3. Results
Micrographs of TiO
2-based nanoheterostructures are shown at
Figure 1. The figure shows that, in general, the micrographs of different samples have a certain similarity: all the studied structures are nanoparticles grouped into agglomerates of an asymmetric form.
The size of the nanoparticles and their specific surface area were determined from X-ray diffraction (XRD) and low-temperature nitrogen adsorption (BET) data, respectively. The results of these measurements for TiO
2 nanoparticles are presented as an example in
Table 1.
We have carefully investigated, in another work [
20], photocatalysis on the structures prepared by the same method as that used in the present work. The highest rate of photocatalysis in the visible spectral region was observed in the TiO
2−MoO
3−V
2O
5 and TiO
2−MoO
3−WO
3 nanoheterostructures. As it was revealed in [
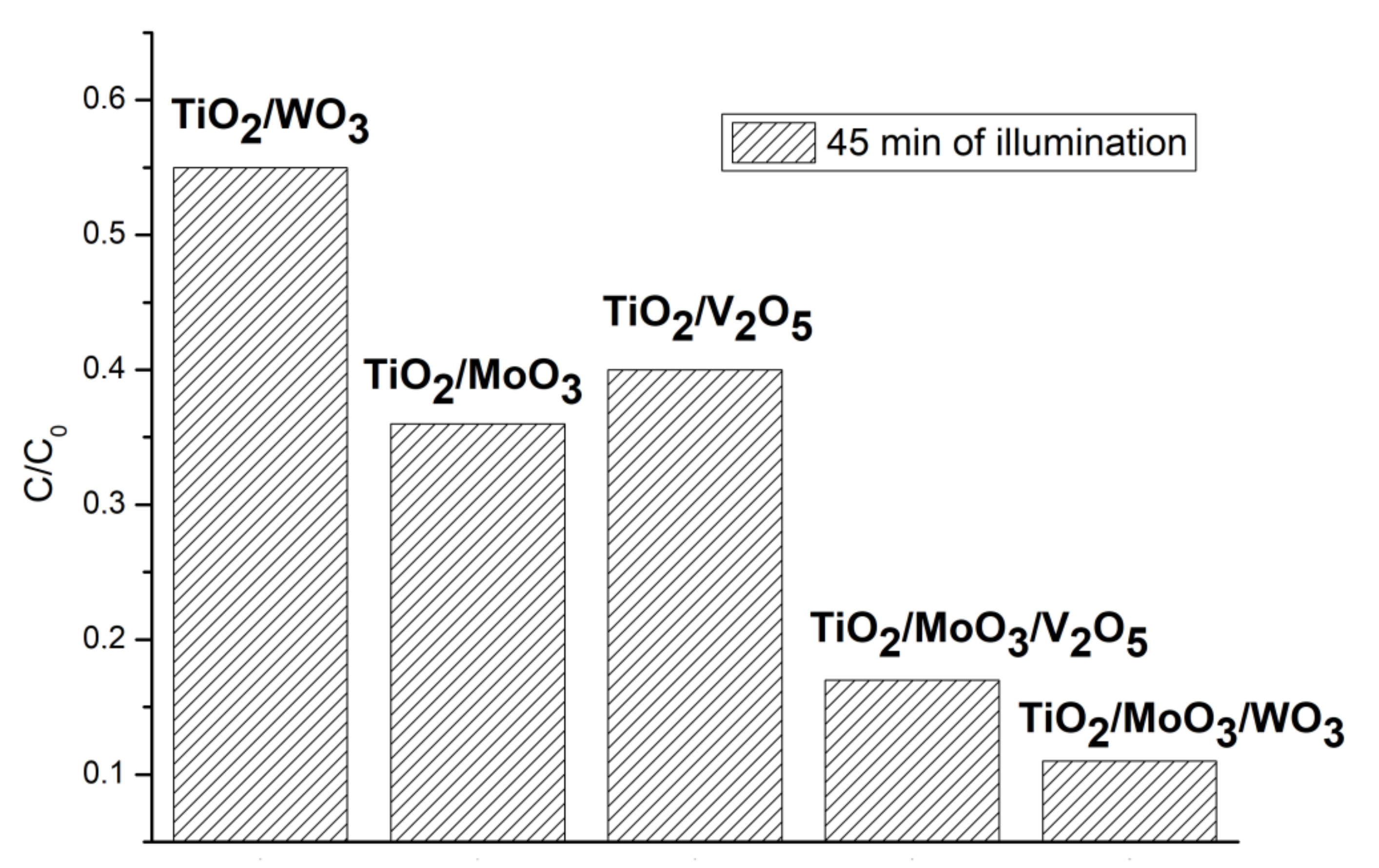
20], this is due to the higher concentrations of radicals in these structures. After the morphology and structure studies, we repeated part of the photocatalysis measurements with a new batch of samples in the current study. The brief results of the photocatalytic activity of the new samples are presented at
Figure 2. The lower is the residual dye concentration C/C
0 after the sample exposition to the visible light – the higher is the sample photocatalytic activity. Our new results are very consistent with our previous findings, which allows us to declare a good replicability of the synthesis method and material properties.
Kinetic curves of photocatalysis for MoO
3 and TiO
2 nanooxides, as well as for nanoheterostructures TiO
2−WO
3, TiO
2−MoO
3, TiO
2−MoO
3−V
2O
5, and TiO
2−MoO
3−WO
3 under photoexcitation in the visible spectral region (450–750 nm) are shown in
Figure S2 of our Supplementary Materials. We have found that, because of asymmetric charge separation in nanoheterostructures, catalysis lasts on their surface long after light is extinguished.
Since defects at the semiconductors’ surface actively interacts with photoexcited charge carriers, we studied the prepared samples by the ESR method. ESR spectra of the previous batch of nanoheterostructures, TiO
2−MoO
3−V
2O
5, TiO
2−MoO
3−WO
3, TiO
2−WO
3, TiO
2−V
2O
5, TiO
2−MoO
3, and their analyses can be found in
Figure S1 of our Supplementary Material.
In the investigated samples, an anticorrelation was found between the residual concentration (after photocatalysis) of the test dye and the concentration of radicals in the nanoheterostructures. Another correlation was found between light absorption in the visible region, the concentration of radicals and the photocatalytic activity [
20].
To determine the position of the radicals’ energy levels in the band gaps of the used samples, we have applied our original method, developed earlier, which is based on ESR spectroscopy [
12]. The dependence of the ESR signal intensity on the photon energy (
hν) was measured when the samples were illuminated directly in the cavity of the spectrometer. A BRUKER ELEXSYS ER 202 UV high-pressure mercury lamp (∆λ = 270 − 1000 nm, 50 W power), equipped with the diffraction monochromator, was used for this purpose. The light intensity was approximately 40 mW cm
−2.
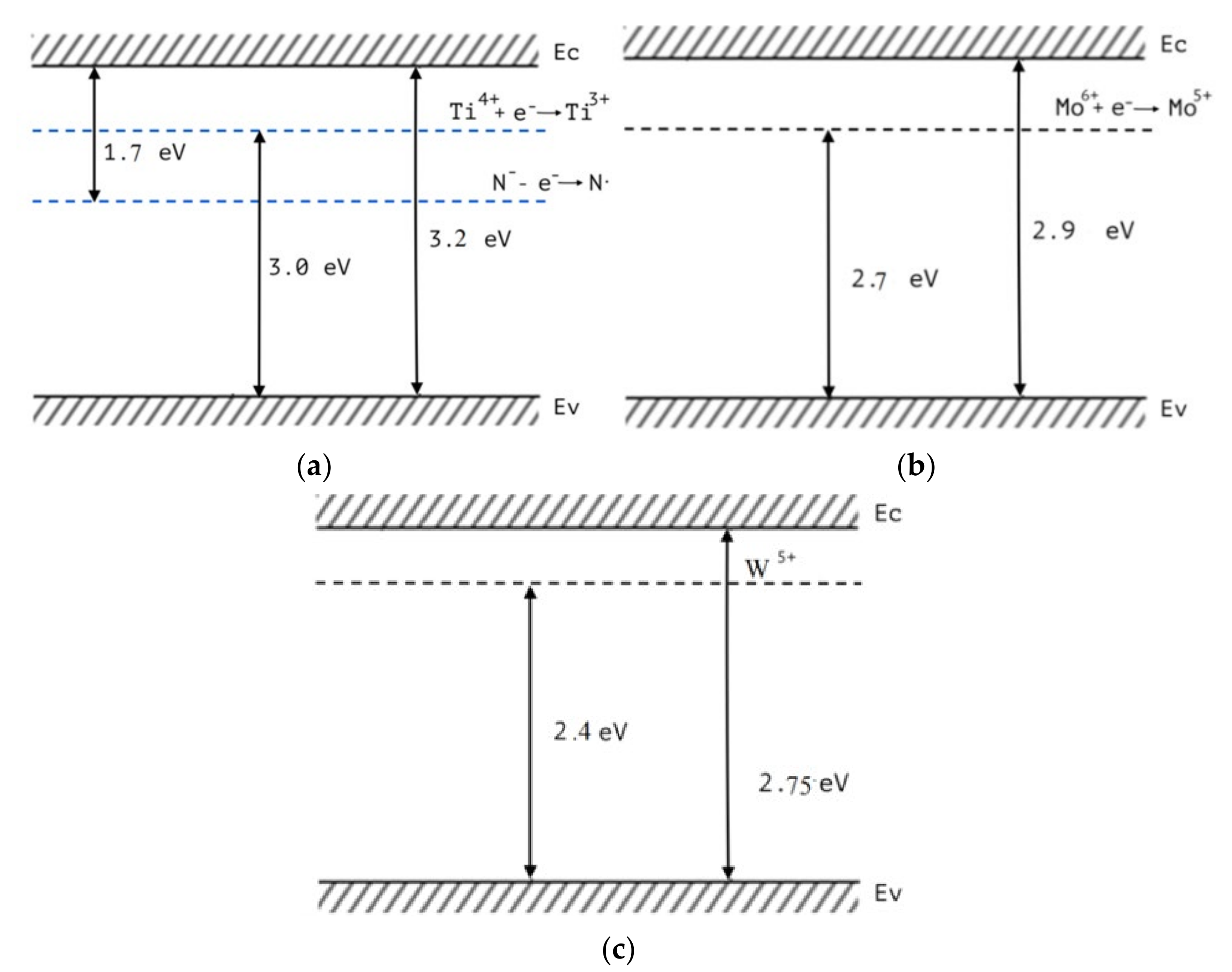
The study was performed for the nanoheterostructures TiO2−MoO3, TiO2−WO3, TiO2−V2O5, TiO2−MoO3−WO3, TiO2−MoO3−V2O5. The change in the magnitude of the ESR signal of their radicals occurs when a certain energy of the incident photons is reached (specific for each type of radical). It is a very important observation, meaning that, under illumination, defects absorb light and defect recharging occurs. We have found that the increase of the ESR signal in TiO2, for nitrogen radicals, occurs at the photon energy ℎν ≥ 1.5 eV, i.e., starting from this energy, the electron moves from the radical level to the TiO2 conduction band as a result of light absorption by the dopant. This allows us to estimate the position of this defect level in the band gap of TiO2, as separated from the bottom of the conduction band by a value of 1.5 eV. When illumination was used with the quanta energy ℎν ≥ 3.0 eV an increase in the ESR signal was observed for Ti3+ centers that can be explained by the transition of an electron from the valence band to the initially nonparamagnetic centers of the Ti4+/oxygen vacancy, which form electron states in the band gap near the bottom of the TiO2 conduction band and are known from literature as «Urbach tails».
In the same way, the observed increase in the ESR signal intensity for the Mo
5+ centers in MoO
3 under ℎ
ν ≥ 2.6 eV illumination can be attributed to the transition of electrons from the valence band of molybdenum oxide to the originally nonparamagnetic Mo
6+ centers, which also form electron states near the bottom of the conduction band of MoO
3. The ESR signal, increasing for V
4+ centers in V
2O
5 under illumination by ℎ
ν ≥ 2.1 eV, in turn can be explained by electrons’ transition from the valence band of V
2O
5 to the initially nonparamagnetic electron states V
5+, localized near the V
2O
5 conduction band’s bottom. Thus, it is possible to construct band diagrams for the nanooxides that make up the studied nanoheterostructures (
Figure 3).
In this study, as in previous studies, the photocatalytic activity (oxidative capacity) of the nanocatalyst samples was investigated using the photodegradation reaction of the rhodamine dye (as it is described in the Materials and Methods section). To do this, the dye was applied to the surface of the samples from the solution. Measurements were conducted after the samples’ drying. The symmetric possibility of transferring the photoexcitation energy from the adsorbed dye molecules to the semiconductor was not taken into account during our previous studies. We have carefully studied the possibility of energy transfer in the other structures, and it is also known from the literature ([
22,
23] for example). This energy transfer can lead to the formation of additional free charge carriers. These additional charge carriers may in turn increase photocatalytic efficiency and affect the rhodamine dye’s degradation rate. In this way, the photosensitization of semiconductor oxides by dye molecules would promote these dye molecules’ faster degradation.
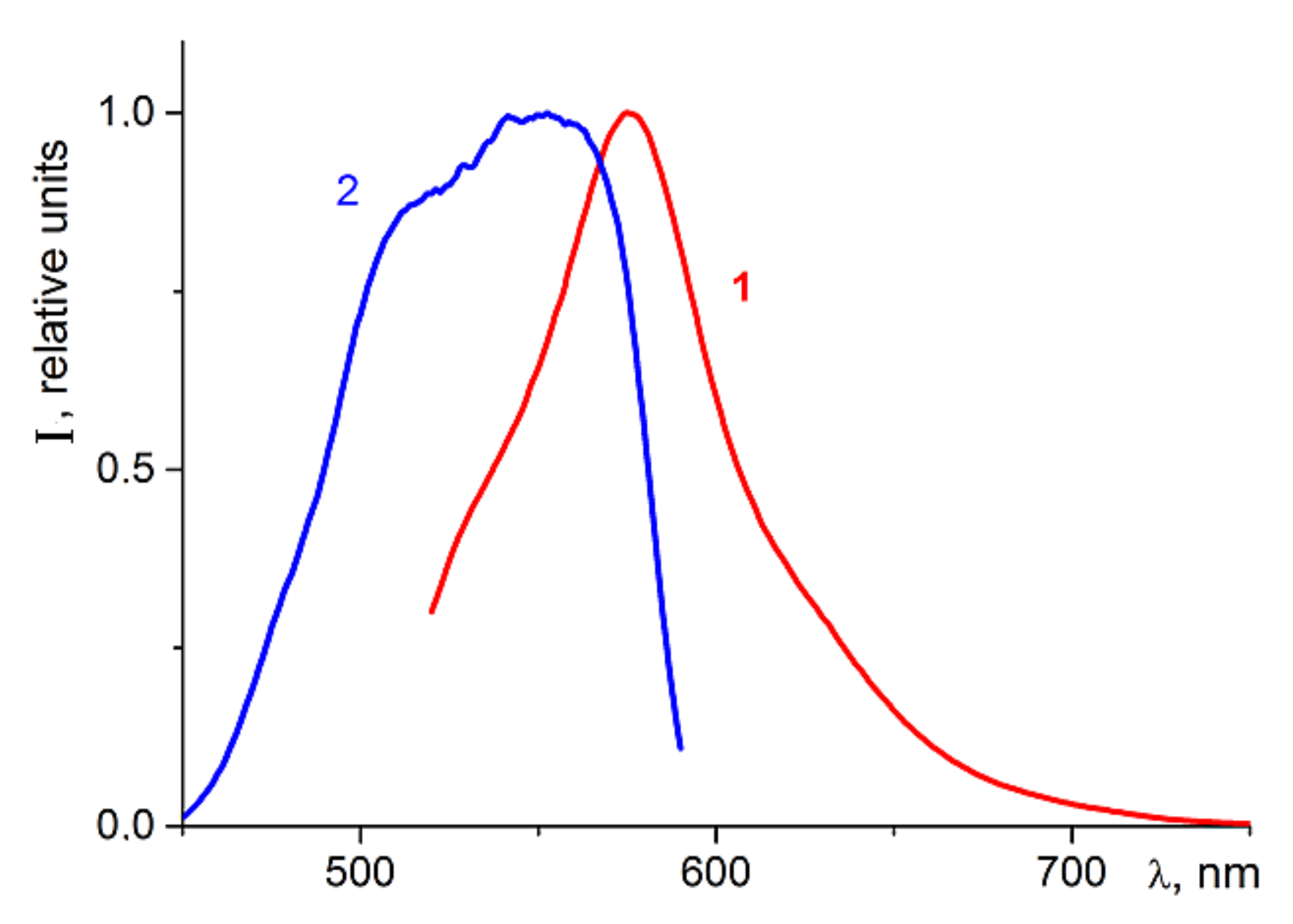
Therefore, in the present work we conducted a special study of the energy transfer efficiency from adsorbed rhodamine-type dye molecules to charge carriers trapped in semiconductor nanoheterostructures possessing photocatalytic activity. For this study, rhodamine dye was adsorbed onto the surface of the photocatalysts’ surface at a concentration providing maximal fluorescence intensity. The fluorescence spectra of the rhodamine dye adsorbed on the TO
2 surface are shown at
Figure 4.
It can be seen from the fluorescence excitation spectrum that the effective excitation of rhodamine molecules does not occur when illuminated at wavelengths shorter than 450 nm.
The remaining research cycle was carried out as follows. For each type of the samples, two series of measurements of the time-intensity dependence I(t) at the fluorescence maximum were performed. The first series of measurements was carried out when the sample was illuminated (between the registrations of the spectra) by radiation at a 333-nm wavelength, outside the effective absorption band of the dye, but in the absorption band of the photocatalysts. The second series was carried out with constant illumination inside the dye absorption band at a 500-nm wavelength. The fluorescence spectra of the dye were always recorded with excitation at a 500-nm wavelength.
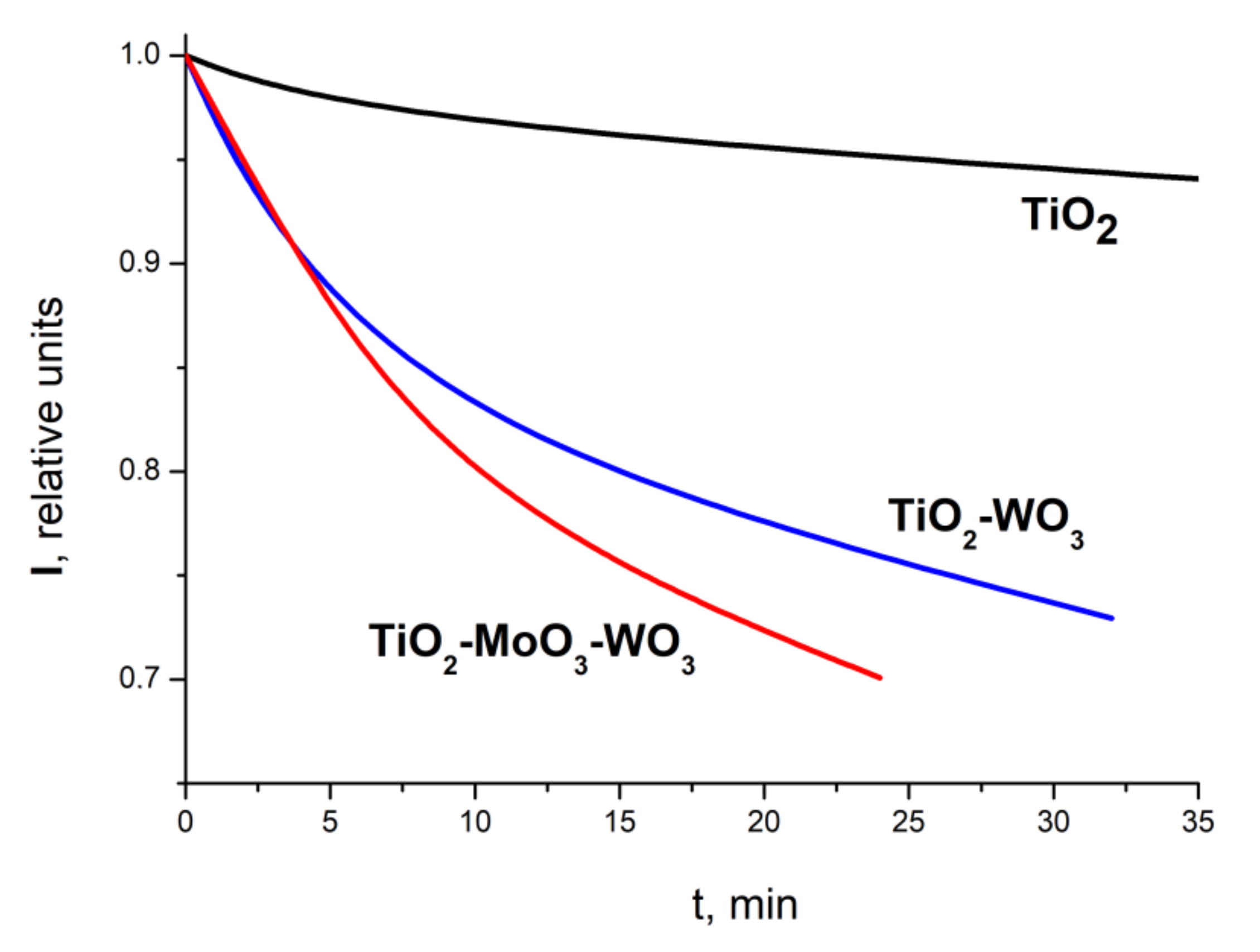
Let us first compare the efficiency of photocatalysis, obtained in symmetric conditions, by measuring the quenching of the dye’s fluorescence on different nanoheterostructures when illuminated at the same 333-nm wavelength.
The time dependencies of the maximum fluorescence intensity of rhodamine dye molecules adsorbed at the photocatalysts surface are shown at
Figure 5. The curves reflect the time dependence of the fluorescence intensity of the adsorbed dye when the structures were illuminated at a 333-nm wavelength between short runs of fluorescence measurements. The graphs show that the luminescence-quenching rate of TiO
2–MoO
3–WO
3 exceeds the result for TiO
2–WO
3 and very significantly exceeds the quenching rate obtained for TiO
2. This corresponds to our data (obtained by an independent method) on the rate of photocatalysis on these structures. Thus, it becomes clear that both methods are symmetric and it is possible to estimate the rate of photocatalysis by quenching the luminescence of adsorbed organic dyes.
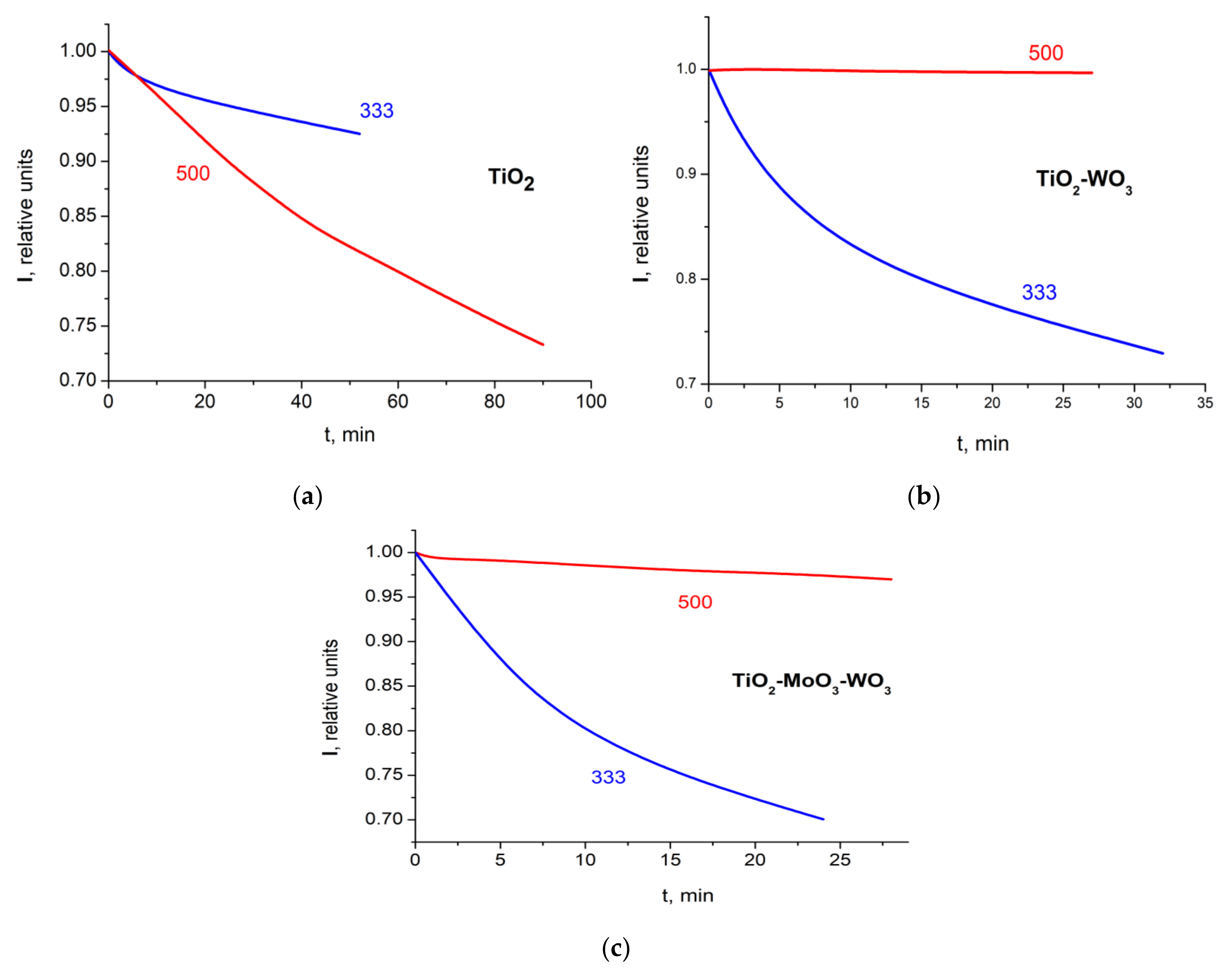
Let us now compare the symmetric photocatalysis results obtained by illumination at two different wavelengths. The results for TiO
2 are shown in
Figure 6a. As it is known, rhodamine dye is able to transfer photoexcitation energy to a semiconductor, leading to the release of charge carriers from the traps to the conduction band [
22,
23]. Probably, this process is very effective in the TiO
2–rhodamine system.
As a result, despite the relatively low illumination intensity, the photocatalytic destruction of the dye molecules leads to a noticeable quenching of the fluorescence over time. The efficiency of the process for TiO2 is asymmetric; under illumination in the dye absorption band it is significantly higher than that under ultraviolet illumination.
The
Figure 6b shows that green light illumination (500 nm) in the dye-absorption band practically does not lead to degradation of rhodamine molecules adsorbed on nanoheterostructures TiO
2–WO
3, while ultraviolet illumination leads to a fairly rapid degradation of the dye. It should be noted that the initial fluorescence intensity of rhodamine on the surface of TiO
2–WO
3 structures was 5–7 times lower than on TiO
2, despite their same dye concentrations. Such a significant quenching (right after the dye deposition) of luminescence most likely indicates a noticeable energy transfer from the dye to the semiconductor. However, this does not lead to additional degradation of the dye over time when the structure is illuminated at the wavelength of 500 nm.
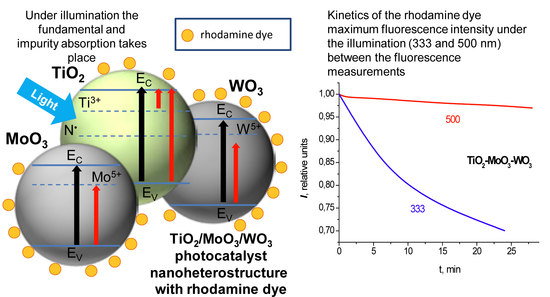
The time dependencies of the fluorescence intensity of rhodamine dye on the TiO
2–MoO
3–WO
3 surface, under illumination at 500-nm and 333-nm wavelengths, are shown in
Figure 6c. The initial fluorescence intensity of rhodamine dye on the surface of these structures was 5–7 times lower than that on the TiO
2 surface, the same as in the previous case. When the structure was illuminated at a 500-nm wavelength, the degradation of the dye fluorescence was observed to be approximately twice slower than on the TiO
2 surface—and, when illuminated at the wavelength of 333 nm, the rate of the dye degradation was quite high.
The initial fluorescence intensity of rhodamine at the surface of TiO
2–MoO
3 samples was approximately 20 times lower than that for the TiO
2 surface, and it practically did not change during irradiation at the wavelengths of 500 and 333 nm. At the same time, the dye fluorescence maximum was shifted by 10 nm to the short-wave region, as compared with the rest of the samples. This may indicate a strong bonding between the dye molecules and the substrate for TiO
2–MoO
3 structures. The similar significant shift in the emission spectrum of rhodamine molecules is usually observed in the presence of strong local electric fields of negatively charged particles near the adsorbed molecules [
24]. It is possible that the observed luminescence of the dye is associated with those of its molecules that were adsorbed far from the active centers on the surface of the catalyst, and that those dye molecules that were located close to the active centers experienced the action of the latter and did not contribute to the luminescence.
At the surfaces of the TiO
2–V
2O
5 and TiO
2–MoO
3–V
2O
5 samples, the initial fluorescence intensity of rhodamine dye was lower than the equipment noise level, which indicates an exceptionally strong quenching (similar to what we have previously observed for vanadium oxides [
25]).
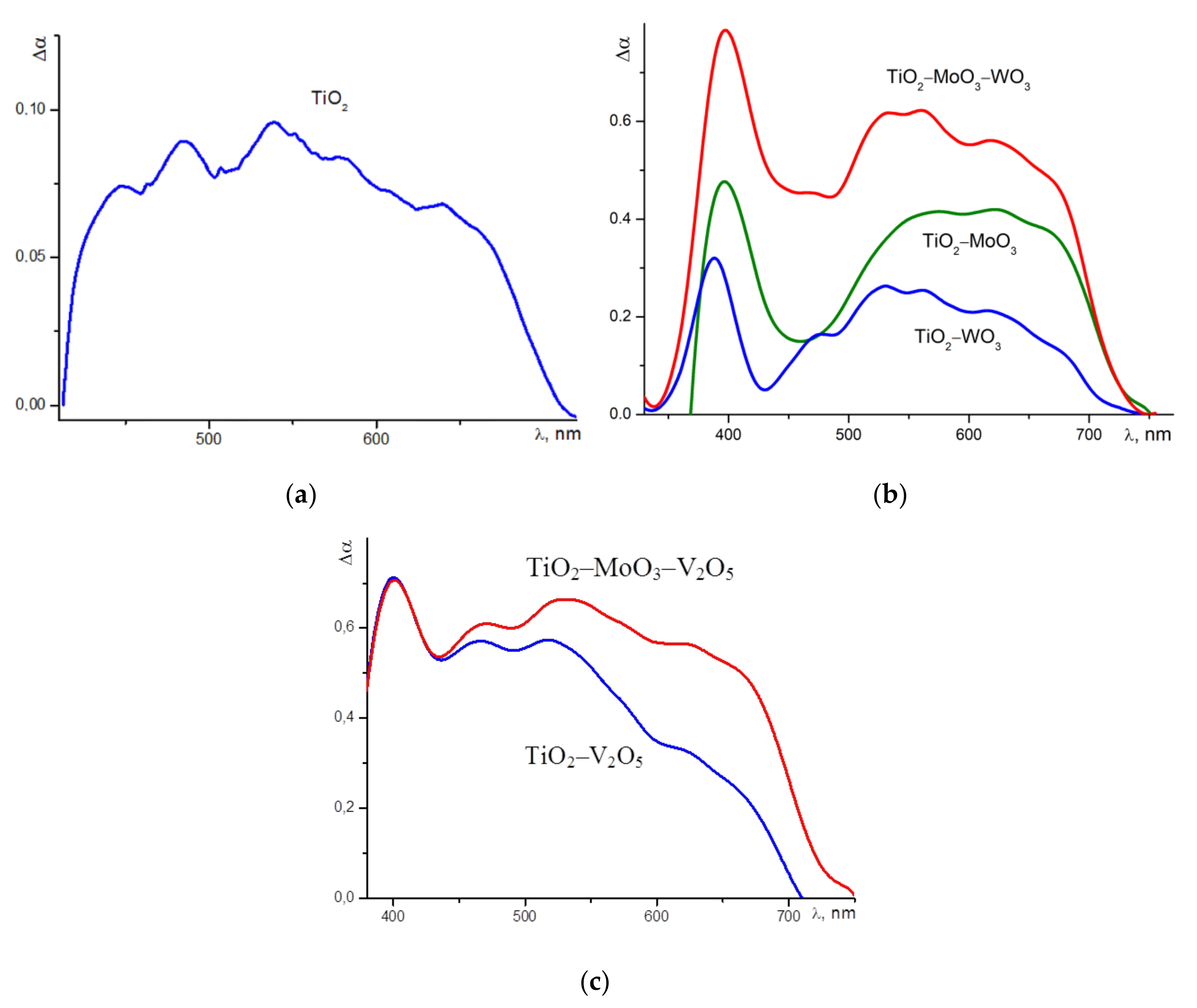
The explanation of the catalysis results obtained by illumination at the 500-nm wavelength can be obtained by comparing them with the absorption spectra of the studied nanoheterostructures in the visible region. To do this, we considered the difference of absorption spectra in the visible region between the studied nanoheterostructures and pure nanocrystalline TiO
2. The heterostructures used in this study were produced on the basis of N-doped TiO
2. The difference absorption spectrum for N-doped TiO
2 is represented at
Figure 7a. Such doping creates an additional wide, structureless absorption band for titanium dioxide nanocrystals, from about 420 to 720 nm. This may correspond to a fairly uniform spectrum of electronic states in the band gap of TiO
2. Therefore, the adsorbed molecules of the rhodamine dye (when illuminated at 500 nm) can efficiently transfer the photoexcitation energy on the order of 2.16 eV (≈575 nm, see
Figure 4) to the semiconductor via the induction-resonance symmetric dipole–dipole mechanism. Using this energy, the electrons are effectively ejected from the local states in the band gap to the conduction band of TiO
2. This can lead to the accelerated photodegradation of the dye on the surface of nitrogen-doped titanium dioxide when illuminated at a 500-nm wavelength. In addition, the doped material of the photocatalyst itself absorbs radiation at the wavelength of 500 nm (used for the measurement), which can also lead to the observed effect.
Samples of TiO
2–WO
3, TiO
2–MoO
3 and TiO
2–MoO
3–WO
3 more effectively absorb light in the visible region than N-doped TiO
2. The difference between the absorption spectra of these nanoheterostructures and pure titanium dioxide in the visible region is shown at
Figure 7b, but there is a clear dip in their absorption spectra in the range of 420 to 540 nm. Therefore, the illumination of such samples at a 500-nm wavelength should not lead to a significant photocatalytic effect. Although all three of these structures can effectively absorb radiation with the wavelength of ~575 nm, this means that in their electronic spectrum there are states that could be acceptors for non-radiative energy transfer from the dye molecules. Moreover, the TiO
2–MoO
3–WO
3 nanoheterostructures have symmetric behaviour regarding their absorption coefficients and photocatalysis. These samples have a higher absorption coefficient at the 500-nm wavelength than the other two structures (
Figure 7b), and their photocatalysis is more effective under such illumination (
Figure 2).
Thus, we were able to show that under the illumination of nanoheterostructures with adsorbed rhodamine dye, at the dye-absorption wavelength (500 nm), photocatalytic reactions are mainly determined by the absorption of light by the nanostructures themselves, and not by the transfer of energy from the dye. Since a strong quenching of the initial fluorescence is observed on the surface of all studied nanoheterostructures, it can be stated that the nonradiative energy transfer from the dye molecules associated with such quenching occurs, but does not lead to a significant additional acceleration of photocatalysis. This means that the determination of the photocatalytic efficiency under illumination in the visible range, using adsorbed rhodamine-type dyes, is a valid procedure.
As for photocatalysis with the use of TiO
2–V
2O
5 and TiO
2–MoO
3–V
2O
5 nanoheterostructures, their absorption in the visible region is very intense and does not have a noticeable dip (
Figure 7c). In addition, the quenching of the dye luminescence on the surfaces of these structures was so strong that it was not possible to measure the kinetics.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}