
Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Promoter Sequences from the Rice Genome
2.2. Multiple Alignment of Promoter Sequences from the Rice Genome by the MAHDS Method
2.3. Creation of Random Matrices from the A Set.
2.4. Global Alignment of PWM and Sequence S1
2.5. Calculations of Multiple Alignment from Two-Dimensional Alignment of the Sequences S1 and S2
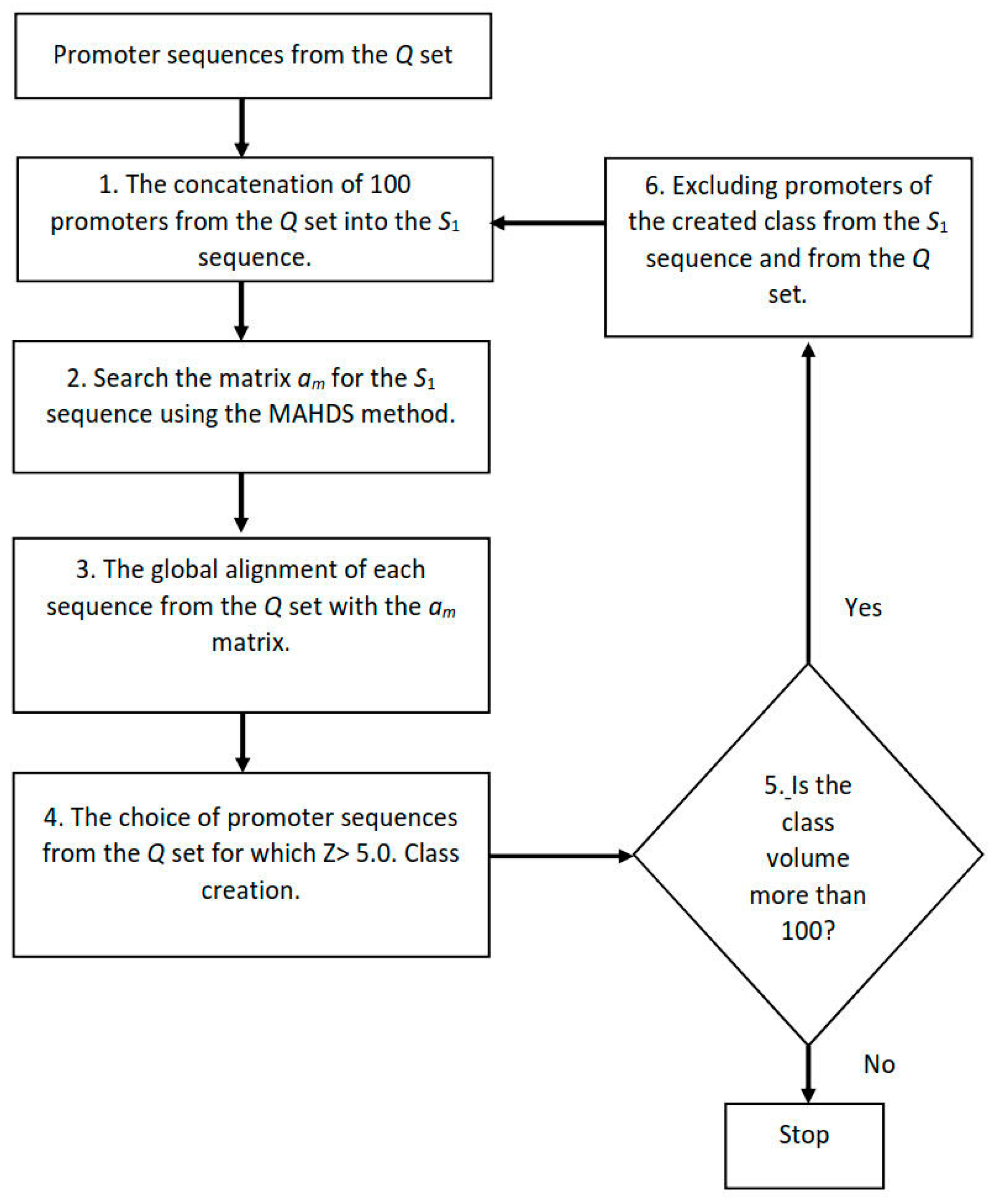
2.6. Creating Classes of Promoter Sequences
2.7. Search for Potential Promoter Sequences in the Rice Genome
3. Results
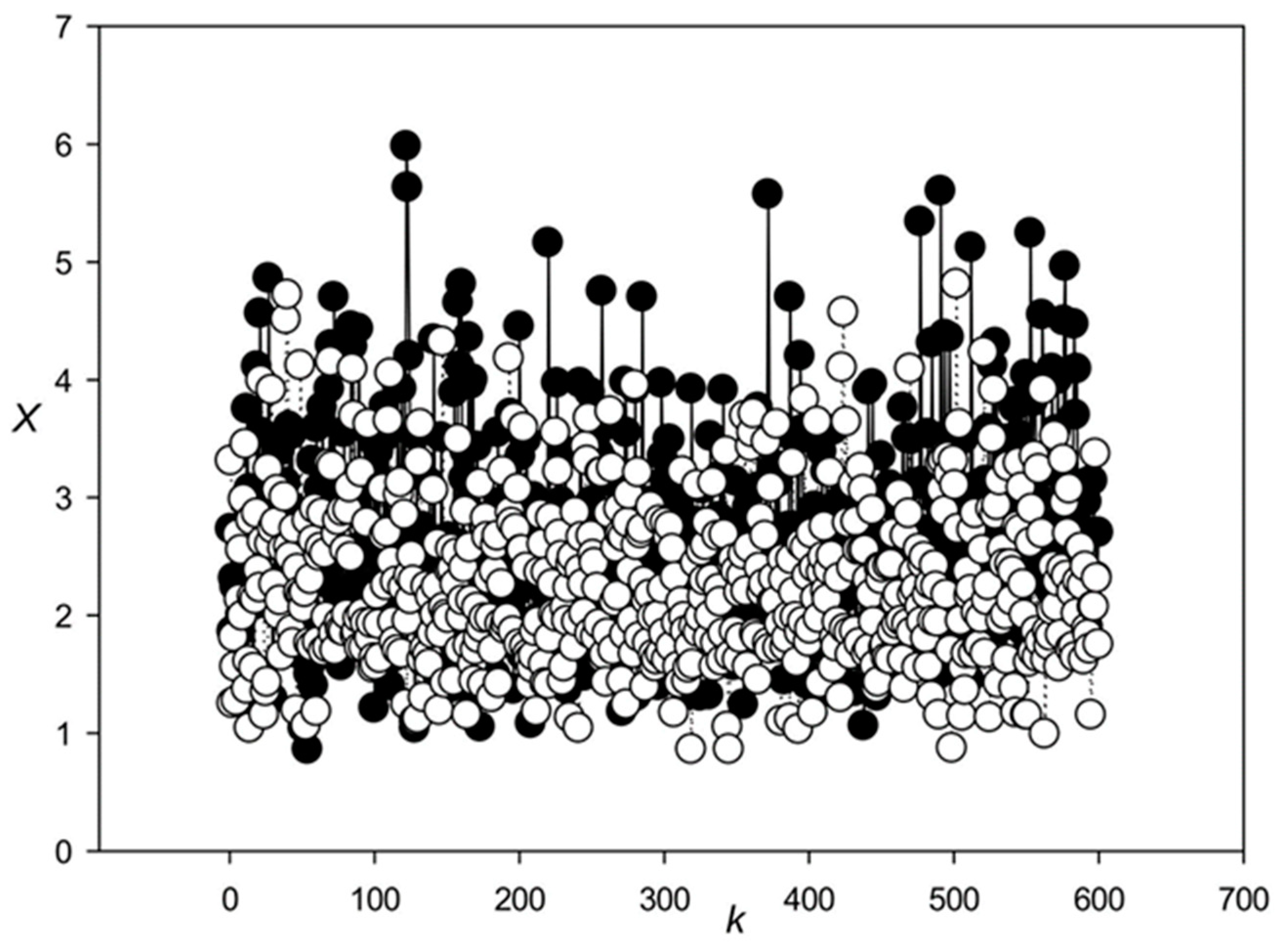
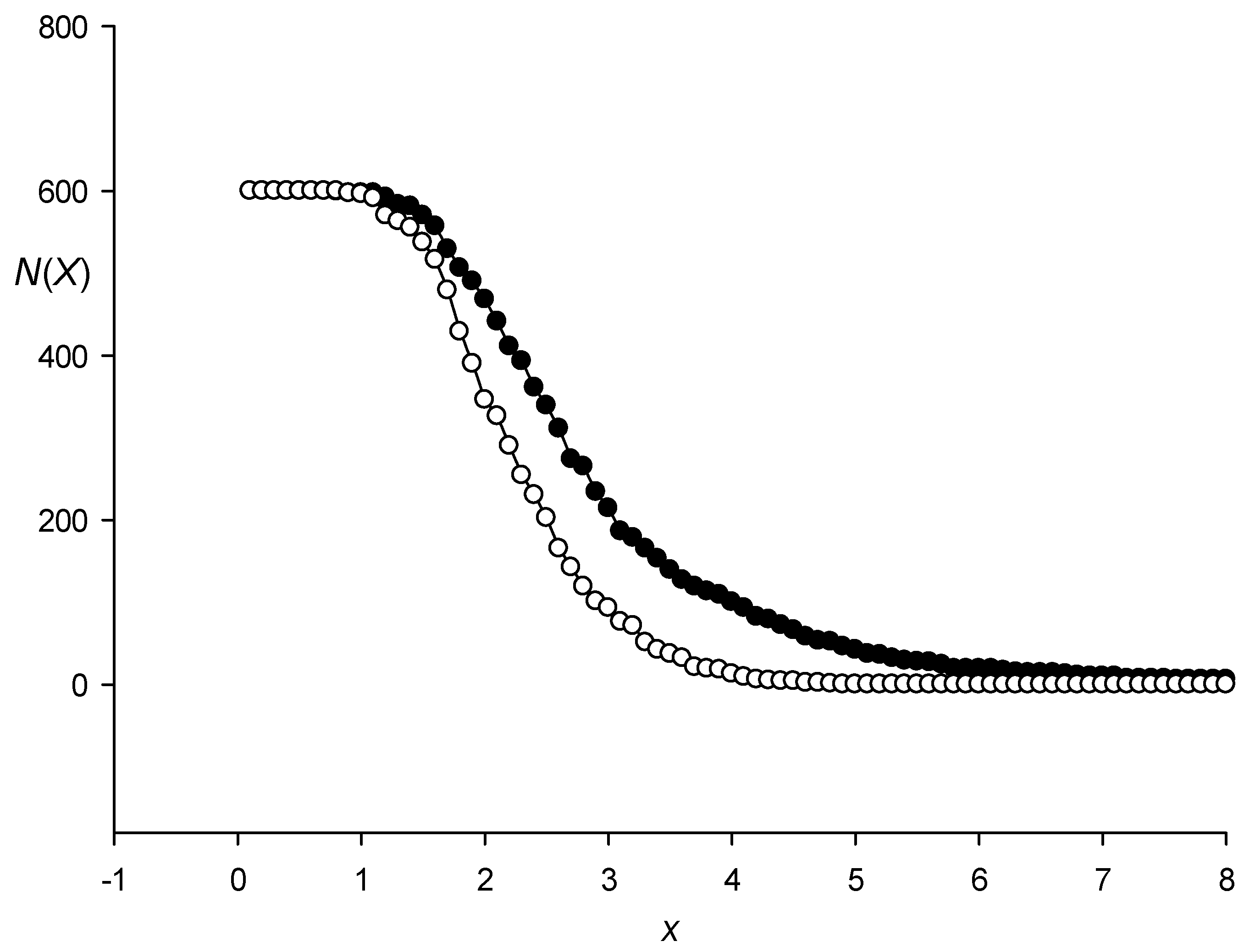
3.1. Classes of Promoter Sequences from the Rice Genome
3.2. PPS in the Rice Genome
3.3. The Intersection of PPS with Known Promoters and Transposons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nogales, E.; Louder, R.K.; He, Y. Structural Insights into the Eukaryotic Transcription Initiation Machinery. Annu. Rev. Biophys. 2017, 46, 59–83. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Hsu, J.-Y.; Theisen, J.W.; Kadonaga, J.T. The RNA polymerase II core promoter—The gateway to transcription. Curr. Opin. Cell Biol. 2008, 20, 253–259. [Google Scholar] [CrossRef]
- Smale, S.T.; Kadonaga, J.T. The RNA Polymerase II Core Promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Dreos, R.; Ambrosini, G.; Groux, R.; Cavin Périer, R.; Bucher, P. The eukaryotic promoter database in its 30th year: Focus on non-vertebrate organisms. Nucleic Acids Res. 2017, 45, D51–D55. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Matsudaira, P.; Kaiser, C.A.; Krieger, M.; Scott, M.P.; Zipursky, L.; Darnell, J. Molecular Cell Biology; Macmillan: New York, NY, USA, 2008; Volume 4, ISBN 0716776014. [Google Scholar]
- Roeder, R. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [CrossRef]
- Korotkova, M.A.; Kamionskya, A.M.; Korotkov, E.V. A mathematical method for the classification of promoter sequences from the A.thaliana genome. In Proceedings of the Journal of Physics: Conference Series; IOP Publishing Ltd.: Bristol, UK, 2020; Volume 1686, p. 012031. [Google Scholar]
- Abeel, T.; Van De Peer, Y.; Saeys, Y. Toward a gold standard for promoter prediction evaluation. Bioinformatics 2009, 25, i313–i320. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.V.; Shahmuradov, I.A.; Salamov, A.A. Identification of promoter regions and regulatory sites. Methods Mol. Biol. 2010, 674, 57–83. [Google Scholar] [CrossRef]
- Abe, H.; Gemmell, N.J. Abundance, arrangement, and function of sequence motifs in the chicken promoters. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcription of Eukaryotic Protein-Coding Genes. Annu. Rev. Genet. 2000, 34, 77–137. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhu, S.; Yan, H. Towards accurate human promoter recognition: A review of currently used sequence features and classification methods. Brief. Bioinform. 2009, 10, 498–508. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.; Pietersma, H.; Cordes, M.; Kuipers, O.P.; Kok, J. PePPER: A webserver for prediction of prokaryote promoter elements and regulons. BMC Genom. 2012, 13, 299. [Google Scholar] [CrossRef]
- Di Salvo, M.; Pinatel, E.; Talà, A.; Fondi, M.; Peano, C.; Alifano, P. G4PromFinder: An algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs. BMC Bioinform. 2018, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Umarov, R.; Kuwahara, H.; Li, Y.; Gao, X.; Solovyev, V.; Hancock, J. Promoter analysis and prediction in the human genome using sequence-based deep learning models. Bioinformatics 2019, 35, 2730–2737. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, X.; Li, Y.; Wu, M.; Zhao, Y. Image-based promoter prediction: A promoter prediction method based on evolutionarily generated patterns. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, E.V.; Suvorova, Y.M.; Kostenko, D.O.; Korotkova, M.A. Multiple Alignment of Promoter Sequences from the Arabidopsis thaliana L. Genome. Genes 2021, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, E.V.; Yakovleva, I.V.; Kamionskaya, A.M. Use of Mathematical Methods for the Biosafety Assessment of Agricultural Crops. Appl. Biochem. Microbiol. 2021, 57, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Patikoglou, G.A.; Kim, J.L.; Sun, L.; Yang, S.H.; Kodadek, T.; Burley, S.K. TATA element recognition by the TATA box-binding protein has been conserved throughout evolution. Genes Dev. 1999, 13, 3217–3230. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Laskin, A.A.; Korotkov, E.V.; Chalei, M.B.; Kudryashov, N.A. The locally optimal method of cyclic alignment to reveal latent periodicities in genetic texts. The NAD-binding protein sites. Mol. Biol. 2003, 37, 663–673. [Google Scholar] [CrossRef]
- Pugacheva, V.; Korotkov, A.; Korotkov, E. Search of latent periodicity in amino acid sequences by means of genetic algorithm and dynamic programming. Stat. Appl. Genet. Mol. Biol. 2016, 15, 381–400. [Google Scholar] [CrossRef]
- Gagniuc, P.; Ionescu-Tirgoviste, C. Eukaryotic genomes may exhibit up to 10 generic classes of gene promoters. BMC Genom. 2012, 13, 512. [Google Scholar] [CrossRef] [PubMed]
- Hellen, C.U.T.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001, 15, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.C.; Matthews, J.M. Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr. Opin. Struct. Biol. 2016, 38, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Pelechano, V.; Järvelin, A.I.; Steinmetz, L.M. Functional consequences of bidirectional promoters. Trends Genet. 2011, 27, 267–276. [Google Scholar] [CrossRef]
- Jin, Y.; Eser, U.; Struhl, K.; Churchman, L.S. The Ground State and Evolution of Promoter Region Directionality. Cell 2017, 170, 889–898.e10. [Google Scholar] [CrossRef]
- Korotkov, E.V.; Kamionskya, A.M.; Korotkova, M.A. Detection of Highly Divergent Tandem Repeats in the Rice Genome. Genes 2021, 12, 473. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Zhou, X.; Ruan, J.; Wang, G.; Zhang, W. Characterization and Identification of MicroRNA Core Promoters in Four Model Species. PLoS Comput. Biol. 2007, 3, e37. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- Solovyev, V.V.; Shahmuradov, I.A. PromH: Promoters identification using orthologous genomic sequences. Nucleic Acids Res. 2003, 31, 3540–3545. [Google Scholar] [CrossRef] [PubMed]
- RegSite Database of Plant Regulatory Elements. Available online: http://linux1.softberry.com/berry.phtml?topic=regsite (accessed on 18 April 2020).
- Knudsen, S. Promoter 2.0: For the recognition of PolII promoter sequences. Bioinformatics 1999, 15, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Mitsis, T.; Efthimiadou, A.; Bacopoulou, F.; Vlachakis, D.; Chrousos, G.; Eliopoulos, E. Transcription factors and evolution: An integral part of gene expression (Review). World Acad. Sci. J. 2020, 2, 3–8. [Google Scholar] [CrossRef]
- Korotkov, E.V.; Kamionskaya, A.M.; Korotkova, M.A. Multiple Alignment of Promoter Sequences from the Human Genome. Biotekhnologiya 2020, 36, 7–14. [Google Scholar] [CrossRef]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.; Lock, A.; Harris, M.A.; Rutherford, K.; Bähler, J.; Oliver, S.G. Hidden in plain sight: What remains to be discovered in the eukaryotic proteome? Open Biol. 2019, 9, 180241. [Google Scholar] [CrossRef] [PubMed]
- Miwa, H.; Itoh, N. Unknown genes, Cebelin and Cebelin-like, predominantly expressed in mouse brain. Heliyon 2018, 4, e00773. [Google Scholar] [CrossRef]
- Warren, A.S.; Archuleta, J.; Feng, W.-C.; Setubal, J.C. Missing genes in the annotation of prokaryotic genomes. BMC Bioinform. 2010, 11, 131. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Number of Genes | Number PPS | ++ | +− | −+ | −− | R | M |
|---|---|---|---|---|---|---|---|---|
| 1 | 5350 | 21,960 | 1271 | 246 | 109 | 1485 | 0 | 62 |
| 2 | 4296 | 13,497 | 1116 | 106 | 121 | 1058 | 0 | 17 |
| 3 | 4648 | 13,121 | 1337 | 61 | 170 | 817 | 1 | 54 |
| 4 | 3429 | 14,775 | 963 | 46 | 170 | 640 | 0 | 39 |
| 5 | 3070 | 11,034 | 479 | 150 | 18 | 981 | 1 | 40 |
| 6 | 3204 | 11,381 | 713 | 87 | 51 | 849 | 0 | 31 |
| 7 | 2917 | 10,016 | 824 | 55 | 62 | 697 | 0 | 18 |
| 8 | 2636 | 10,223 | 849 | 23 | 97 | 481 | 0 | 33 |
| 9 | 2144 | 7575 | 579 | 15 | 87 | 275 | 0 | 6 |
| 10 | 2184 | 7943 | 204 | 124 | 11 | 664 | 0 | 3 |
| 11 | 2663 | 11,648 | 630 | 39 | 91 | 512 | 0 | 33 |
| 12 | 2215 | 12,104 | 597 | 69 | 83 | 542 | 0 | 11 |
| All | 38,756 | 145,277 | 9562 | 1021 | 1070 | 9001 | 2 | 347 |
| Class Number | ++ | +− | −+ | −− |
|---|---|---|---|---|
| 1 | 6133 | 456 | 561 | 4955 |
| 2 | 792 | 195 | 145 | 958 |
| 3 | 1169 | 144 | 163 | 1218 |
| 4 | 755 | 101 | 112 | 700 |
| 5 | 713 | 125 | 89 | 1170 |
| N | Name of Dispersed Repeat OT Transposon | Number of Intersections | The Expected Number of Intersections | X1 |
|---|---|---|---|---|
| 1 | DNAnona/Helitron | 7466 | 7044 | 5.03 |
| 2 | DNAnona/unknown | 1501 | 870 | 21.39 |
| 3 | MITE/Tourist | 10,507 | 8955 | 16.40 |
| 4 | MITE/Stow | 9891 | 8189 | 18.81 |
| 5 | DNAauto/MULE | 2140 | 2792 | −12.34 |
| 6 | DNAnona/MULE | 12,288 | 9531 | 28.24 |
| 7 | LINE/unknown | 2153 | 4045 | −29.75 |
| 8 | LTR/Gypsy | 18,043 | 22,837 | −31.72 |
| 9 | DNAnona/hAT | 3824 | 3616 | 3.46 |
| 10 | DNAnona/MULEtir | 3328 | 1793 | 36.25 |
| 11 | DNAnona/Tourist | 917 | 463 | 21.10 |
| 12 | LTR/Copia | 2675 | 4736 | −29.95 |
| 13 | DNAauto/CACTA | 1265 | 1967 | −15.83 |
| 14 | SINE/unknown | 1252 | 1666 | −10.14 |
| 15 | DNAnona/CACTA | 3736 | 2395 | 27.40 |
| 16 | DNAauto/hAT | 438 | 479 | −1.87 |
| 17 | DNAnona/PILE | 426 | 403 | 1.15 |
| 18 | DNAauto/PILE | 259 | 251 | 0.50 |
| 19 | LTR/TRIM | 190 | 705 | −19.40 |
| 20 | DNAauto/Helitron | 226 | 487 | −11.83 |
| 21 | Evirus/ERTBV-C | 39 | 45 | −0.89 |
| 22 | LTR/unknown | 119 | 232 | −7.42 |
| 23 | DNAnona/CACTG | 1141 | 675 | 17.94 |
| 24 | DNAauto/CACTG | 2614 | 2503 | 2.22 |
| 25 | LTR/Solo | 36 | 15 | 5.42 |
| 26 | DNAauto/MLE | 154 | 182 | −2.08 |
| 27 | Evirus/ERTBV-B | 21 | 59 | −4.95 |
| 28 | Evirus/ERTBV-A | 22 | 45 | −3.43 |
| 29 | Evirus/ERTBV | 23 | 20 | 0.67 |
| 30 | DNAauto/POLE | 161 | 168 | −0.54 |
| 31 | DNAnona/POLE | 253 | 168 | 6.56 |
| 32 | DNAnona/MLE | 32 | 44 | −1.81 |
| 33 | Centro/tandem | 93 | 298 | −11.88 |
| N | Name of Dispersed Repeat ot Transposon | M1 | M2 | M3 | M4 | M5 |
|---|---|---|---|---|---|---|
| 1 | DNAnona/Helitron | 3015 | 1540 | 795 | 1089 | 1027 |
| 2 | DNAnona/unknown | 670 | 268 | 176 | 136 | 251 |
| 3 | MITE/Tourist | 4507 | 1850 | 1329 | 1216 | 1605 |
| 4 | MITE/Stow | 5376 | 1208 | 894 | 944 | 1469 |
| 5 | DNAauto/MULE | 948 | 370 | 311 | 240 | 271 |
| 6 | DNAnona/MULE | 6201 | 1982 | 1362 | 1377 | 1366 |
| 7 | LINE/unknown | 813 | 328 | 383 | 293 | 336 |
| 8 | LTR/Gypsy | 9877 | 2610 | 2333 | 1903 | 1320 |
| 9 | DNAnona/hAT | 1883 | 615 | 411 | 438 | 477 |
| 10 | DNAnona/MULEtir | 1528 | 629 | 486 | 323 | 362 |
| 11 | DNAnona/Tourist | 441 | 154 | 151 | 80 | 91 |
| 12 | LTR/Copia | 839 | 447 | 484 | 522 | 383 |
| 13 | DNAauto/CACTA | 564 | 170 | 204 | 187 | 140 |
| 14 | SINE/unknown | 653 | 170 | 132 | 140 | 157 |
| 15 | DNAnona/CACTA | 1823 | 542 | 492 | 482 | 397 |
| 16 | DNAauto/hAT | 241 | 37 | 61 | 57 | 42 |
| 17 | DNAnona/PILE | 211 | 53 | 54 | 38 | 70 |
| 18 | DNAauto/PILE | 91 | 49 | 48 | 40 | 31 |
| 19 | LTR/TRIM | 85 | 24 | 30 | 22 | 29 |
| 20 | DNAauto/Helitron | 100 | 34 | 24 | 23 | 45 |
| 21 | Evirus/ERTBV-C | 1 | 12 | 12 | 11 | 3 |
| 22 | LTR/unknown | 50 | 17 | 15 | 18 | 19 |
| 23 | DNAnona/CACTG | 670 | 169 | 107 | 121 | 74 |
| 24 | DNAauto/CACTG | 757 | 611 | 353 | 654 | 239 |
| 25 | LTR/Solo | 15 | 6 | 3 | 8 | 4 |
| 26 | DNAauto/MLE | 65 | 22 | 35 | 10 | 22 |
| 27 | Evirus/ERTBV-B | 5 | 5 | 1 | 6 | 4 |
| 28 | Evirus/ERTBV-A | 10 | 3 | 2 | 4 | 3 |
| 29 | Evirus/ERTBV | 2 | 8 | 2 | 1 | 10 |
| 30 | DNAauto/POLE | 59 | 22 | 37 | 19 | 24 |
| 31 | DNAnona/POLE | 108 | 38 | 45 | 29 | 33 |
| 32 | DNAnona/MLE | 16 | 5 | 6 | 1 | 4 |
| 33 | Centro/tandem | 6 | 31 | 1 | 43 | 12 |
| Total: | 41,630 | 14,029 | 10,779 | 10,475 | 10,320 |
| N | Name of Dispersed Repeat ot Transposon | M1 | M2 | M3 | M4 | M5 |
|---|---|---|---|---|---|---|
| 1 | DNAnona/Helitron | −9.3738 | 9.8600 | −4.2215 | 6.4616 | 4.8613 |
| 3 | MITE/Tourist | −7.3783 | 3.9365 | 0.8583 | −1.2957 | 10.3410 |
| 4 | MITE/Stow | 9.8136 | −9.6839 | −9.4540 | −7.1205 | 8.7958 |
| 7 | LINE/unknown | −6.7306 | −0.9827 | 7.1819 | 2.1467 | 5.1011 |
| 8 | LTR/Gypsy | 14.3752 | −5.5078 | 2.2207 | −5.7351 | −17.8504 |
| 12 | LTR/Copia | −12.3378 | 0.8120 | 8.4570 | 11.2236 | 3.7471 |
| 24 | DNAauto/CACTG | −13.9871 | 9.3190 | 1.6723 | 19.2314 | −4.0017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korotkov, E.V.; Suvorova, Y.M.; Nezhdanova, A.V.; Gaidukova, S.E.; Yakovleva, I.V.; Kamionskaya, A.M.; Korotkova, M.A. Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences. Symmetry 2021, 13, 917. https://doi.org/10.3390/sym13060917
Korotkov EV, Suvorova YM, Nezhdanova AV, Gaidukova SE, Yakovleva IV, Kamionskaya AM, Korotkova MA. Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences. Symmetry. 2021; 13(6):917. https://doi.org/10.3390/sym13060917
Chicago/Turabian StyleKorotkov, Eugene V., Yulia. M. Suvorova, Anna V. Nezhdanova, Sofia E. Gaidukova, Irina V. Yakovleva, Anastasia M. Kamionskaya, and Maria A. Korotkova. 2021. "Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences" Symmetry 13, no. 6: 917. https://doi.org/10.3390/sym13060917
APA StyleKorotkov, E. V., Suvorova, Y. M., Nezhdanova, A. V., Gaidukova, S. E., Yakovleva, I. V., Kamionskaya, A. M., & Korotkova, M. A. (2021). Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences. Symmetry, 13(6), 917. https://doi.org/10.3390/sym13060917