
Progress on the Stereoselective Synthesis of Chiral Molecules Based on Metal-Catalyzed Dynamic Kinetic Resolution of Alcohols with Lipases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
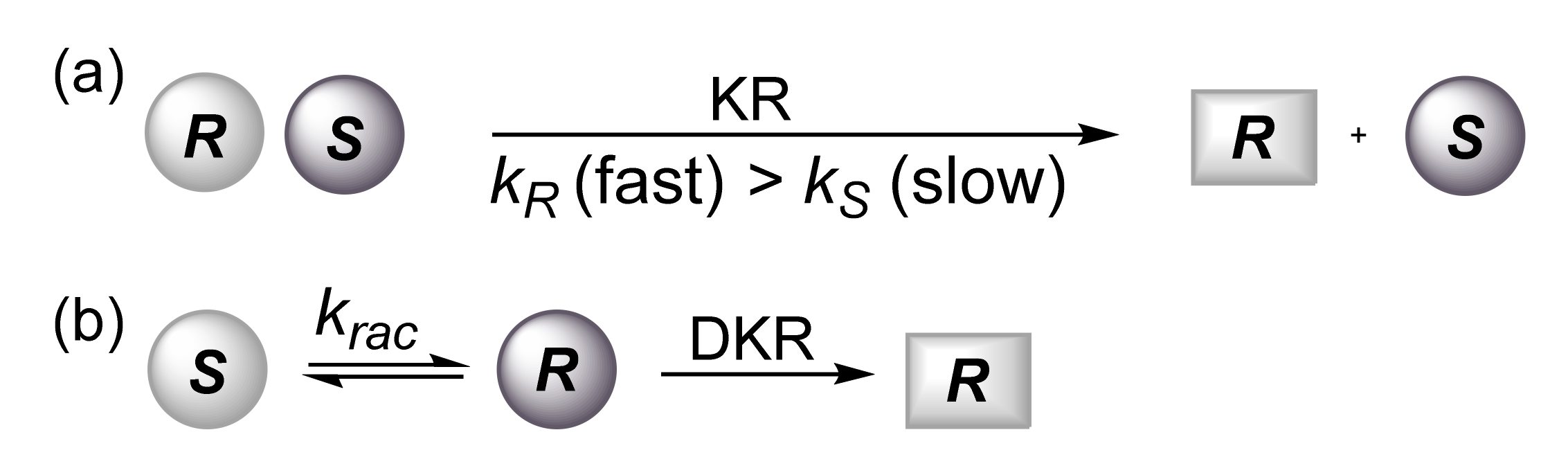
:1. Introduction
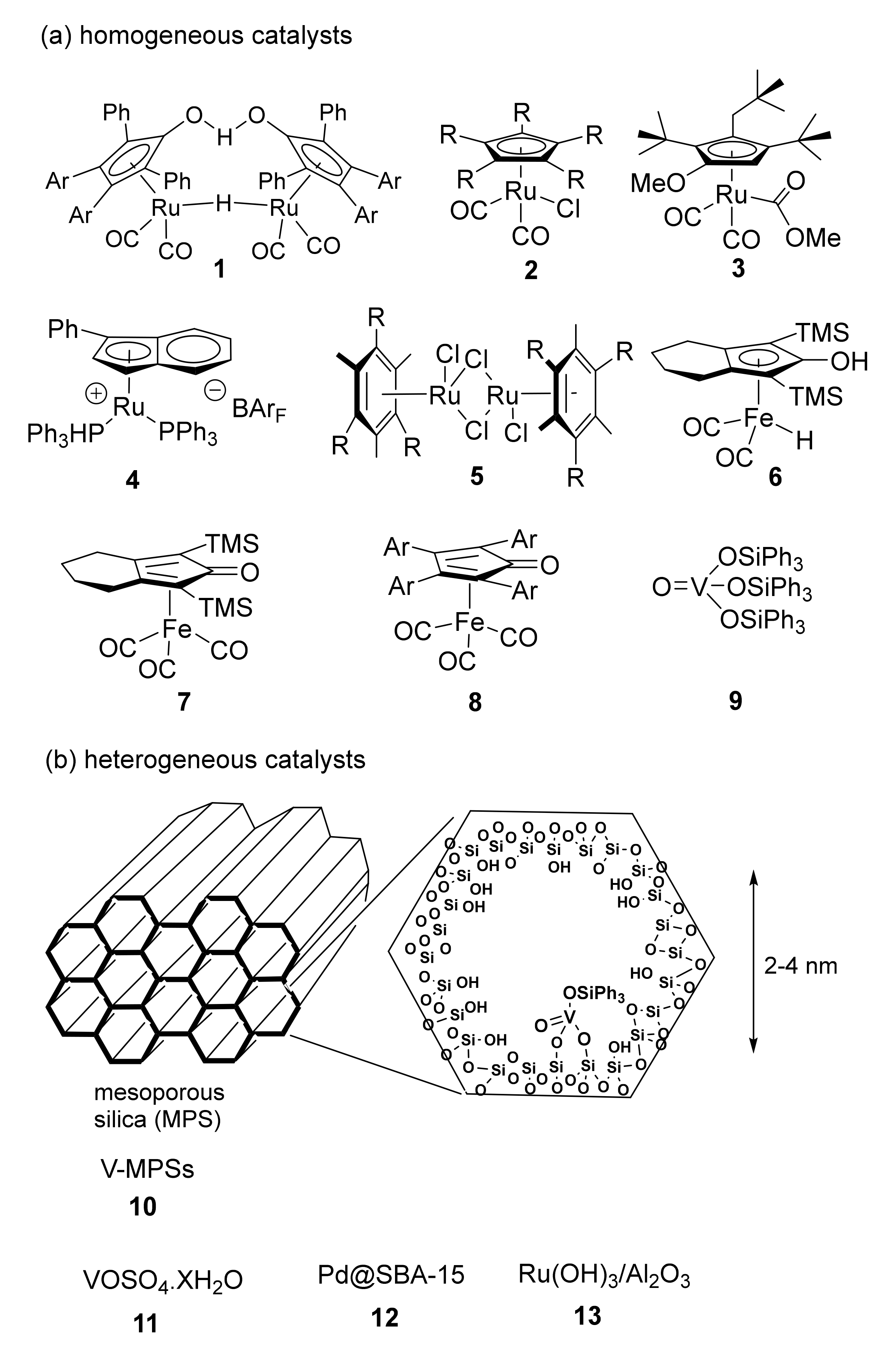
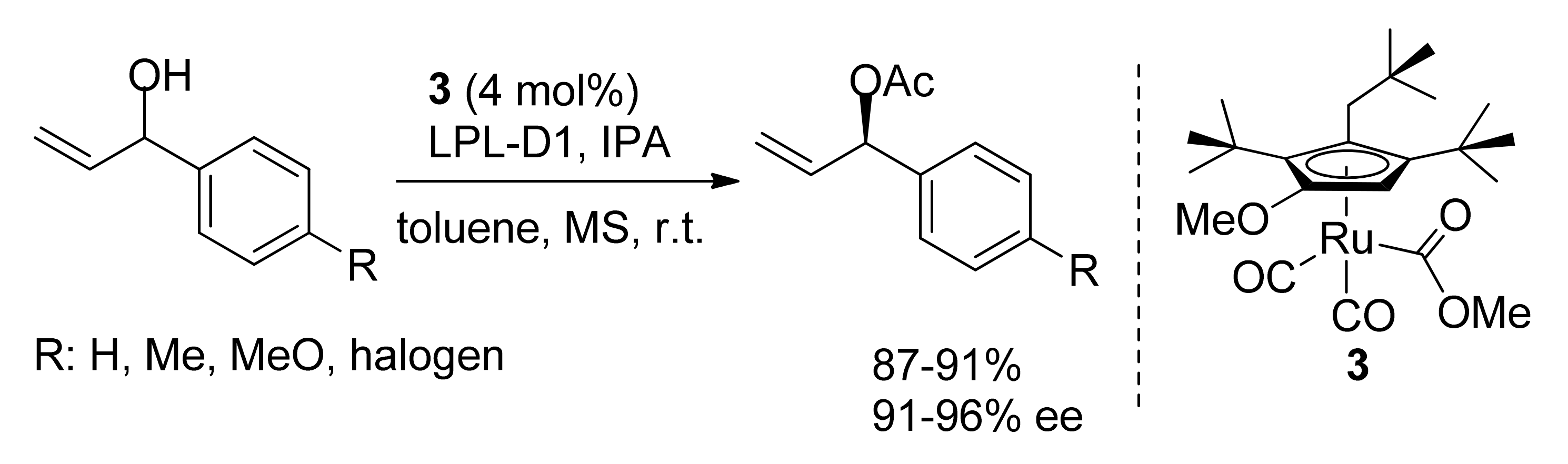
2. DKR of Alcohols Co-Catalyzed by Homogeneous Catalysts (Ru, Fe)
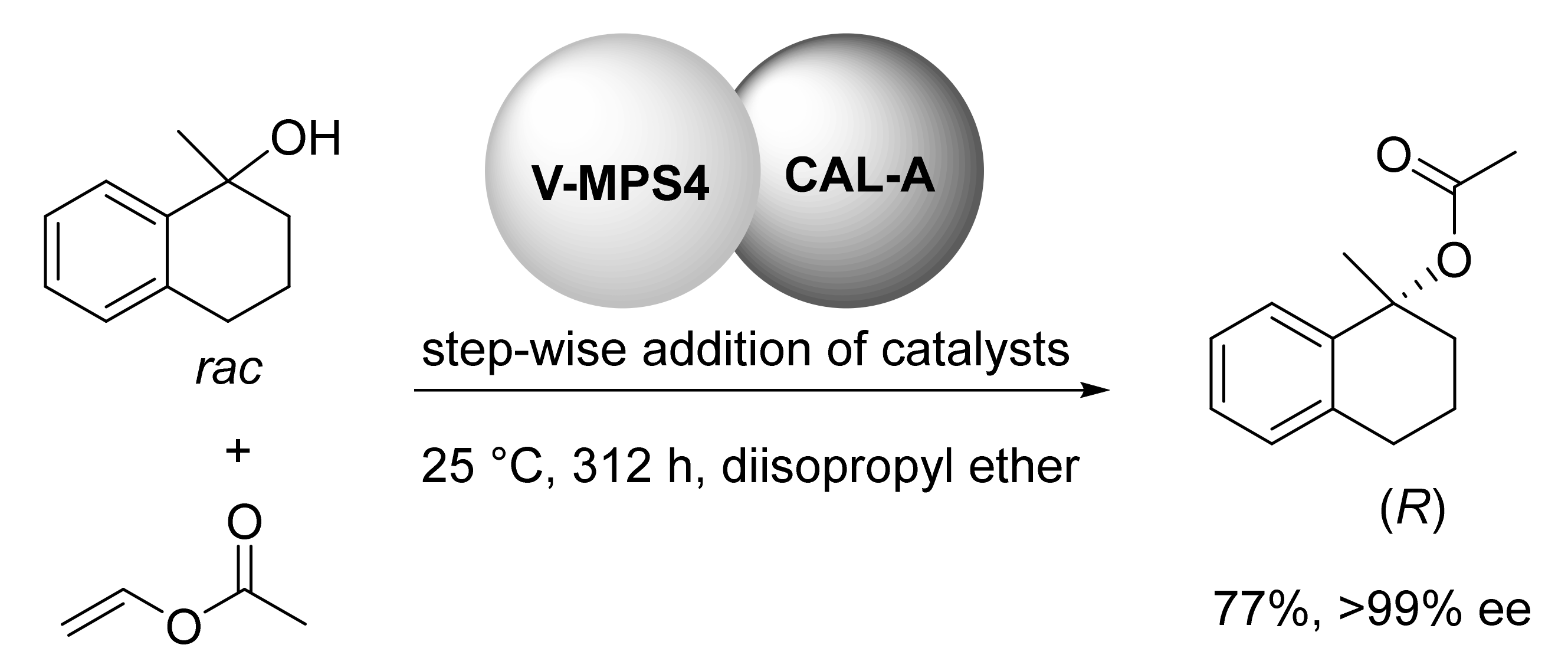
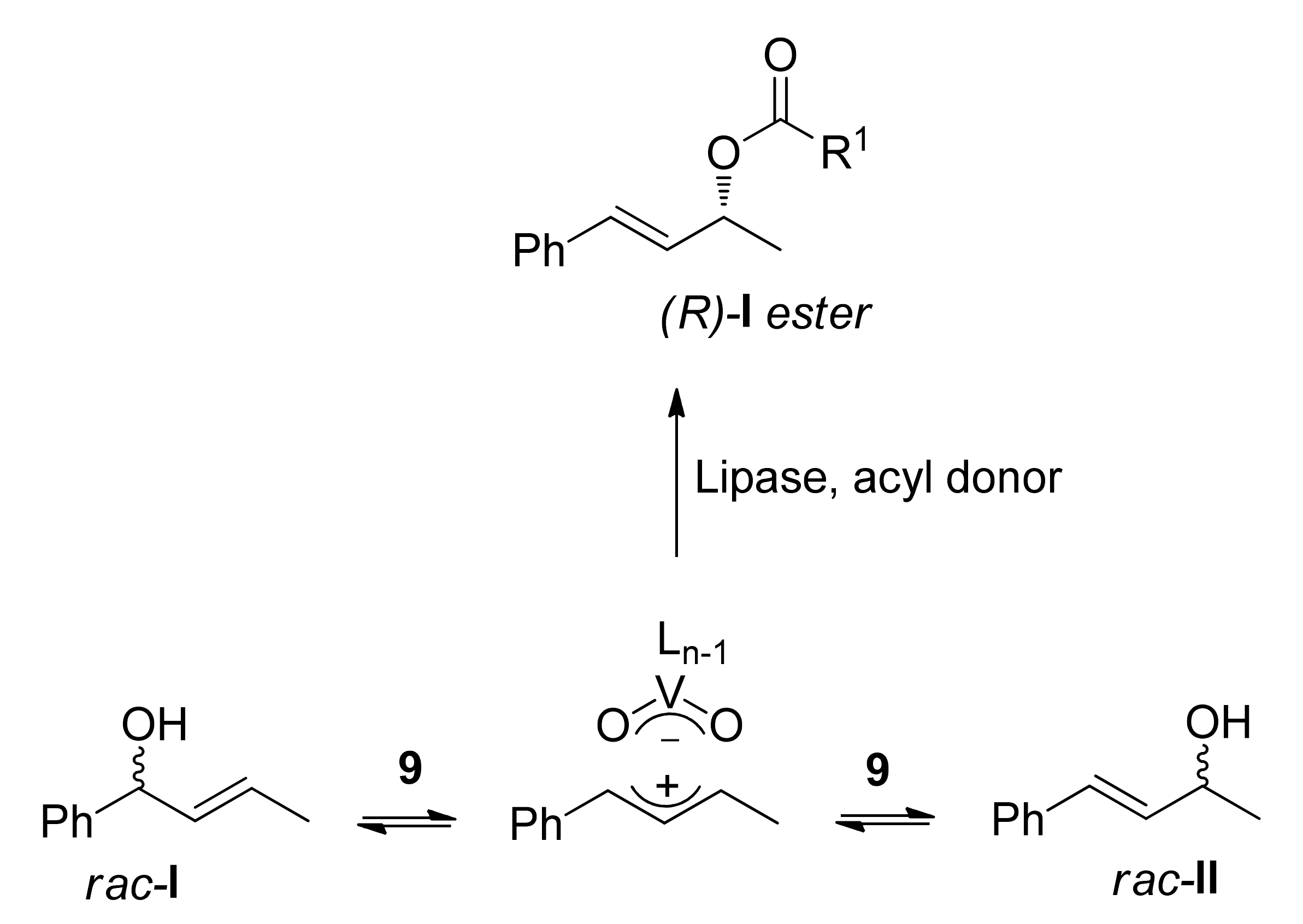
3. DKR of Alcohols Co-Catalyzed by Heterogeneous Catalysts (V, Pd)
4. Cascade Synthesis of Complex Molecules including DKR
4.1. Reductive Acylation of Ketones
4.2. Combining Aldol Reaction and DKR
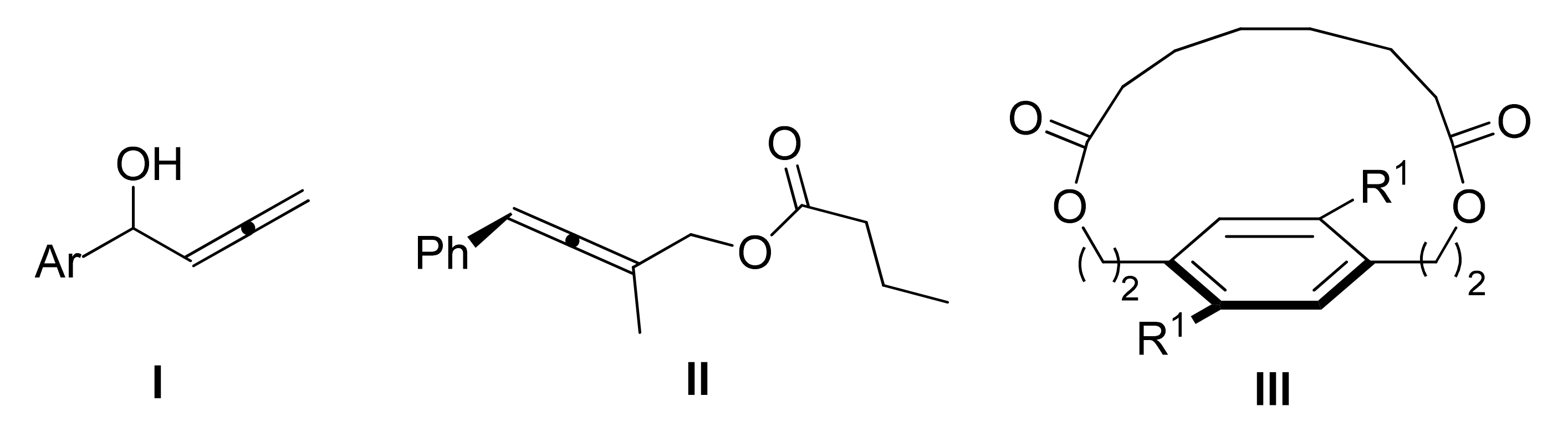
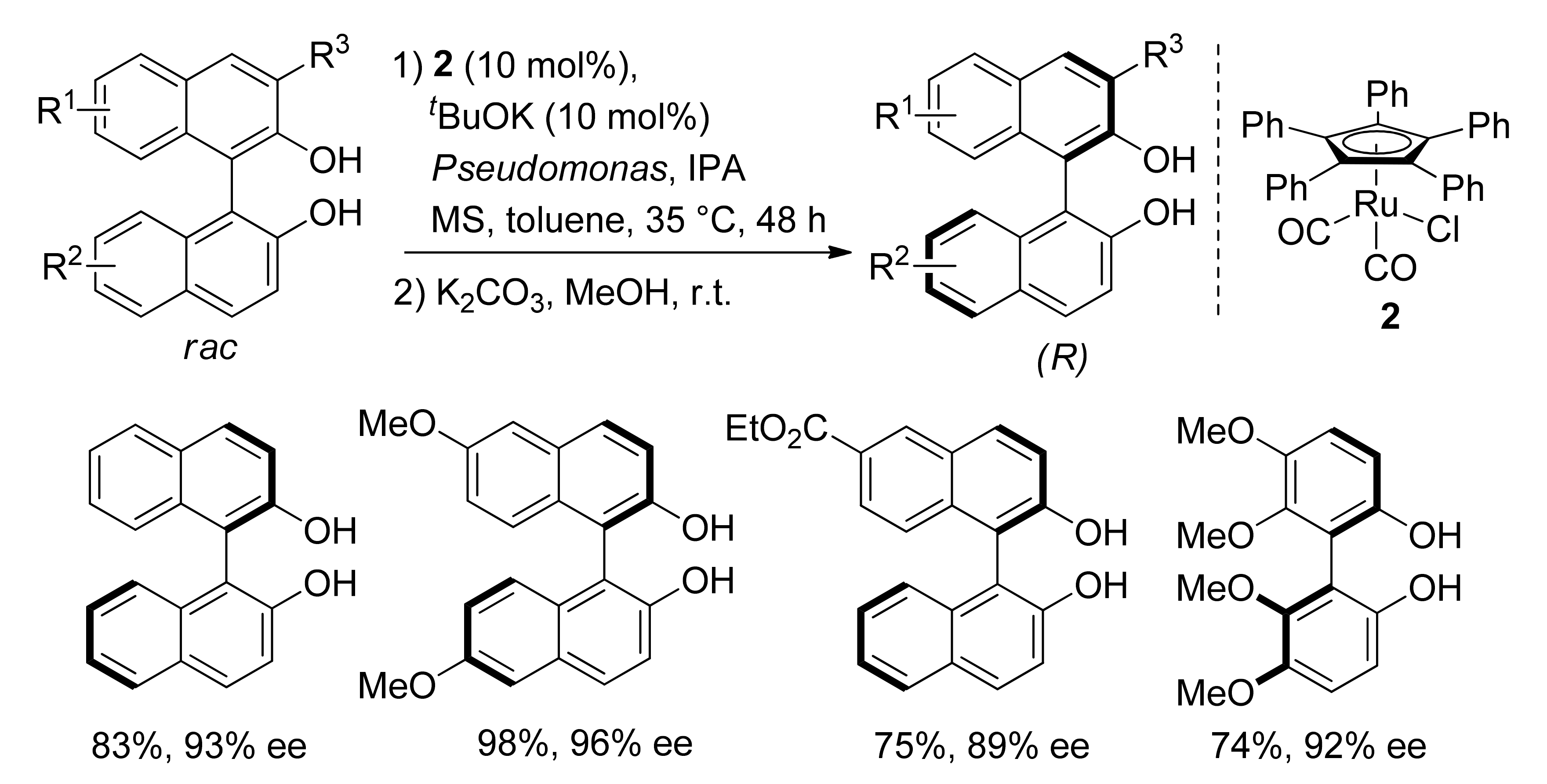
4.3. Combining DKR and Intramolecular Cyclizations: Synthesis of Enantiopure Carbo- and Heterocycles
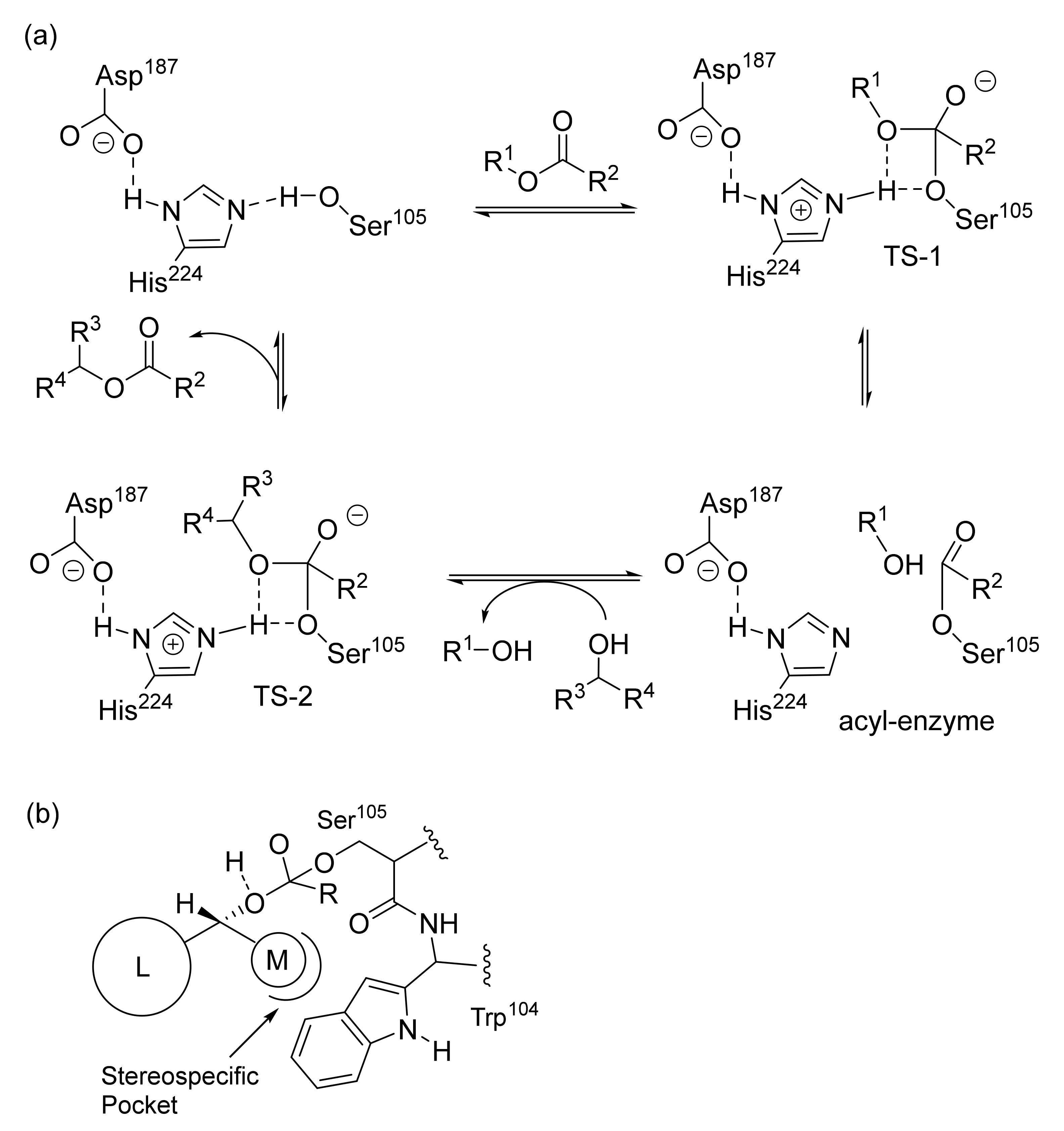
5. Metal Catalysts: Racemization Mechanism
5.1. Ru-Catalysts 2–5
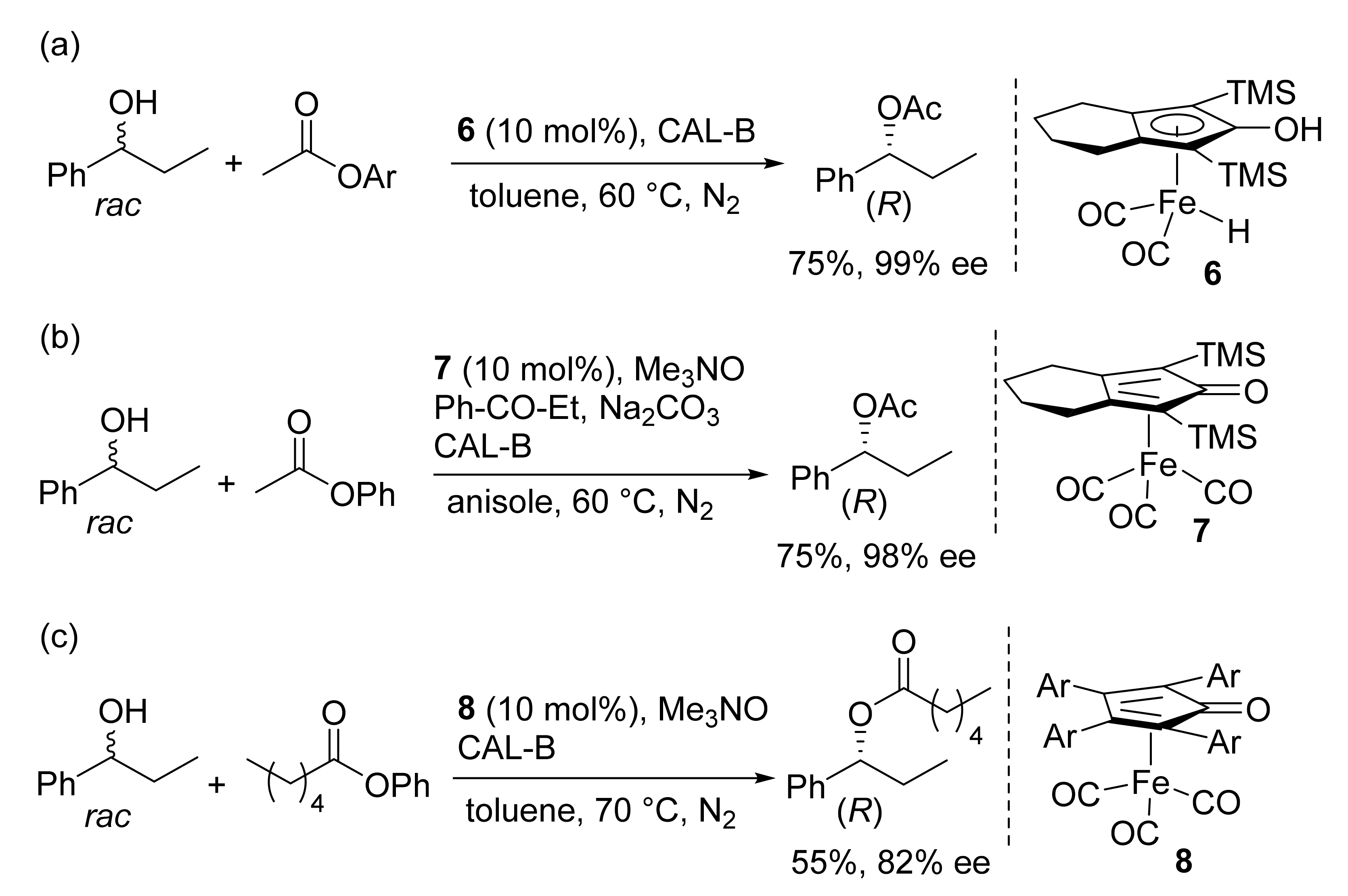
5.2. Ru- and Fe-Complexes 1, 6–8
5.3. Vanadium Catalysts
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAL-A | Candida antartica lipase A |
| CAL-B | Candida antartica lipase B |
| DKR | dynamic kinetic resolution |
| DYKAT | dynamic kinetic asymmetric transformation |
| DET | direct enantio-convergent transformation |
| KR | kinetic resolution |
| IL | ionic liquid |
| IPA | isopropenyl acetate |
| LDA | lithium diisopropylamide |
| LPL | lipoprotein lipase from Burkholderia species |
| MPS | mesoporous silica |
| MS | molecular sieves |
| PCPA | p-chlorophenylacetate. |
| PS | lipase from Burkholderia cepacia |
| PSIM | immobilized Burkholderia cepacia lipase |
| SET | single electron transfer |
| TBME | tert-butyl methyl ether |
| THF | tetrahydrofuran |
| TMS | trimethylsilyl group |
References
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lu, B.; Li, W.; Zhang, Z. Coordination determined chemo- and enantioselectivities in asymmetric hydrogenation of multi-functionalized ketones. Coord. Chem. Rev. 2018, 355, 39–53. [Google Scholar] [CrossRef]
- Wang, H.; Wen, J.; Zhang, X. Chiral tridentate ligands in transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 2021, 121, 7530–7567. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Tokunaga, M.; Kitamura, M. Stereoselective organic synthesis via dynamic kinetic resolution. Bull. Chem. Soc. Jpn. 1995, 68, 36–56. [Google Scholar] [CrossRef]
- Seddigi, Z.S.; Malik, M.S.; Ahmed, S.A.; Babalghith, A.O.; Kamal, A. Lipases in asymmetric transformations: Recent advances in classical kinetic resolution and lipase–metal combinations for dynamic processes. Coord. Chem. Rev. 2017, 348, 54–70. [Google Scholar] [CrossRef]
- De Miranda, A.S.; Miranda, L.S.M.; de Souza, R.O.M.A. Lipases: Valuable catalysts for dynamic kinetic resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Weissfloch, A.N.E.; Rappaport, A.T.; Cuccia, L.A. A rule to predict which enantiomer of a secondary alcohol reacts faster in reactions catalyzed by cholesterol esterase, lipase from Pseudomonas cepacia, and lipase from Candida rugosa. J. Org. Chem. 1991, 56, 2656–2665. [Google Scholar] [CrossRef]
- Orrenius, C.; Haffner, F.; Rotticci, D.; Öhrner, N.; Norin, T.; Hult, K. Chiral recognition of alcohol enantiomers in acyl transfer reactions catalysed by Candida Antarctica lipase B. Biocatal. Biotransform. 1998, 16, 1–15. [Google Scholar] [CrossRef]
- Ferrario, V.; Nitti, P.; Ebert, C.; Gardossi, L. Modelling and predicting enzyme enantioselectivity: The aid of computational methods for the rational use of lipase B from Candida Antarctica. Curr. Biotechnol. 2015, 4, 87–99. [Google Scholar] [CrossRef]
- Dinh, P.M.; Howarth, J.A.; Hudnott, A.R.; Williams, J.M.J.; Harris, W. Catalytic racemisation of alcohols: Applications to enzymatic resolution reactions. Tetrahedron Lett. 1996, 37, 7623–7626. [Google Scholar] [CrossRef]
- Larsson, A.L.E.; Persson, B.A.; Bäckvall, J.-E. Enzymatic resolution of alcohols coupled with ruthenium-catalyzed racemization of the substrate alcohol. Angew. Chem. Int. Ed. 1997, 36, 1211–1212. [Google Scholar] [CrossRef]
- Ahn, Y.; Ko, S.-B.; Mahn-Joo Kim, M.-J.; Park, J. Racemization catalysts for the dynamic kinetic resolution of alcohols and amines. Coord. Chem. Rev. 2008, 252, 647–658. [Google Scholar] [CrossRef]
- Warner, M.C.; Bäckvall, J.-E. Mechanistic aspects on cyclopentadienylruthenium complexes in catalytic racemization of alcohols. Acc. Chem. Res. 2013, 46, 2545–2555. [Google Scholar] [CrossRef]
- Lee, H.; Han, K.; Kim, M.-J.; Park, J. Chemoenzymatic dynamic kinetic resolution of alcohols and amines. Eur. J. Org. Chem. 2010, 2010, 999–1015. [Google Scholar] [CrossRef]
- Hoyos, P.; Pace, V.; Alcántara, A.R. Dynamic kinetic resolution via hydrolase-metal combo catalysis in stereoselective synthesis of bioactive compounds. Adv. Synth. Catal. 2012, 354, 2585–2611. [Google Scholar] [CrossRef]
- Verho, O.; Bäckvall, J.-E. Chemoenzymatic dynamic kinetic resolution: A powerful tool for the preparation of enantiomerically pure alcohols and amines. J. Am. Chem. Soc. 2015, 137, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Långvik, O.; Saloranta, T.; Murzin, D.Y.; Leino, R. Heterogeneous chemoenzymatic catalyst combinations for one-pot dynamic kinetic resolution applications. ChemCatChem 2015, 7, 4004–4015. [Google Scholar] [CrossRef]
- Bath, V.; Welin, E.R.; Guo, X.; Stoltz, B.M. Advances in stereoconvergent catalysis from 2005 to 2015: Transition-metal-mediated stereoablative reactions, dynamic kinetic resolutions, and dynamic kinetic asymmetric transformations. Chem. Rev. 2017, 117, 4528–4561. [Google Scholar]
- Trost, B.M.; Toste, F.D. Palladium-catalyzed kinetic and dynamic kinetic asymmetric transformation of 5-acyloxy-2-(5H)-furanone. Enantioselective synthesis of (-)-Aflatoxin B lactone. J. Am. Chem. Soc. 1999, 121, 3543–3544. [Google Scholar] [CrossRef]
- Trost, B.M.; Fandrick, D.R. Palladium-catalyzed dynamic kinetic asymmetric allylic alkylation with the DPPBA ligands. Aldrichchimica Acta 2007, 40, 59–71. [Google Scholar]
- Ito, H.; Kunii, S.; Sawamura, M. Direct enantio-convergent transformation of racemic substrates without racemization or symmetrization. Nat. Chem. 2010, 2, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Roush, W.R. Enantioconvergent hydroboration of a racemic allene: Enantioselective synthesis of (E)-δ-stannyl-anti-homoallylic alcohols via aldehyde crotylboration. J. Am. Chem. Soc. 2011, 133, 5744–5747. [Google Scholar] [CrossRef] [Green Version]
- Yun, I.; Park, J.Y.; Park, J.; Kim, M.-J. Base-free dynamic kinetic resolution of secondary alcohols with a ruthenium−lipase couple. J. Org. Chem. 2019, 84, 16293–16298. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Choi, Y.K.; Kim, Y.H.; Park, E.S.; Kim, E.J.; Kim, M.-J.; Park, J. Aminocyclopentadienyl ruthenium complexes as racemization catalysts for dynamic kinetic resolution of secondary alcohols at ambient temperature. J. Org. Chem. 2004, 69, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, A.; Jezowska, M.M.; Laymand, K.; Möbus, J.; Martín-Matute, B. Synthesis of β-hydroxy and β-amino ketones from allylic alcohols catalyzed by Ru(η5-C5Ph5)(CO)2Cl. Eur. J. Inorg. Chem. 2012, 2012, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Holzwarth, M.S.; Plietker, B. Biorelevant metals in sustainable metal catalysis—A survey. ChemCatChem 2013, 5, 1650–1679. [Google Scholar] [CrossRef]
- Filonenko, G.A.; van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Catalytic (de)hydrogenation promoted by non-precious metals—Co, Fe and Mn: Recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornschein, C.; Gustafson, K.P.J.; Verho, O.; Beller, M.; Bäckvall, J.-E. Evaluation of Fe and Ru pincer-type complexes as catalysts for the racemization of secondary benzylic alcohols. Chem. Eur. J. 2016, 22, 11583–11586. [Google Scholar] [CrossRef]
- El-Sepelgy, O.; Alandini, N.; Rueping, M. Merging iron catalysis and biocatalysis—Iron carbonyl complexes as efficient hydrogen autotransfer catalysts in dynamic kinetic resolutions. Angew. Chem. Int. Ed. 2016, 55, 13602–13605. [Google Scholar] [CrossRef]
- Gustafson, K.P.J.; Guðmundsson, A.; Lewis, K.; Bäckvall, J.-E. Chemoenzymatic dynamic kinetic resolution of secondary alcohols using an air-and moisture-stable iron racemization catalyst. Chem. Eur. J. 2017, 23, 1048–1051. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zhang, N.; Liu, M.; Zhou, S. New air-stable iron catalyst for efficient dynamic kinetic resolution of secondary benzylic and aliphatic alcohols. Tetrahedron Lett. 2017, 58, 2487–2489. [Google Scholar] [CrossRef]
- Yang, B.; Zhu, C.; Qiu, Y.; Bäckvall, J.-E. Enzyme- and ruthenium-catalyzed enantioselective transformation of α-allenic alcohols into 2,3-dihydrofurans. Angew. Chem. Int. Ed. 2016, 55, 5568–5572. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, C.; Godin, É.; Minozzi, C.; Sosoe, J.; Pochet, C.; Collins, S.K. Biocatalytic synthesis of planar chiral macrocycles. Science 2020, 367, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Deska, J.; del Pozo Ochoa, C.; Backvall, J.-E. Chemoenzymatic dynamic kinetic resolution of axially chiral allenes. Chem. Eur. J. 2010, 16, 4447–4451. [Google Scholar] [CrossRef] [PubMed]
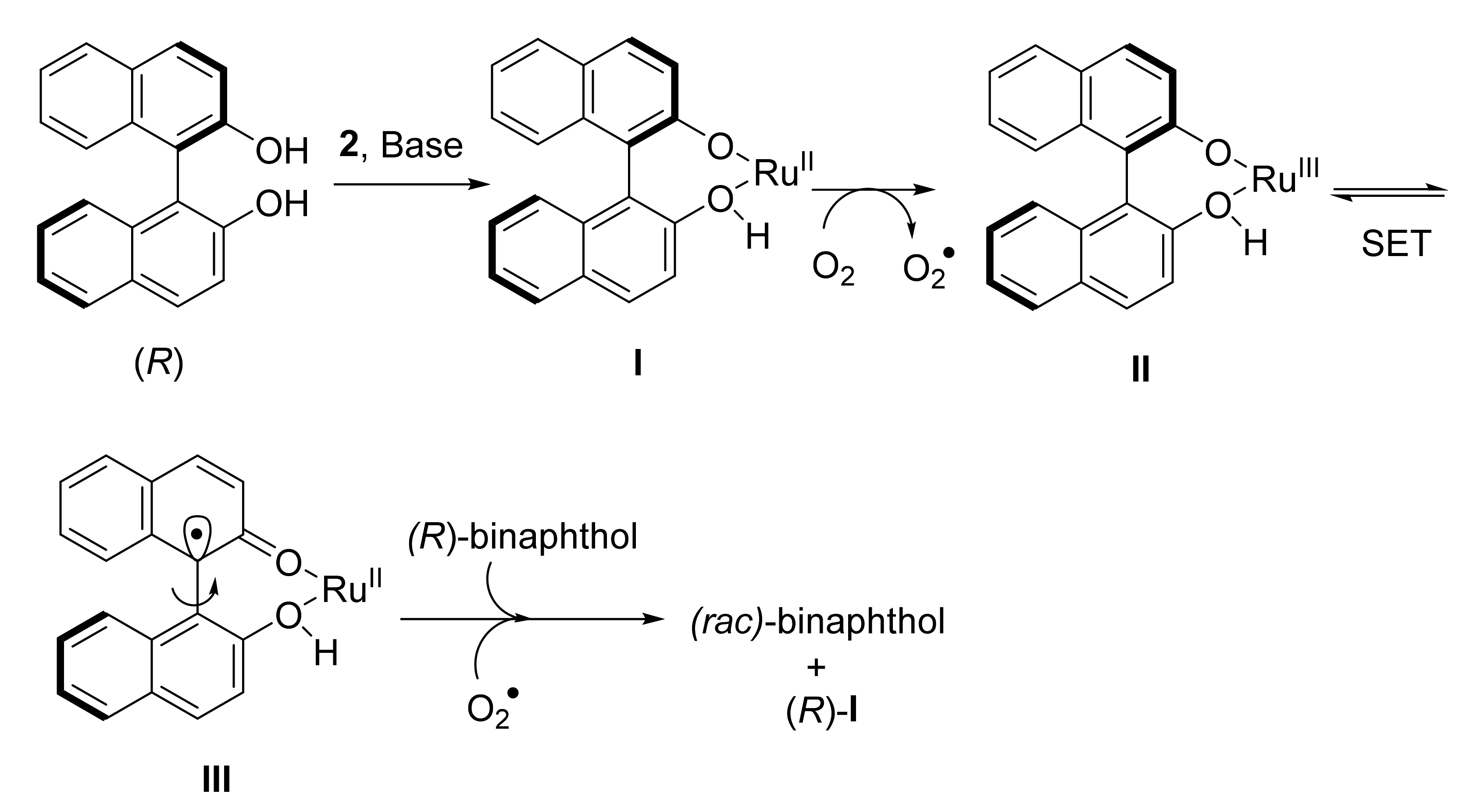
- Moustafa, G.A.I.; Oki, Y.; Akai, S. Lipase-catalyzed dynamic kinetic resolution of C1 and C2-symmetric racemic axially chiral 2,2′-dihydroxy-1,1′-biaryls. Angew. Chem. Int. Ed. 2018, 57, 10278–10282. [Google Scholar] [CrossRef]
- Kozlowski, M.C.; Morgan, B.J.; Linton, E.C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 2009, 38, 3193–3207. [Google Scholar] [CrossRef]
- Takizawa, S.; Gröger, H.; Sasai, H. Vanadium in asymmetric synthesis: Emerging concepts in catalyst design and applications. Chem. Eur. J. 2015, 21, 8992–8997. [Google Scholar] [CrossRef]
- Akai, S.; Tanimoto, K.; Kanao, Y.; Egi, M.; Yamamoto, T.; Kita, Y. A dynamic kinetic resolution of allyl alcohols by the combined use of lipases and [VO(OSiPh3)3]. Angew. Chem. Int. Ed. 2006, 45, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Egi, M.; Sugiyama, K.; Saneto, M.; Hanada, R.; Kato, K.; Akai, S. A Mesoporous-silica-immobilized oxovanadium cocatalyst for the lipase-catalyzed dynamic kinetic resolution of racemic alcohols. Angew. Chem. Int. Ed. 2013, 52, 3654–3658. [Google Scholar] [CrossRef]
- Akai, S. Dynamic kinetic resolution of racemic allylic alcohols via hydrolase metal combo catalysis: An effective method for the synthesis of optically active compounds. Chem. Lett. 2014, 43, 746–754. [Google Scholar] [CrossRef]
- Kourist, R.; de María, P.D.; Bornscheuer, U.T. Enzymatic synthesis of optically active tertiary alcohols: Expanding the biocatalysis toolbox. ChemBioChem 2008, 9, 491–498. [Google Scholar] [CrossRef]
- Görbe, T.; Lihammar, R.; Bäckvall, J.-E. Heterogeneous acid-catalyzed racemization of tertiary alcohols. Chem. Eur. J. 2018, 24, 77–80. [Google Scholar] [CrossRef]
- Kuhn, F.; Katsuragi, S.; Oki, Y.; Scholz, C.; Akai, S.; Gröger, H. Dynamic kinetic resolution of a tertiary alcohol. Chem. Commun. 2020, 56, 2885–2888. [Google Scholar] [CrossRef]
- Kawanishi, S.; Oki, S.; Kundu, D.; Akai, S. Lipase/oxovanadium co-catalyzed dynamic kinetic resolution of propargyl alcohols: Competition between racemization and rearrangement. Org. Lett. 2019, 21, 2978–2982. [Google Scholar] [CrossRef]
- Kawanishi, S.; Sugiyama, K.; Oki, Y.; Ikawa, T.; Akai, S. Preparation of optically active cycloalkenes bearing all-carbon quaternary stereogenic centres via lipase–oxovanadium combocatalysed dynamic kinetic resolution. Green Chem. 2017, 19, 411–417. [Google Scholar] [CrossRef]
- Britton, J.; Raston, C.L. Multi-step continuous-flow synthesis. Chem. Soc. Rev. 2017, 46, 1250–1271. [Google Scholar] [CrossRef] [Green Version]
- Foley, A.M.; Maguire, A.R. The impact of recent developments in technologies which enable the increased use of biocatalysts. Eur. J. Org. Chem. 2019, 2019, 3713–3734. [Google Scholar] [CrossRef]
- Higashio, K.; Katsuragi, S.; Kundu, D.; Adebar, N.; Plass, C.; Kühn, F.; Gröger, H.; Akai, S. Continuous-flow dynamic kinetic resolution of racemic alcohols by lipase–oxovanadium cocatalysis. Eur. J. Org. Chem. 2020, 1961–1967. [Google Scholar] [CrossRef]
- De Almeida, L.A.; Marcondes, T.H.; Milagre, C.D.F.; Milagre, H.M.S. Lipase-oxovanadium heterogeneous catalysis system: A robust protocol for the dynamic kinetic resolution of sec-alcohols. ChemCatChem 2020, 12, 2849–2858. [Google Scholar] [CrossRef]
- De França, A.S.; Silva, M.V.M.; Neves, R.V.; de Souza, S.P.; Leão, R.A.C.; Monteiro, C.M.; Rocha, Â.; Afonso, C.A.M.; de Souza, R.O.M.A. Studies on the dynamic resolution of Crizotinib intermediate. Bioorg. Med. Chem. 2018, 26, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, M.; Feng, B.; Li, Z.; Li, Y.; Li, H.; Li, H. Dynamic kinetic resolution of aromatic sec-alcohols by using a heterogeneous palladium racemization catalyst and lipase. Catal. Sci. Technol. 2017, 7, 5838–5842. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, P.; Gao, S.; Wang, Z.; Luan, P.; González-Sabín, J.; Jiang, J. Construction of chemoenzymatic cascade reactions for bridging chemocatalysis and biocatalysis: Principles, strategies and prospective. Chem. Eng. J. 2021, 420, 127659–127676. [Google Scholar] [CrossRef]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the incompatibility challenge in chemoenzymatic and multi-catalytic cascade reactions. Chem. Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Jung, H.M.; Koh, J.H.; Kim, M.-J.; Park, J. Concerted catalytic reactions for conversion of ketones or enol acetates to chiral acetates. Org. Lett. 2000, 2, 409–411. [Google Scholar] [CrossRef]
- Jung, H.M.; Koh, J.H.; Kim, M.-J.; Park, J. Practical ruthenium/lipase-catalyzed asymmetric transformations of ketones and enol acetates to chiral acetates. Org. Lett. 2000, 2, 2487–2490. [Google Scholar] [CrossRef]
- Millet, R.; Träff, A.M.; Petrus, M.L.; Bäckvall, J.-E. Enantioselective synthesis of syn- and anti-1,3-aminoalcohols via β-aminoketones and subsequent reduction/dynamic kinetic asymmetric transformation. J. Am. Chem. Soc. 2010, 132, 15182–15184. [Google Scholar] [CrossRef]
- Fernandez-Salas, J.A.; Manzini, S.; Nolan, S.P. A cationic ruthenium complex for the dynamic kinetic resolution of secondary alcohols. Chem. Eur. J. 2014, 20, 13132–13135. [Google Scholar] [CrossRef] [PubMed]
- El-Sepelgy, O.; Brzozowska, A.; Rueping, M. Asymmetric chemoenzymatic reductive acylation of ketones by a combined iron-catalyzed hydrogenation–racemization and enzymatic resolution cascade. ChemSusChem 2017, 10, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Långvik, O.; Sandberg, T.; Wärnå, J.; Murzin, D.Y.; Leino, R. One-pot synthesis of (R)-2-acetoxy-1-indanone from 1,2-indanedione combining metal catalyzed hydrogenation and chemoenzymatic dynamic kinetic resolution. Catal. Sci. Technol. 2015, 5, 150–160. [Google Scholar] [CrossRef]
- Huerta, F.F.; Bäckvall, J.-E. Enantioselective synthesis of β-hydroxy acid derivatives via a one-pot aldol reaction−dynamic kinetic resolution. Org. Lett. 2001, 3, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Akai, S.; Tanimoto, K.; Kita, Y. Lipase-catalyzed domino dynamic kinetic resolution of racemic 3-vinylcyclohex-2-en-1-ols/intramolecular Diels–Alder reaction: One-pot synthesis of optically active polysubstituted decalins. Angew. Chem. Int. Ed. 2004, 43, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Kawanishi, S.; Oki, Y.; Kamiyab, M.; Hanadab, R.; Egi, M.; Akai, S. Lipase-catalyzed asymmetric synthesis of naphtho[2,3-c]furan-1(3H)-one derivatives by a one-pot dynamic kinetic resolution/intramolecular Diels–Alder reaction: Total synthesis of (-)-himbacine. Bioorg. Med. Chem. 2018, 26, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimochi, I.; Hori, S.; Takeuchi, Y.; Egi, M.; Satoh, T.; Kanomata, K.; Ikawa, T.; Akai, S. Four-step one-pot catalytic asymmetric synthesis of polysubstituted tricyclic compounds: Lipase-catalyzed dynamic kinetic resolution followed by an intramolecular Diels–Alder reaction. Synlett 2021, 32, 822–828. [Google Scholar]
- Koszelewski, D.; Borys, F.; Brodzka, A.; Ostaszewski, R. Synthesis of enantiomerically pure 5,6-dihydropyran-2-ones via chemoenzymatic sequential DKR-RCM reaction. Eur. J. Org. Chem. 2018, 2019, 1653–1658. [Google Scholar] [CrossRef]
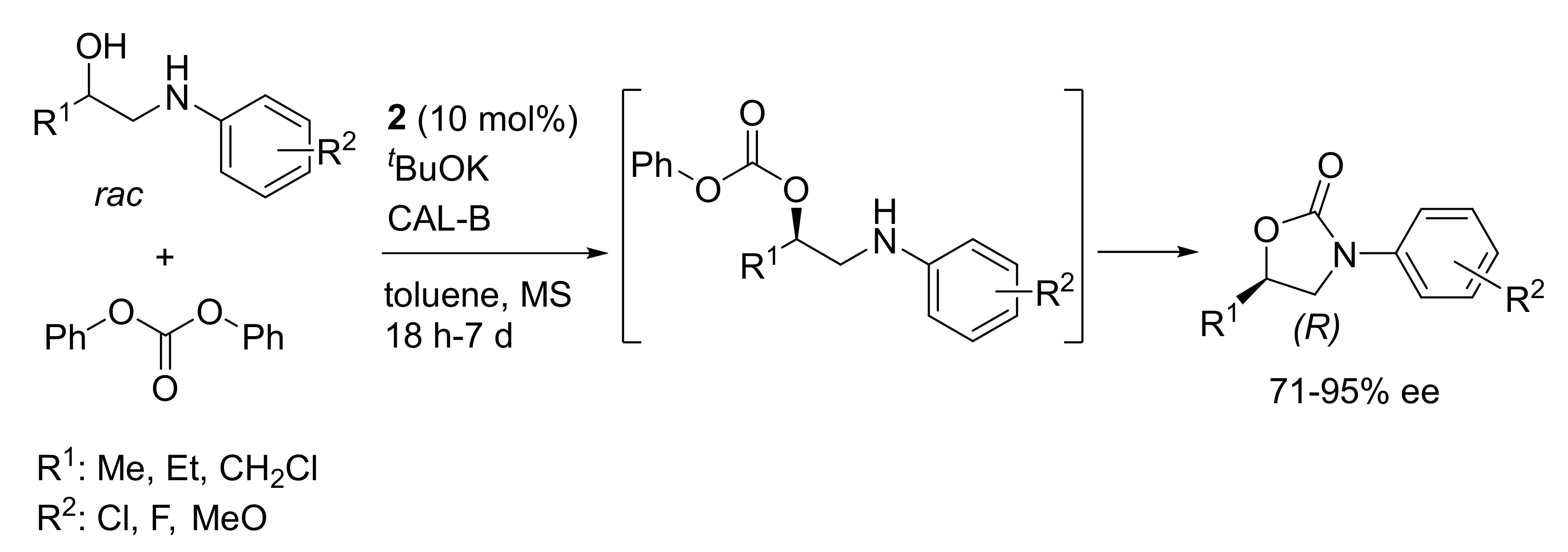
- Zhang, Y.; Xie, S.; Yan, M.; Ramström, O. Enzyme- and ruthenium-catalyzed dynamic kinetic resolution involving cascade alkoxycarbonylations for asymmetric synthesis of 5-substituted N-Aryloxazolidinones. Mol. Catal. 2019, 470, 138–144. [Google Scholar] [CrossRef]
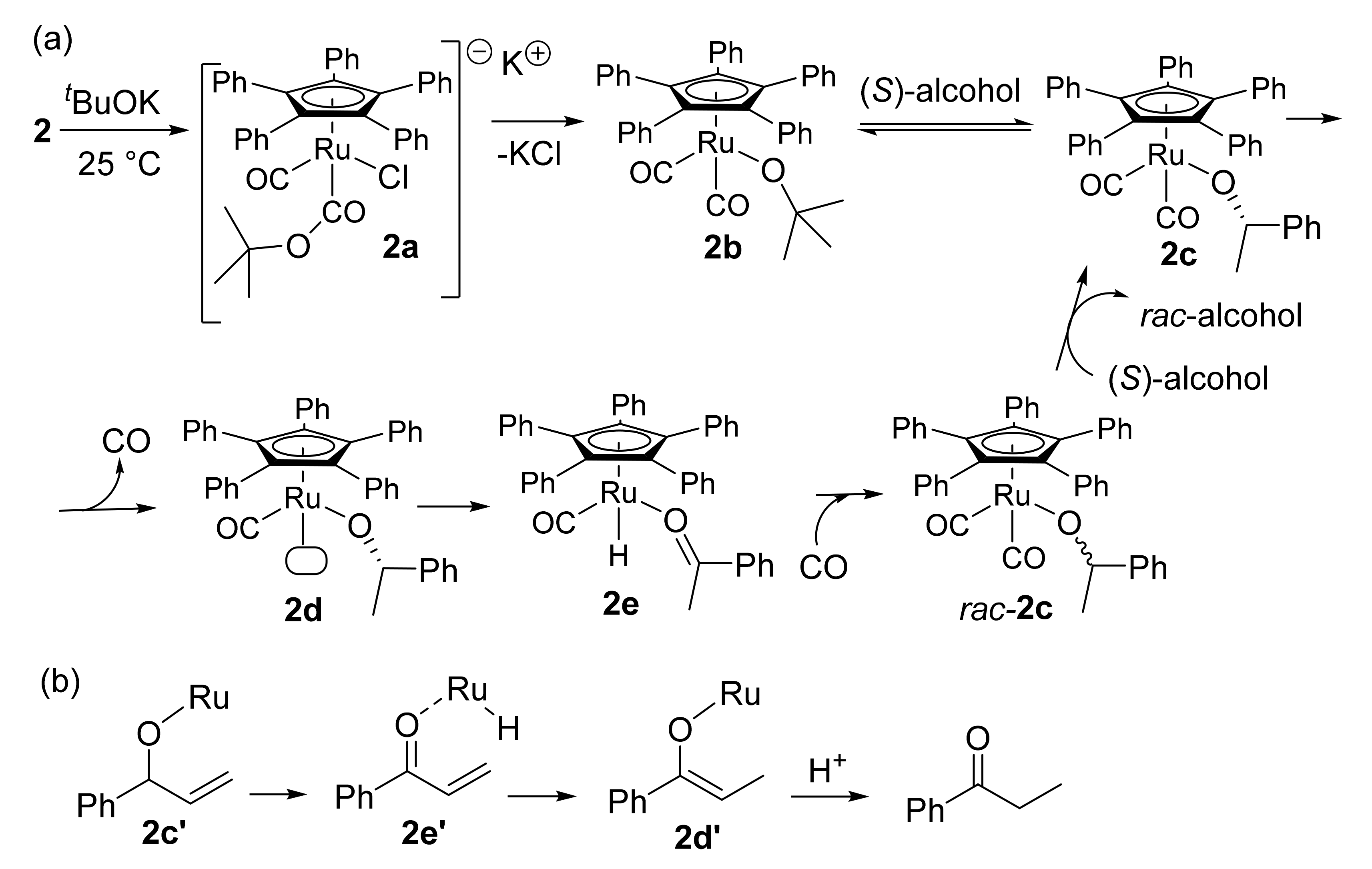
- Gustafson, K.P.J.; Guðmundsson, A.; Bajnóczi, É.G.; Yuan, N.; Zou, X.; Persson, I.; Bäckvall, J.-E. In situ structural determination of a homogeneous ruthenium racemization catalyst and its activated intermediates using X-ray absorption spectroscopy. Chem. Eur. J. 2020, 26, 3411–3419. [Google Scholar] [CrossRef]
- Bäckvall, J.-E.; Andreasson, U. Ruthenium-catalyzed isomerization of allylic alcohols to saturated ketones. Tetrahedron Lett. 1993, 34, 5459–5462. [Google Scholar] [CrossRef]
- Koh, J.H.; Jeong, H.M.; Park, J. Efficient catalytic racemization of secondary alcohols. Tetrahedron Lett. 1998, 39, 5545–5548. [Google Scholar] [CrossRef]
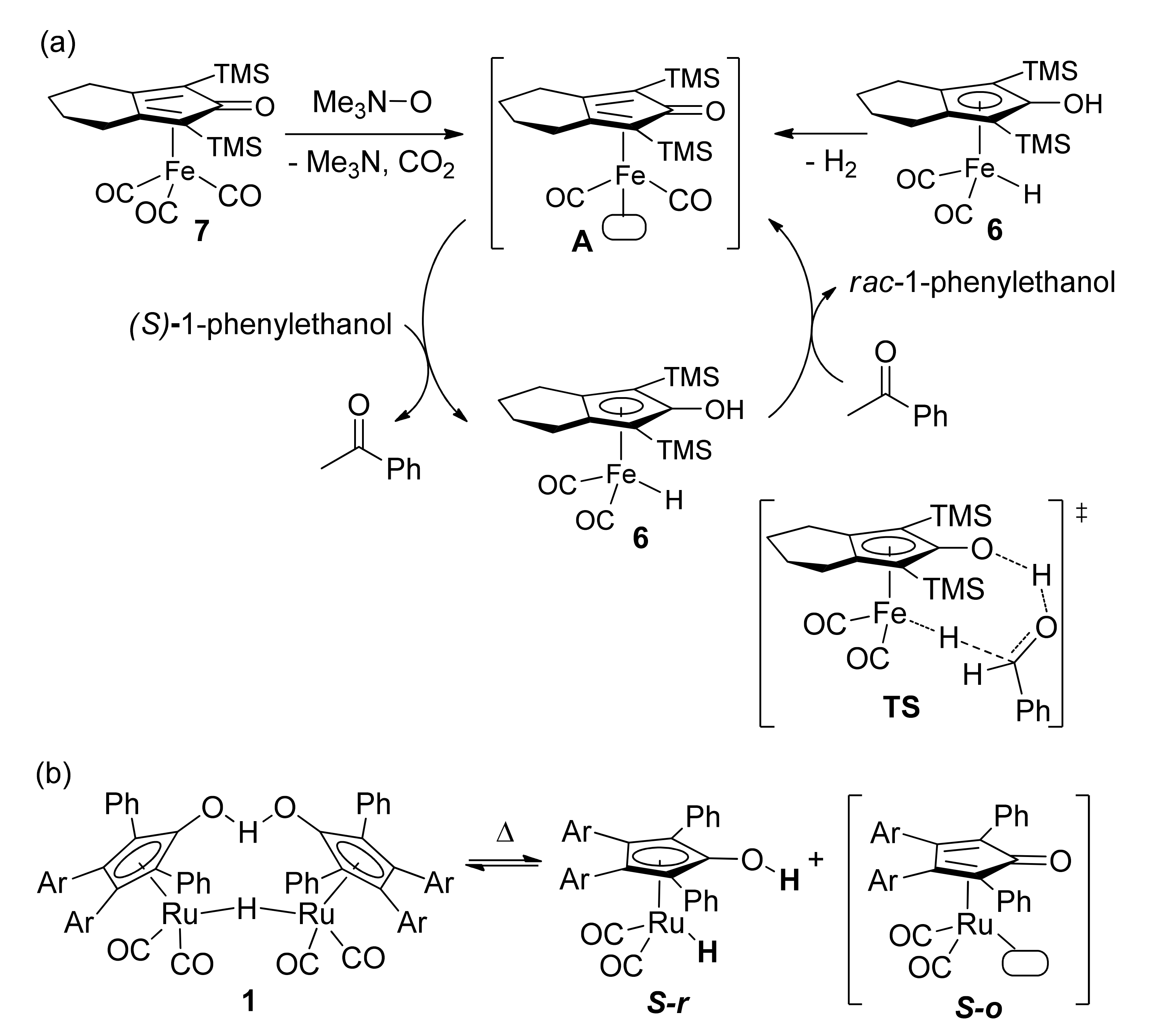
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Casey, C.P.; Guan, H. An efficient and chemoselective iron catalyst for the hydrogenation of ketones. J. Am. Chem. Soc. 2007, 129, 5816–5817. [Google Scholar] [CrossRef]
- Conley, B.L.; Pennington-Boggio, M.K.; Boz, E.; Williams, T.J. Discovery, applications, and catalytic mechanisms of Shvo’s catalyst. Chem. Rev. 2010, 110, 2294–2312. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, S.; Wahlen, J.; Jacobs, P.A.; De Vos, D.E. Heterogeneous vanadium catalysts for racemization and chemoenzymatic dynamic kinetic resolution of benzylic alcohols. Green Chem. 2007, 9, 1104–1108. [Google Scholar] [CrossRef]
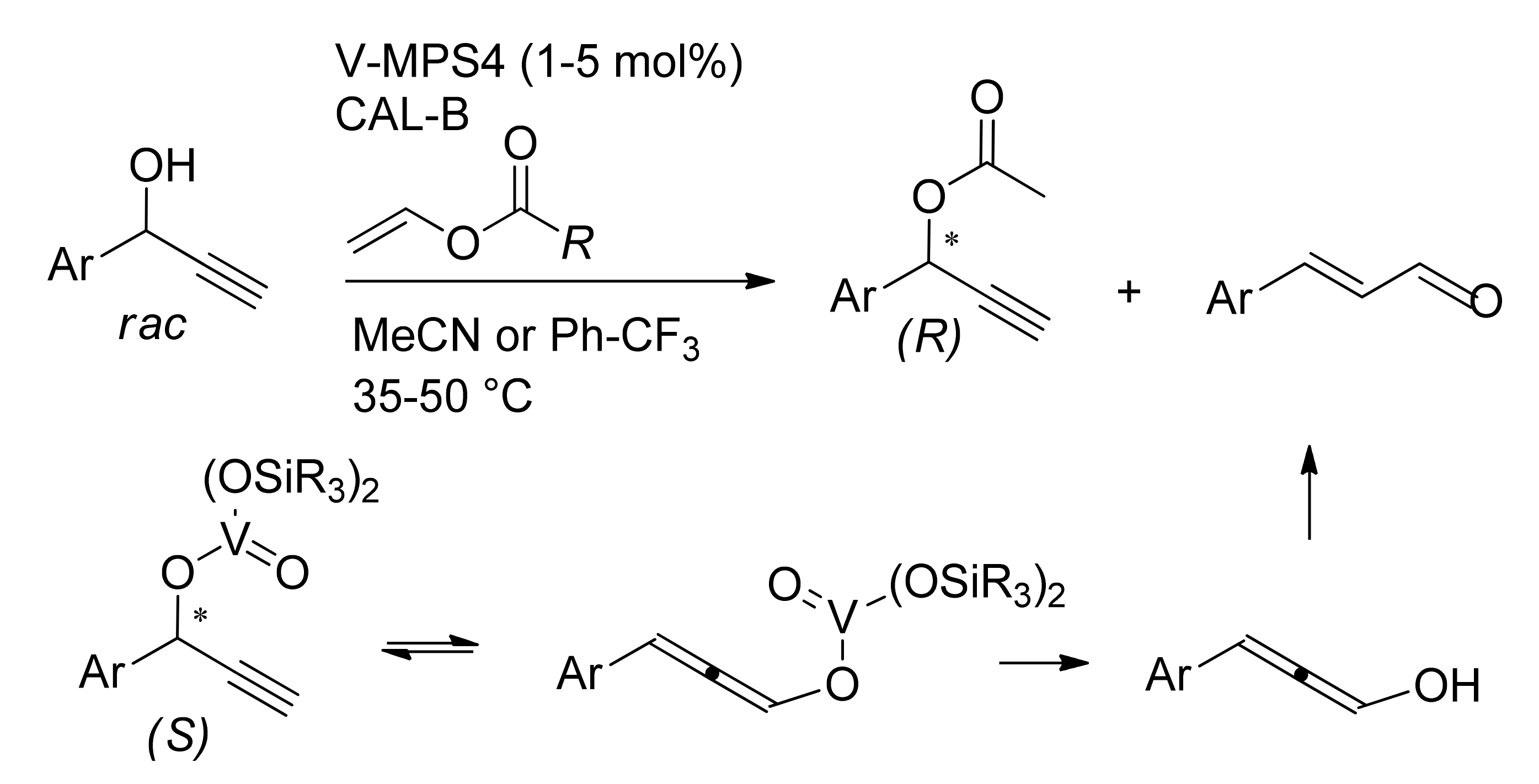
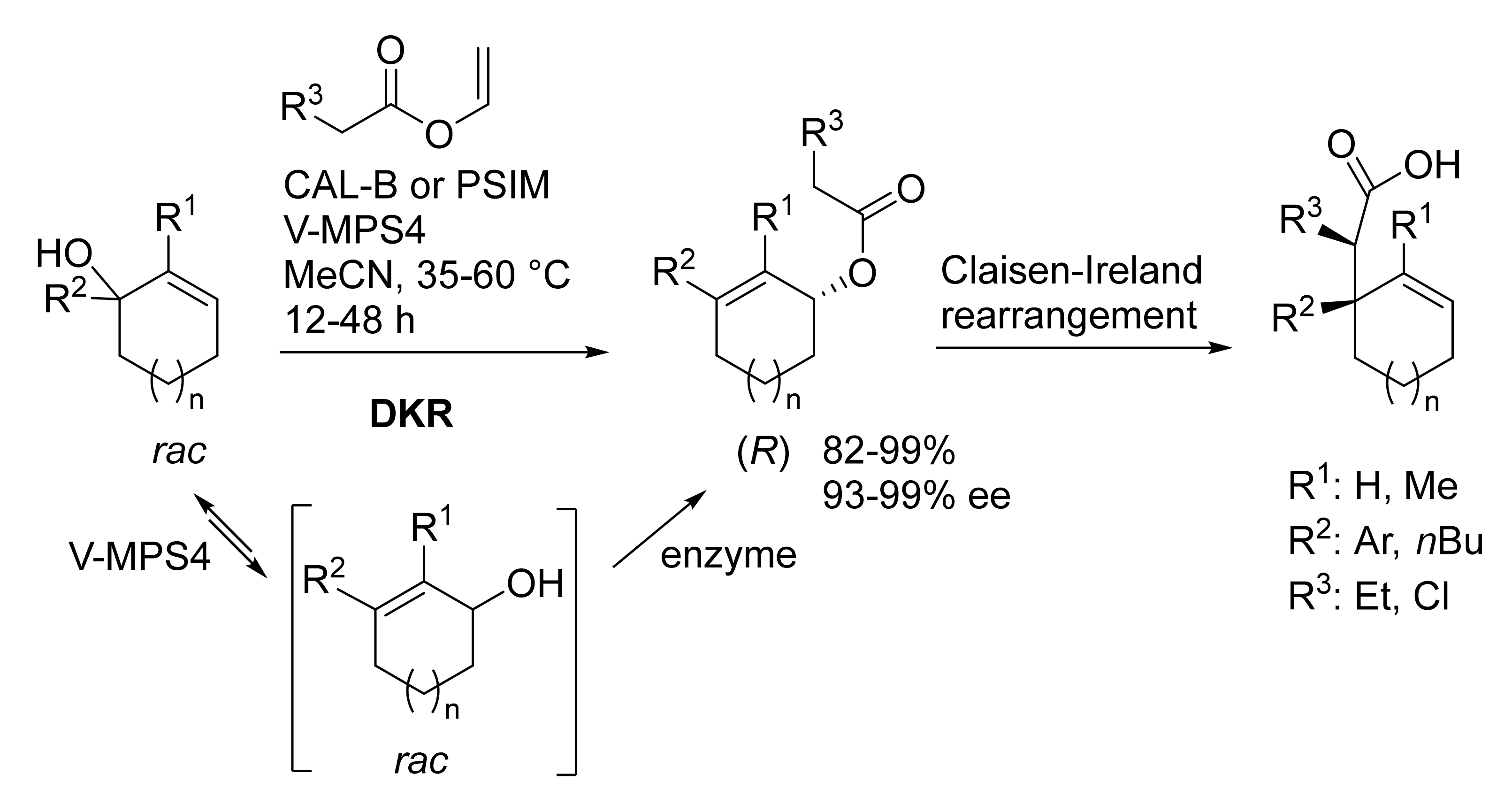
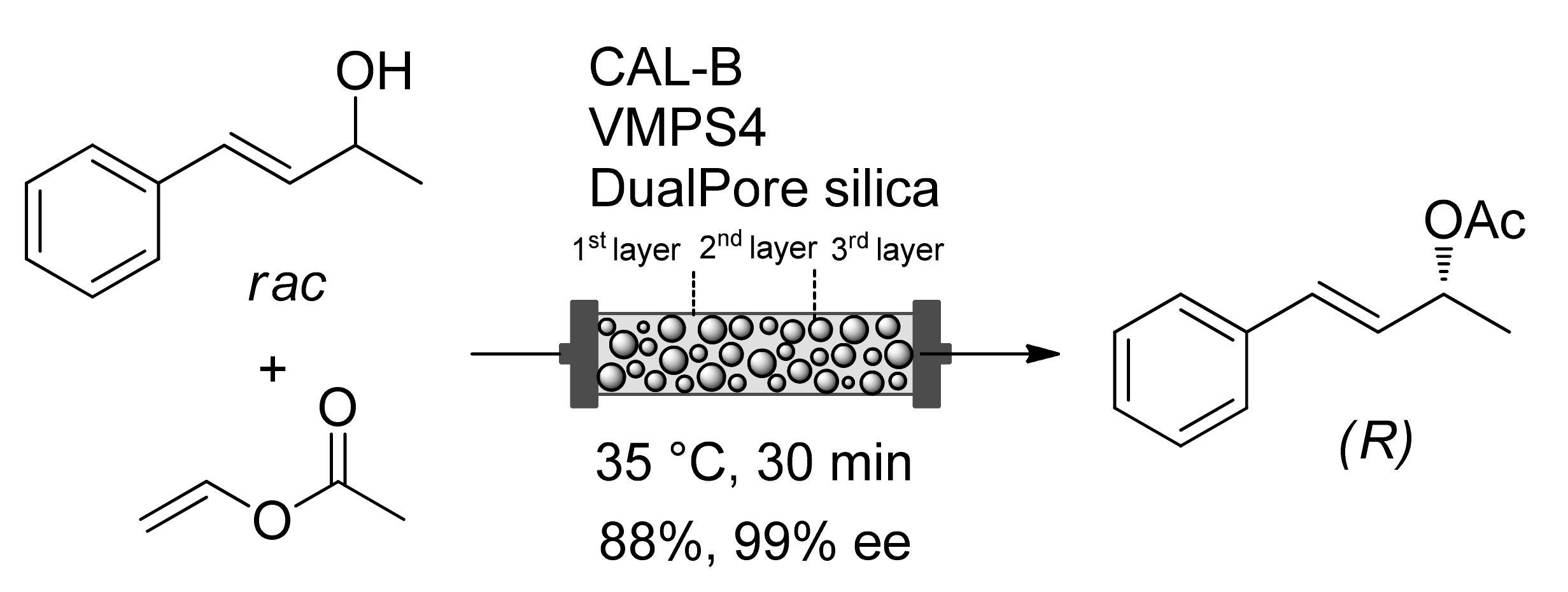
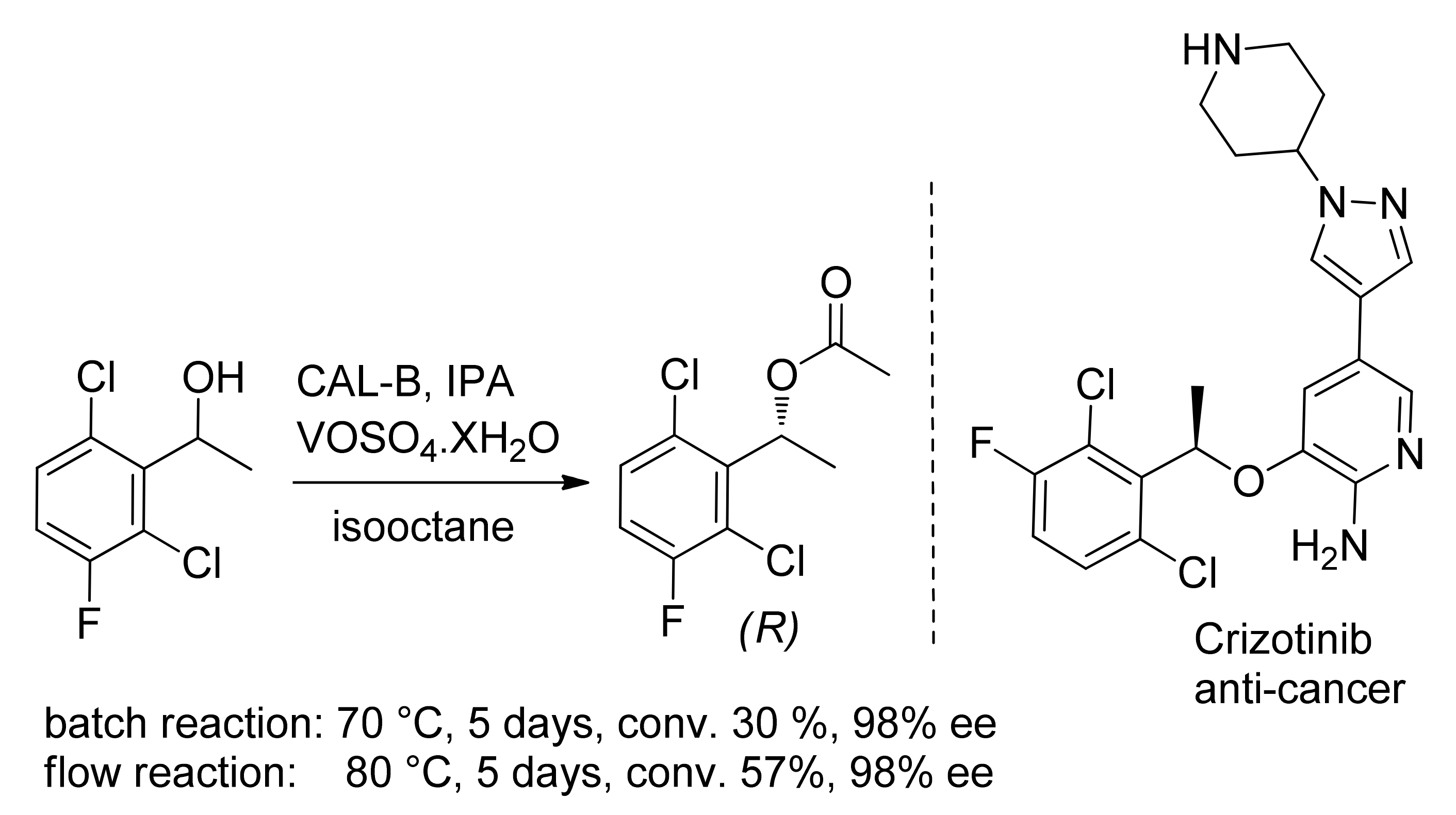
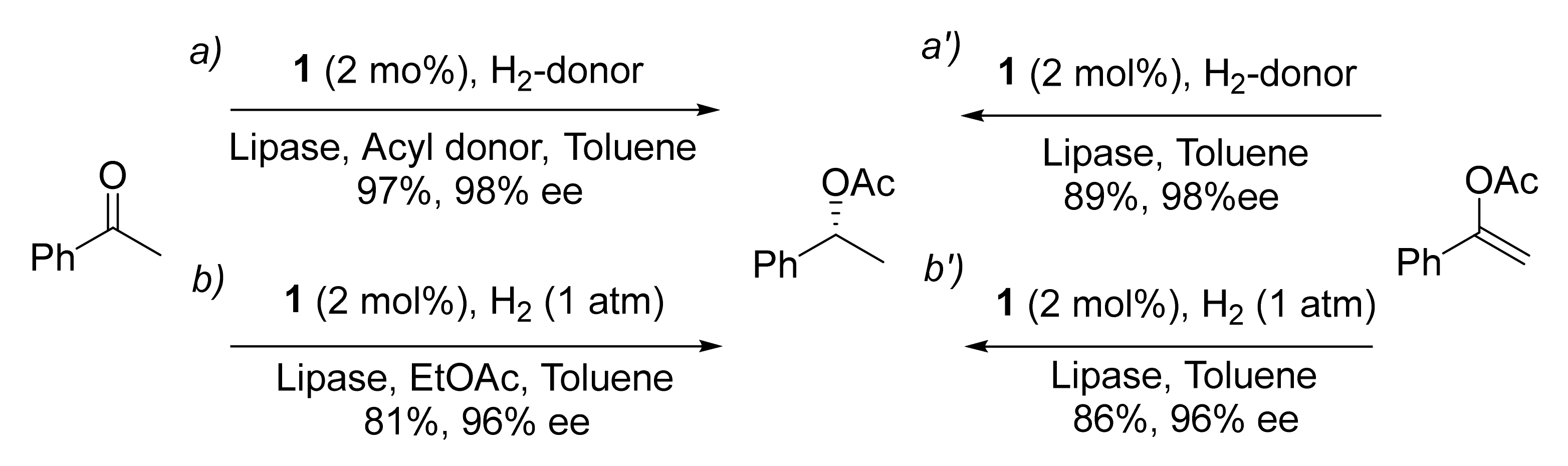
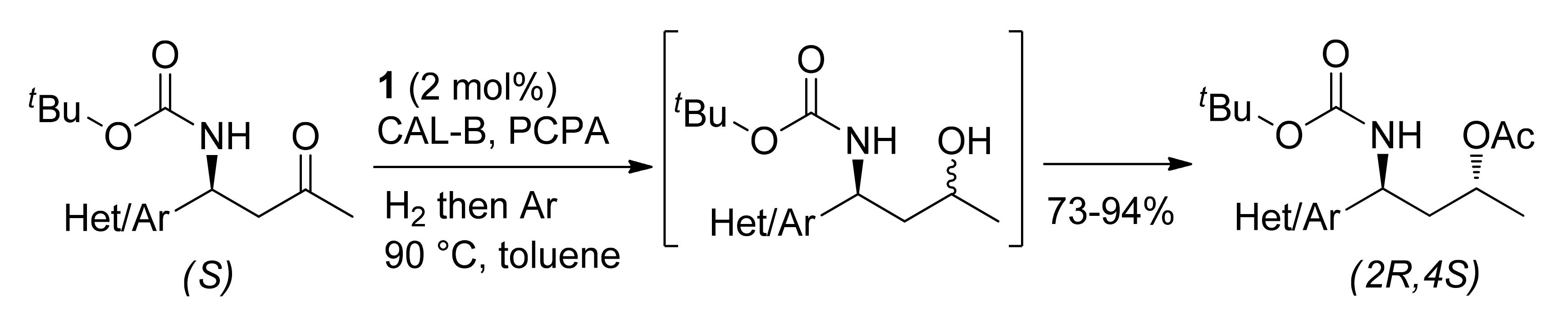
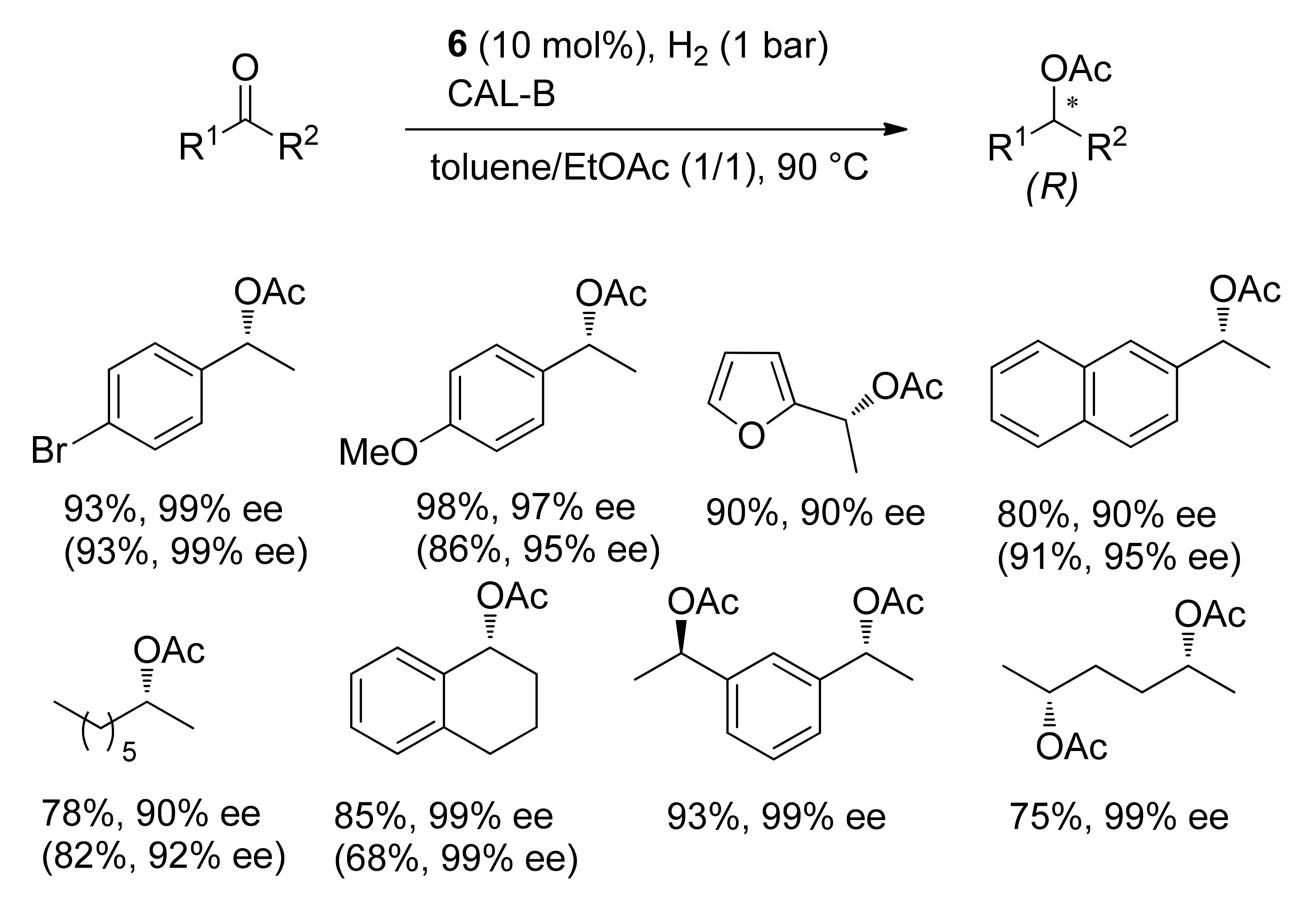
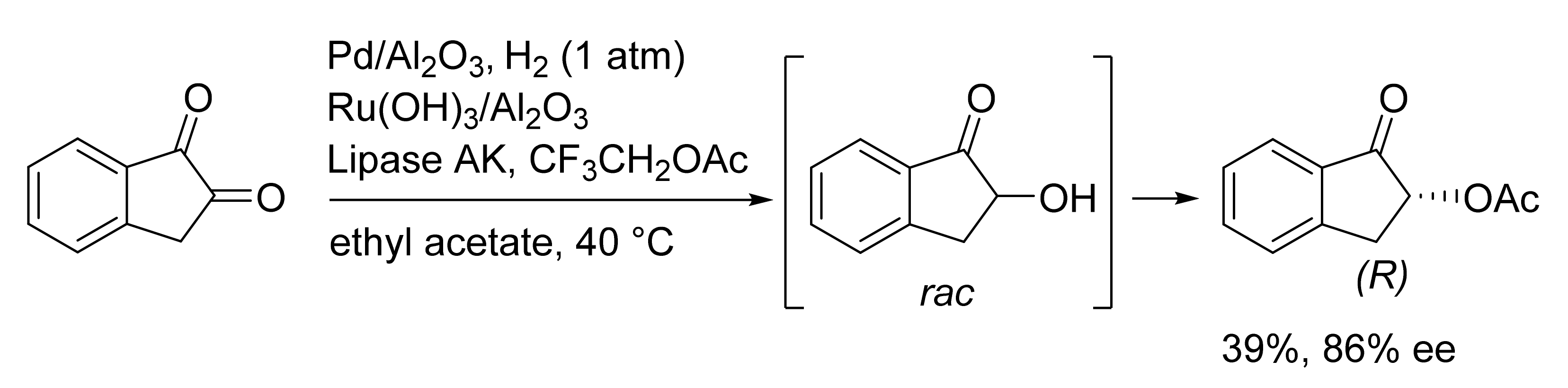
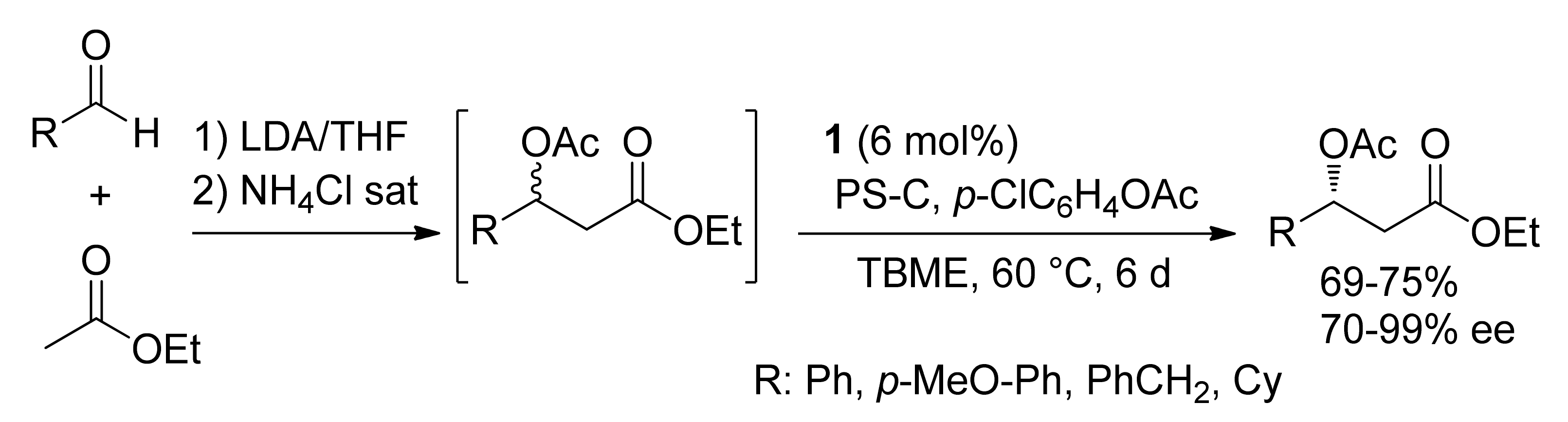
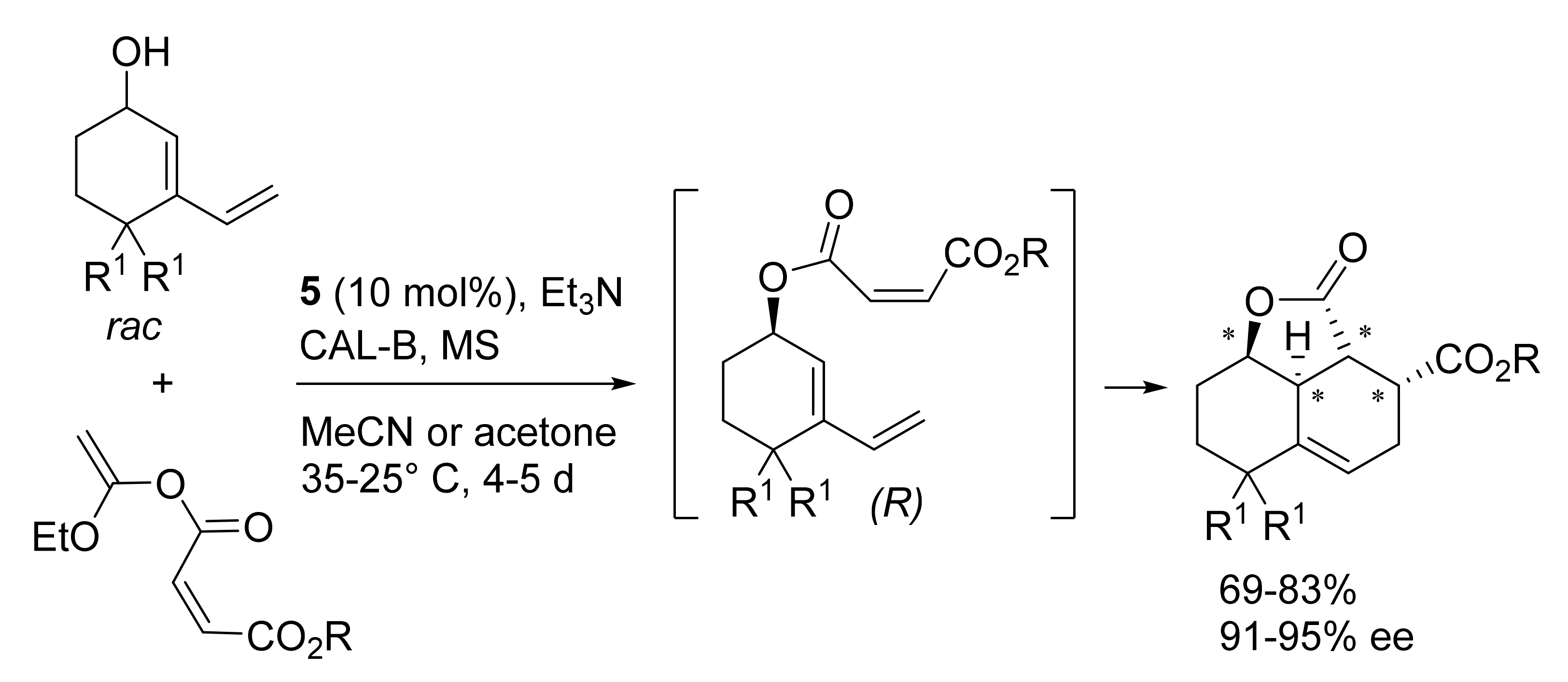
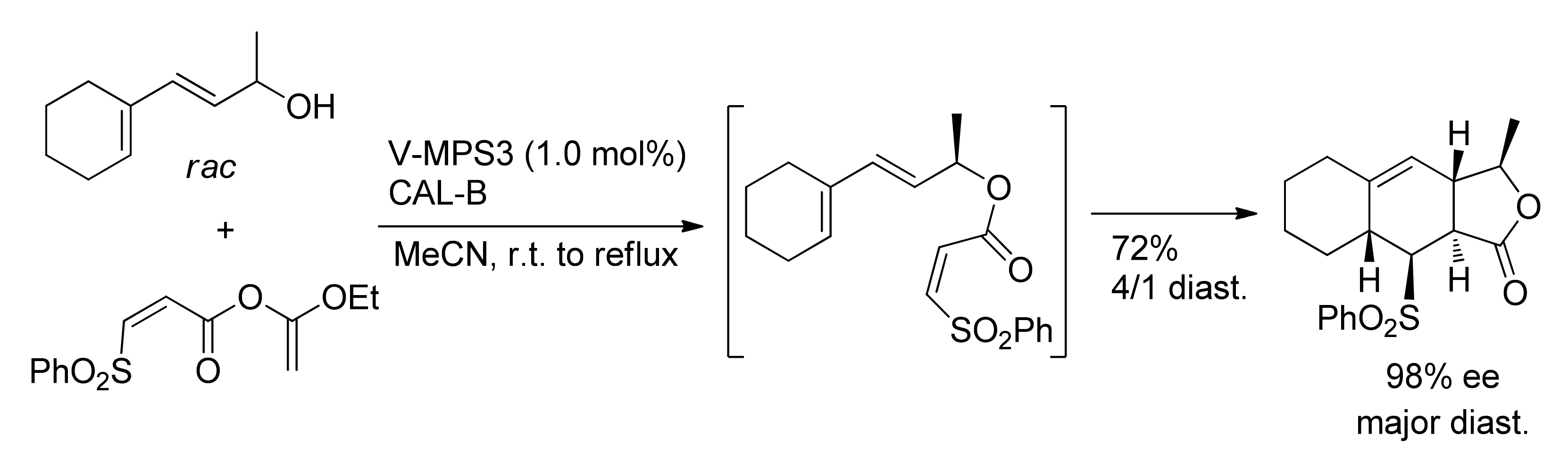
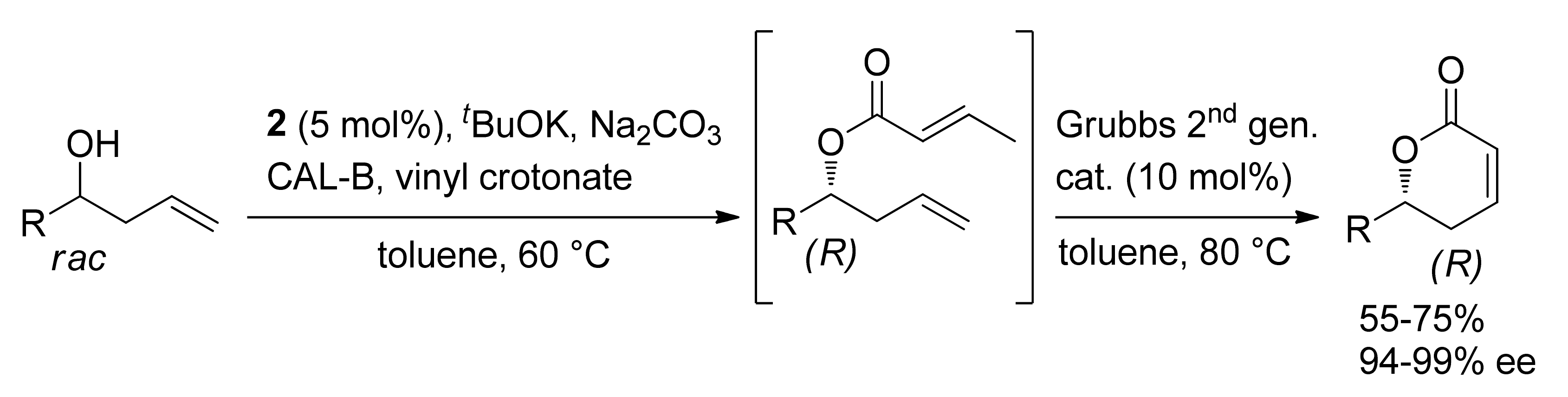

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraccioli, R. Progress on the Stereoselective Synthesis of Chiral Molecules Based on Metal-Catalyzed Dynamic Kinetic Resolution of Alcohols with Lipases. Symmetry 2021, 13, 1744. https://doi.org/10.3390/sym13091744
Ferraccioli R. Progress on the Stereoselective Synthesis of Chiral Molecules Based on Metal-Catalyzed Dynamic Kinetic Resolution of Alcohols with Lipases. Symmetry. 2021; 13(9):1744. https://doi.org/10.3390/sym13091744
Chicago/Turabian StyleFerraccioli, Raffaella. 2021. "Progress on the Stereoselective Synthesis of Chiral Molecules Based on Metal-Catalyzed Dynamic Kinetic Resolution of Alcohols with Lipases" Symmetry 13, no. 9: 1744. https://doi.org/10.3390/sym13091744
APA StyleFerraccioli, R. (2021). Progress on the Stereoselective Synthesis of Chiral Molecules Based on Metal-Catalyzed Dynamic Kinetic Resolution of Alcohols with Lipases. Symmetry, 13(9), 1744. https://doi.org/10.3390/sym13091744