19 | Lung | |||||
| 40 | 0.56 | 7129 | 97.13 | Leukaemia-2 | |||||
| GA | 18 | 0.9 | 2000 | 93.38 | Colon | ||||
| 17 | 0.24 | 7129 | 96.76 | Leukemia | |||||
| 27 | 0.21 | 12,600 | 95.32 | Prostate | |||||
| 18 | 0.14 | 12,625 | 95.23 | Glioma | |||||
| 27 | 0.22 | 12,533 | 91.68 | Lung | |||||
| 35 | 0.49 | 7129 | 94.12 | Leukemia-2 | |||||
| 20 | [83] | mRMR | CS | KNN | 8 | 0.11 | 7129 | 71.43 | Central Nervous |
| 82 | 4.1 | 2000 | 85.48 | Colon | |||||
| 206 | 2.89 | 7129 | 100 | Lung | |||||
| 8 | 0.06 | 12,600 | 71.43 | Prostate | |||||
| 8 | 0.11 | 7129 | 71.43 | Central Nervous | |||||
| SVM | 335 | 16.75 | 2000 | 87.1 | Colon | ||||
| 333 | 4.67 | 7129 | 100 | Lung | |||||
| 8 | 0.06 | 12,600 | 71.43 | Prostate | |||||
| 21 | [26] | Laplacian or Fisher Score | GA | KNN | - | - | 2308 | 91.6 | SRBCT |
| - | - | 4026 | 97.9 | DLBCL | |||||
| - | - | 7129 | 97.2 | Leukemia | |||||
| - | - | 12,600 | 95.6 | Prostate | |||||
| SVM | 18 | 0.78 | 2308 | 100 | SRBCT | ||||
| 9 | 0.22 | 4026 | 100 | DLBCL | |||||
| 15 | 0.21 | 7129 | 100 | Leukemia | |||||
| 14 | 0.11 | 12,600 | 96.3 | Prostate | |||||
| NB | - | - | 2308 | 89.2 | SRBCT | ||||
| - | - | 4026 | 95.8 | DLBCL | |||||
| - | - | 7129 | 93.1 | Leukemia | |||||
| - | - | 12,600 | 93.4 | Prostate | |||||
| 22 | [84] | IG | GA | GP | 3 | 0.04 | 7129 | 97.06 | Leukemia |
| 60 | 3 | 2000 | 85.48 | Colon | |||||
| 38 | 0.53 | 7120 | 86.67 | Central nervous sys | |||||
| 11 | 0.38 | 2880 | 74.4 | Lung-Ontario | |||||
| 9 | 0.13 | 7129 | 100 | Lung-Michigan | |||||
| 110 | 1.54 | 7129 | 94.8 | DLBCL | |||||
| 26 | 0.21 | 12,600 | 100 | Prostate | |||||
| 23 | [55] | SU | HS | NB | 26 | 0.36 | 7129 | 100 | ALL-AML |
| 9 | 0.45 | 2000 | 87.53 | Colon | |||||
| 17 | 0.24 | 7129 | 81.42 | CNS | |||||
| 9 | 0.22 | 4026 | 100 | Lymphoma | |||||
| 12 | 0.08 | 15,154 | 99.65 | Ovarian | |||||
| 37 | 1.6 | 2308 | 99.89 | SRBCT | |||||
| 24 | mRMR | GBC | SVM | 10 | 0.5 | 2000 | 98.38 | Colon | |
| 4 | 0.06 | 7129 | 100 | Leukemia-1 | |||||
| [85] | 4 | 0.06 | 7129 | 100 | Lung | ||||
| 6 | 0.26 | 2308 | 100 | SRBCT | |||||
| 4 | 0.1 | 4026 | 100 | Lymphoma | |||||
| 8 | 0.11 | 7129 | 100 | Leukemia-2 | |||||
| 25 | [87] | mRMR | ABC | SVM | 15 | 0.75 | 2000 | 96.77 | Colon |
| 14 | 0.19 | 7129 | 100 | Leukemia-1 | |||||
| 8 | 0.11 | 7129 | 100 | Lung | |||||
| 10 | 0.43 | 2308 | 100 | SRBCT | |||||
| 5 | 0.12 | 4026 | 100 | Lymphoma | |||||
| 20 | 0.28 | 7129 | 100 | Leukemia-2 | |||||
| 26 | [56] | Statistical | GA | SVM | 8 | 0.4 | 2000 | 100 | Colon |
| X2 test | 8 | 0.35 | 2308 | 100 | SRBCT | ||||
| 5 | 0.07 | 7129 | 100 | ALL/AML | |||||
| 6 | 0.15 | 4026 | 100 | DLBCL | |||||
| 26 | 0.16 | 16,306 | 87.04 | GCM | |||||
| 27 | [88] | CFS | GA | KNN | 24 | 0.42 | 5726 | 90.5 | 9_Tumors |
| 137 | 1.09 | 12,533 | 100 | 11_Tumors | |||||
| 53 | 0.35 | 15,009 | 74.39 | 14_Tumors | |||||
| 44 | 0.74 | 5920 | 99.45 | Brain_1 | |||||
| 33 | 0.32 | 10,367 | 100 | Brain_2 | |||||
| 22 | 0.41 | 5327 | 100 | Leukemia-1 | |||||
| 35 | 0.31 | 11,225 | 100 | Leukemia-2 | |||||
| 195 | 1.55 | 12,600 | 98.42 | Lung | |||||
| 29 | 1.26 | 2308 | 100 | SRBCT | |||||
| 24 | 0.23 | 10,509 | 99.22 | Prostate | |||||
| 17 | 0.31 | 5469 | 100 | DLBCL | |||||
| 28 | Fisher Criterion | AC | NB | 5 | 0.07 | 7129 | 97.6 | ALL-AML Leukemia | |
| [89] | 6 | 0.05 | 12,600 | 99.1 | Prostate | ||||
| 12 | 0.1 | 12,582 | 98.95 | MLL | |||||
| 12 | 0.17 | 7129 | 86.3 | ALL-AML-4 | |||||
| SVM | 2 | 0.03 | 7129 | 95.95 | ALL-AML Leukemia | ||||
| 14 | 0.11 | 12,600 | 98.35 | Prostate | |||||
| - | - | 12,582 | - | MLL | |||||
| - | - | 7129 | - | ALL-AML-4 | |||||
| KNN | 3 | 0.04 | 7129 | 95.95 | ALL-AML Leukemia | ||||
| 9 | 0.07 | 12,600 | 99.4 | Prostate | |||||
| 18 | 0.14 | 12,582 | 97.55 | MLL | |||||
| 15 | 0.21 | 7129 | 80.99 | ALL-AML-4 | |||||
6. Analysis and Discussion
7. Future Trends
7.1. Understanding the Biological Associated Genes Using Intelligent Systems
7.2. Genome Data Will Exponentially Increase
7.3. Comprehensive and Delicate Approaches for the Discovery of Potentially Clinically Relevant Genes
7.4. Stratifying the Studies Related to the Transcription of Genetic Codes into Messenger RNA
7.5. RNA-Seq, Leading to Extensive and More Complex Bioinformatic Analysis
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 70, 313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Tiwari, R.K. Application of microarray in breast cancer: An overview. J. Pharm. Bioallied Sci. 2012, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Reimer, T.; Gerber, B. Management of early invasive breast cancer in very young women (<35 years). Clin. Breast Cancer 2011, 11, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Breastcancer.org. U.S. Breast Cancer Statistics. 2019. Available online: https://www.breastcancer.org/facts-statistics (accessed on 6 December 2021).
- Brekelmans, C.T.M.; Seynaeve, C.; Menke-Pluymers, M.; Brüggenwirth, H.T.; Tilanus-Linthorst, M.M.A.; Bartels, C.C.M.; Kriege, M.; van Geel, A.N.; Crepin, C.M.G.; Blom, J.C. Survival and prognostic factors in BRCA1-associated breast cancer. Ann. Oncol. 2006, 17, 391–400. [Google Scholar] [CrossRef]
- Hisham, A.N.; Yip, C.-H. Overview of breast cancer in Malaysian women: A problem with late diagnosis. Asian J. Surg. 2004, 27, 130–133. [Google Scholar] [CrossRef]
- IARC CancerBase. Section of Cancer Surveillance. In GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012; IARC: Lyon, France, 2012; pp. 1–7. [Google Scholar]
- Lipscombe, L.L.; Goodwin, P.J.; Zinman, B.; McLaughlin, J.R.; Hux, J.E. The impact of diabetes on survival following breast cancer. Breast Cancer Res. Treat. 2008, 109, 389–395. [Google Scholar] [CrossRef]
- Yang, L.; Parkin, D.M.; Ferlay, J.; Li, L.; Chen, Y. Estimates of cancer incidence in China for 2000 and projections for 2005. Cancer Epidemiol. Biomark. Prev. 2005, 14, 243–250. [Google Scholar] [CrossRef]
- Sotiriou, C.; Neo, S.-Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef]
- Mount, D.W.; Putnam, C.W.; Centouri, S.M.; Manziello, A.M.; Pandey, R.; Garland, L.L.; Martinez, J.D. Using logistic regression to improve the prognostic value of microarray gene expression data sets: Application to early-stage squamous cell carcinoma of the lung and triple negative breast carcinoma. BMC Med. Genom. 2014, 7, 33. [Google Scholar] [CrossRef]
- Alexe, G.; Alexe, S.; Axelrod, D.E.; Bonates, T.O.; Lozina, I.I.; Reiss, M.; Hammer, P.L. Breast cancer prognosis by combinatorial analysis of gene expression data. Breast Cancer Res. 2006, 8, R41. [Google Scholar] [CrossRef]
- Narendra, P.M.; Fukunaga, K. A branch and bound algorithm for feature subset selection. IEEE Comput. Archit. Lett. 1977, 26, 917–922. [Google Scholar] [CrossRef]
- Abd-Elnaby, M.; Alfonse, M.; Roushdy, M. Classification of breast cancer using microarray gene expression data: A survey. J. Biomed. Inform. 2021, 117, 103764. [Google Scholar] [CrossRef]
- Schena, M. DNA Microarrays: A Practical Approach; No. 205; Practical approach series; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Rew, D.A. DNA microarray technology in cancer research. Eur. J. Surg. Oncol. 2001, 27, 504–508. [Google Scholar] [CrossRef]
- Govindarajan, R.; Duraiyan, J.; Kaliyappan, K.; Palanisamy, M. Microarray and its applications. J. Pharm. Bioallied Sci. 2012, 4 (Suppl. S2), S310. [Google Scholar]
- Zhang, J.-G.; Deng, H.-W. Gene selection for classification of microarray data based on the Bayes error. BMC Bioinform. 2007, 8, 1–9. [Google Scholar] [CrossRef]
- Dawany, N.B.; Tozeren, A. Asymmetric microarray data produces gene lists highly predictive of research literature on multiple cancer types. BMC Bioinform. 2010, 11, 1–14. [Google Scholar] [CrossRef]
- Guyon, I.; Gunn, S.; Nikravesh, M.; Zadeh, L.A. Feature Extraction: Foundations and Applications; Springer: New York, NY, USA, 2008; Volume 207. [Google Scholar]
- Cai, J.; Luo, J.; Wang, S.; Yang, S. Feature selection in machine learning: A new perspective. Neurocomputing 2018, 300, 70–79. [Google Scholar] [CrossRef]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature selection: A data perspective. ACM Comput. Surv. 2018, 50, 94. [Google Scholar] [CrossRef]
- Miao, J.; Niu, L. A survey on feature selection. Procedia Comput. Sci. 2016, 91, 919–926. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef]
- Shukla, A.K.; Singh, P.; Vardhan, M. A hybrid gene selection method for microarray recognition. Biocybern. Biomed. Eng. 2018, 38, 975–991. [Google Scholar] [CrossRef]
- Dashtban, M.; Balafar, M. Gene selection for microarray cancer classification using a new evolutionary method employing artificial intelligence concepts. Genomics 2017, 109, 91–107. [Google Scholar] [CrossRef]
- Lu, H.; Chen, J.; Yan, K.; Jin, Q.; Xue, Y.; Gao, Z. A hybrid feature selection algorithm for gene expression data classification. Neurocomputing 2017, 256, 56–62. [Google Scholar] [CrossRef]
- McCall, J. Genetic algorithms for modelling and optimisation. J. Comput. Appl. Math. 2005, 184, 205–222. [Google Scholar] [CrossRef]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the ICNN’95-International Conference on Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar]
- Geem, Z.W.; Kim, J.H.; Loganathan, G.V. A new heuristic optimization algorithm: Harmony search. Simulation 2001, 76, 60–68. [Google Scholar] [CrossRef]
- Dorigo, M.; Maniezzo, V.; Colorni, A. Ant system: Optimization by a colony of cooperating agents. IEEE Trans. Syst. Man Cybern. Part B 1996, 26, 29–41. [Google Scholar] [CrossRef]
- Karaboga, D. An Idea Based on Honey Bee Swarm for Numerical Optimization; Technical Report-tr06; Erciyes University, Engineering Faculty, Computer Engineering Department: Kayseri, Turkey, 2005. [Google Scholar]
- Yang, X.-S. Firefly algorithms for multimodal optimization. In International Symposium on Stochastic Algorithms; Springer: Berlin/Heidelberg, Germany, 2009; pp. 169–178. [Google Scholar]
- Gandomi, A.H.; Yang, X.-S.; Alavi, A.H. Cuckoo search algorithm: A metaheuristic approach to solve structural optimization problems. Eng. Comput. 2013, 29, 17–35. [Google Scholar] [CrossRef]
- Rashedi, E.; Nezamabadi-Pour, H.; Saryazdi, S. GSA: A gravitational search algorithm. Inf. Sci. 2009, 179, 2232–2248. [Google Scholar] [CrossRef]
- Mirjalili, S.; Mirjalili, S.M.; Lewis, A. Grey wolf optimizer. Adv. Eng. Softw. 2014, 69, 46–61. [Google Scholar] [CrossRef]
- Mirjalili, S.; Lewis, A. The whale optimization algorithm. Adv. Eng. Softw. 2016, 95, 51–67. [Google Scholar] [CrossRef]
- Rana, N.; Latiff, M.S.A.; Abdulhamid, S.M.; Chiroma, H. Whale optimization algorithm: A systematic review of contemporary applications, modifications and developments. Neural Comput. Appl. 2020, 32, 16245–16277. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Taheri, S.; Mammadov, M. Learning the naive Bayes classifier with optimization models. Int. J. Appl. Math. Comput. Sci. 2013, 23, 787–795. [Google Scholar] [CrossRef]
- Kleinbaum, D.G.; Dietz, K.; Gail, M.; Klein, M.; Klein, M. Logistic Regression; Springer: New York, NY, USA, 2002. [Google Scholar]
- Ross, T.J. Fuzzy Logic with Engineering Applications; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- O’Neill, M.C.; Song, L. Neural network analysis of lymphoma microarray data: Prognosis and diagnosis near-perfect. BMC Bioinform. 2003, 4, 13. [Google Scholar]
- Kundu, R.; Chattopadhyay, S.; Cuevas, E.; Sarkar, R. AltWOA: Altruistic Whale Optimization Algorithm for feature selection on microarray datasets. Comput. Biol. Med. 2022, 144, 105349. [Google Scholar] [CrossRef]
- Tahmouresi, A.; Rashedi, E.; Yaghoobi, M.M.; Rezaei, M. Gene selection using pyramid gravitational search algorithm. PLoS ONE 2022, 17, e0265351. [Google Scholar] [CrossRef]
- Hamim, M.; el Moudden, I.; Pant, M.D.; Moutachaouik, H.; Hain, M. A hybrid gene selection strategy based on fisher and ant colony optimization algorithm for breast cancer classification. Int. J. Online Biomed. Eng. (iJOE) 2021, 17, 148–163. [Google Scholar] [CrossRef]
- Afif, G.G.; Astuti, W. Cancer Detection based on Microarray Data Classification Using FLNN and Hybrid Feature Selection. J. RESTI (Rekayasa Sist. Dan Teknol. Inf.) 2021, 5, 794–801. [Google Scholar] [CrossRef]
- Loey, M.; Jasim, M.W.; El-Bakry, H.M.; Taha, M.H.N.; Khalifa, N.E.M. Breast and colon cancer classification from gene expression profiles using data mining techniques. Symmetry 2020, 12, 408. [Google Scholar]
- Han, X.H.; Li, D.A.; Wang, L. A Hybrid Cancer Classification Model Based Recursive Binary Gravitational Search Algorithm in Microarray Data. Procedia Comput. Sci. 2019, 154, 274–282. [Google Scholar] [CrossRef]
- Jain, I.; Jain, V.K.; Jain, R. Correlation feature selection based improved-binary particle swarm optimization for gene selection and cancer classification. Appl. Soft Comput. 2018, 62, 203–215. [Google Scholar] [CrossRef]
- Shukla, A.K.; Singh, P.; Vardhan, M. A two-stage gene selection method for biomarker discovery from microarray data for cancer classification. Chemom. Intell. Lab. Syst. 2018, 183, 47–58. [Google Scholar] [CrossRef]
- Mohapatra, P.; Chakravarty, S.; Dash, P.K. Microarray medical data classification using kernel ridge regression and modified cat swarm optimization based gene selection system. Swarm Evol. Comput. 2016, 28, 144–160. [Google Scholar] [CrossRef]
- Shreem, S.S.; Abdullah, S.; Nazri, M.Z.A. Hybrid feature selection algorithm using symmetrical uncertainty and a harmony search algorithm. Int. J. Syst. Sci. 2016, 47, 1312–1329. [Google Scholar] [CrossRef]
- Lee, C.-P.; Leu, Y. A novel hybrid feature selection method for microarray data analysis. Appl. Soft Comput. 2011, 11, 208–213. [Google Scholar] [CrossRef]
- Alba, E.; Garcia-Nieto, J.; Jourdan, L.; Talbi, E.-G. Gene Selection in Cancer Classification Using PSO/SVM and GA/SVM Hybrid Algorithms. In Proceedings of the 2007 IEEE Congress on Evolutionary Computation, Singapore, 25–28 September 2007; pp. 284–290. [Google Scholar]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Gravier, E.; Pierron, G.; Vincent-Salomon, A.; Gruel, N.; Raynal, V.; Savignoni, A.; De Rycke, Y.; Pierga, J.-Y.; Lucchesi, C.; Reyal, F.; et al. A prognostic DNA signature for T1T2 node-negative breast cancer patients. Genes Chromosom. Cancer 2010, 49, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.-J.; Chang, K.-M.; Hsu, H.-C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Van’T Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530. [Google Scholar] [CrossRef]
- Jinyan, L.; Huiqing, L. Kent Ridge Bio-Medical Data Set Repository; School of Computer Engineering Nanyang Technological University: Nanyang, China, 2002. [Google Scholar]
- Alonso-González, C.J.; Moro-Sancho, Q.I.; Simon-Hurtado, A.; Varela-Arrabal, R. Microarray gene expression classification with few genes: Criteria to combine attribute selection and classification methods. Expert Syst. Appl. 2012, 39, 7270–7280. [Google Scholar] [CrossRef]
- Zhu, Z.; Ong, Y.-S.; Dash, M. Markov blanket-embedded genetic algorithm for gene selection. Pattern Recognit. 2007, 40, 3236–3248. [Google Scholar] [CrossRef]
- Zhu, Z.; Ong, Y.S.; Dash, M. Microarray Datasets in Weka ARFF Format. Pattern Recognit. 2007, 49, 3236–3248. Available online: http://csse.szu.edu.cn/staff/zhuzx/Datasets.html (accessed on 4 May 2021). [CrossRef]
- Hedenfalk, I.; Duggan, D.; Chen, Y.; Radmacher, M.; Bittner, M.; Simon, R.; Meltzer, P.; Gusterson, B.; Esteller, M.; Raffeld, M.; et al. Gene-expression profiles in hereditary breast cancer. N. Engl. J. Med. 2001, 344, 539–548. [Google Scholar] [CrossRef]
- Chen, A.H.; Yang, C. The improvement of breast cancer prognosis accuracy from integrated gene expression and clinical data. Expert Syst. Appl. 2012, 39, 4785–4795. [Google Scholar] [CrossRef]
- Cano, A.; Masegosa, A.; Moral, S. Kent Ridge Bio-Medical Data Repository. 2005. Available online: http://datam.i2r.a-star.edu.sg/datasets/krbd/ (accessed on 24 August 2022).
- Abasabadi, S.; Nematzadeh, H.; Motameni, H.; Akbari, E. Hybrid feature selection based on SLI and genetic algorithm for microarray datasets. J. Supercomput. 2022, 1–29. [Google Scholar] [CrossRef]
- Kowsari, Y.; Nakhodchi, S.; Gholamiangonabadi, D. Gene selection from microarray expression data: A Multi-objective PSO with adaptive K-nearest neighborhood. arXiv Prepr. 2022, arXiv:2205.15020. [Google Scholar]
- Sazzed, S. ANOVA-SRC-BPSO: A Hybrid Filter and Swarm Optimization-Based Method for Gene Selection and Cancer Classification Using Gene Expression Profiles. In Proceedings of the Canadian Conference on AI, Vancouver, BC, Canada, 25–28 May 2021. [Google Scholar]
- Zhang, G.; Hou, J.; Wang, J.; Yan, C.; Luo, J. Feature selection for microarray data classification using hybrid information gain and a modified binary krill herd algorithm. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 288–301. [Google Scholar] [CrossRef]
- Pragadeesh, C.; Jeyaraj, R.; Siranjeevi, K.; Abishek, R.; Jeyakumar, G. Hybrid feature selection using micro genetic algorithm on microarray gene expression data. J. Intell. Fuzzy Syst. 2019, 36, 2241–2246. [Google Scholar] [CrossRef]
- Almugren, N.; Alshamlan, H.M. New Bio-Marker Gene Discovery Algorithms for Cancer Gene Expression Profile. IEEE Access 2019, 7, 136907–136913. [Google Scholar] [CrossRef]
- Baliarsingh, S.K.; Vipsita, S.; Muhammad, K.; Dash, B.; Bakshi, S. Analysis of high-dimensional genomic data employing a novel bio-inspired algorithm. Appl. Soft Comput. 2019, 77, 520–532. [Google Scholar] [CrossRef]
- Musheer, R.A.; Verma, C.K.; Srivastava, N. Novel machine learning approach for classification of high-dimensional microarray data. Soft Comput. 2019, 23, 13409–13421. [Google Scholar] [CrossRef]
- Baliarsingh, S.K.; Vipsita, S.; Dash, B. A new optimal gene selection approach for cancer classification using enhanced Jaya-based forest optimization algorithm. Neural Comput. Appl. 2019, 32, 8599–8616. [Google Scholar] [CrossRef]
- Baliarsingh, S.K.; Ding, W.; Vipsita, S.; Bakshi, S. A memetic algorithm using emperor penguin and social engineering optimization for medical data classification. Appl. Soft Comput. 2019, 85, 105773. [Google Scholar] [CrossRef]
- Vijay, S.A.A.; GaneshKumar, P. Fuzzy expert system based on a novel hybrid stem cell (HSC) algorithm for classification of micro array data. J. Med. Syst. 2018, 42, 61. [Google Scholar] [CrossRef]
- Alshamlan, H.M. Co-ABC: Correlation artificial bee colony algorithm for biomarker gene discovery using gene expression profile. Saudi J. Biol. Sci. 2018, 25, 895–903. [Google Scholar] [CrossRef]
- Motieghader, H.; Najafi, A.; Sadeghi, B.; Masoudi-Nejad, A. A hybrid gene selection algorithm for microarray cancer classification using genetic algorithm and learning automata. Inform. Med. Unlocked 2017, 9, 246–254. [Google Scholar] [CrossRef]
- Aziz, R.; Verma, C.K.; Srivastava, N. A novel approach for dimension reduction of microarray. Comput. Biol. Chem. 2017, 71, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.S.; Zainudin, S.; Othman, Z.A. Metaheuristic approach for an enhanced mRMR filter method for classification using drug response microarray data. Expert Syst. Appl. 2017, 90, 224–231. [Google Scholar] [CrossRef]
- Salem, H.; Attiya, G.; El-Fishawy, N. Classification of human cancer diseases by gene expression profiles. Appl. Soft Comput. 2017, 50, 124–134. [Google Scholar] [CrossRef]
- Alshamlan, H.M.; Badr, G.H.; Alohali, Y.A. Genetic Bee Colony (GBC) algorithm: A new gene selection method for microarray cancer classification. Comput. Biol. Chem. 2015, 56, 49–60. [Google Scholar] [CrossRef]
- Alshamlan, H.; Badr, G.; Alohali, Y. A Comparative Study of Cancer Classification Methods Using Microarray Gene Expression Profile. In Proceedings of the First International Conference on Advanced Data and Information Engineering (DaEng-2013); Springer: New York, NY, USA, 2014; pp. 389–398. [Google Scholar]
- Alshamlan, H.; Badr, G.; Alohali, Y. mRMR-ABC: A hybrid gene selection algorithm for cancer classification using microarray gene expression profiling. Biomed Res. Int. 2015, 2015, 604910. [Google Scholar] [CrossRef]
- Chuang, L.-Y.; Yang, C.-H.; Wu, K.-C.; Yang, C.-H. A hybrid feature selection method for DNA microarray data. Comput. Biol. Med. 2011, 41, 228–237. [Google Scholar] [CrossRef]
- Sharbaf, F.V.; Mosafer, S.; Moattar, M.H. A hybrid gene selection approach for microarray data classification using cellular learning automata and ant colony optimization. Genomics 2016, 107, 231–238. [Google Scholar] [CrossRef]
- Bendtsen, C.; Petrovski, S. How data and AI are helping unlock the secrets of disease. AstraZeneca Blog. 2019. [Google Scholar]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Almugren, N.; Alshamlan, H. A survey on hybrid feature selection methods in microarray gene expression data for cancer classification. IEEE Access 2019, 7, 78533–78548. [Google Scholar] [CrossRef]





| Type/ Studies | Benefits | Limitations | Standard Search | Example | Performance |
|---|---|---|---|---|---|
| (1) Filter | (a) Independent (b) Low computational cost (c) Faster than wrapper (d) Ignores classifier (e) Fast computational time | (a) No interaction with classifier (b) Computational complexity (c) Redundant (d) Ignores useful features | (a) Univariate (b) Multivariate | Stat test: t-test IG Fisher Score ReliefF | (a) Faster than other FS methods (b) Degrades feature relevancy |
| (2) Wrapper | (a) Cooperates with classifier (b) Feature-dependent (c) Computationally intensive (d) Has contact with FS and classifier | (a) Risk of overfitting (b) Classifier-dependent (c) High computation time (d) Complex exponential time | (a) Deterministic (b) Stochastic | AC Fuzzy Inference GA | (a) Better than filter approach (b) High performance |
| (3) Embedded | (a) Combines optimal FS method with classifier (b) Has contact with classifier (c) Low risk of overfitting | (a) Classifier dependent on selection method (b) Predisposed to overfitting | (1) Model classifier preference (2) Built-in model (3) Simplified model | SVM-RFE Lasso | (a) Computational cost less than wrapper |
| (4) Hybrid or Ensemble | (a) Combines more than one FS method. | (a) High processing time and complicated | (a) Exhaustive search (b) Optimum FS | LR-RF | (a) Complex (b) Less error |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Ali, N.; Besar, R.; Ab. Aziz, N.A. Hybrid Feature Selection of Breast Cancer Gene Expression Microarray Data Based on Metaheuristic Methods: A Comprehensive Review. Symmetry 2022, 14, 1955. https://doi.org/10.3390/sym14101955
Mohd Ali N, Besar R, Ab. Aziz NA. Hybrid Feature Selection of Breast Cancer Gene Expression Microarray Data Based on Metaheuristic Methods: A Comprehensive Review. Symmetry. 2022; 14(10):1955. https://doi.org/10.3390/sym14101955
Chicago/Turabian StyleMohd Ali, Nursabillilah, Rosli Besar, and Nor Azlina Ab. Aziz. 2022. "Hybrid Feature Selection of Breast Cancer Gene Expression Microarray Data Based on Metaheuristic Methods: A Comprehensive Review" Symmetry 14, no. 10: 1955. https://doi.org/10.3390/sym14101955
APA StyleMohd Ali, N., Besar, R., & Ab. Aziz, N. A. (2022). Hybrid Feature Selection of Breast Cancer Gene Expression Microarray Data Based on Metaheuristic Methods: A Comprehensive Review. Symmetry, 14(10), 1955. https://doi.org/10.3390/sym14101955