
Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR

 , , and
, , and
Abstract
1. Introduction
- (1)
- Direct measurements: diffusion coefficients (D’s), longitudinal relaxation times (T1s), and transverse relaxation times (T2s) [2,3], although the differences can sometimes be quite small for these measurements and some idea of associate structure may be required if mixed results are obtained for the T1s and T2s.
- (2)
- Variation in sample conditions: plots of enantiomeric titration [2,5], serial dilution, temperature variation, and titration of cofactors (e.g., secondary solvent, acid/base, ionic strength) though some idea of associate structure may be required and care is required in the interpretation of the plots together with appreciation of the δ’s [21].
- (3)
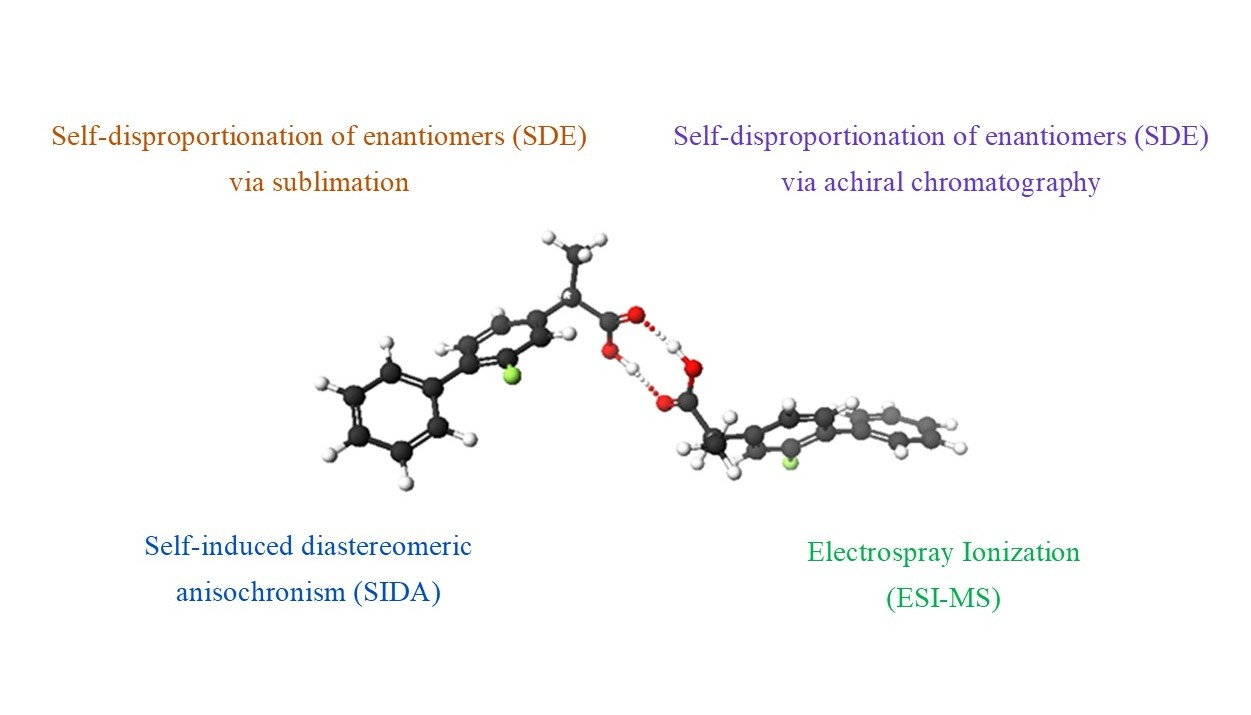
- Other complex approaches: the interpretation of δ’s and intermolecular nuclear Overhauser effects (NOE’s) [3], although intimate knowledge of the structure is likely to be required for δ’s, together with an appreciation of the consequences of varying relative concentrations [21]. Some general structural comprehension at least is also likely to be required in the case of intermolecular NOE’s, and suitable spins are not always available for such an analysis.
2. Results and Discussion
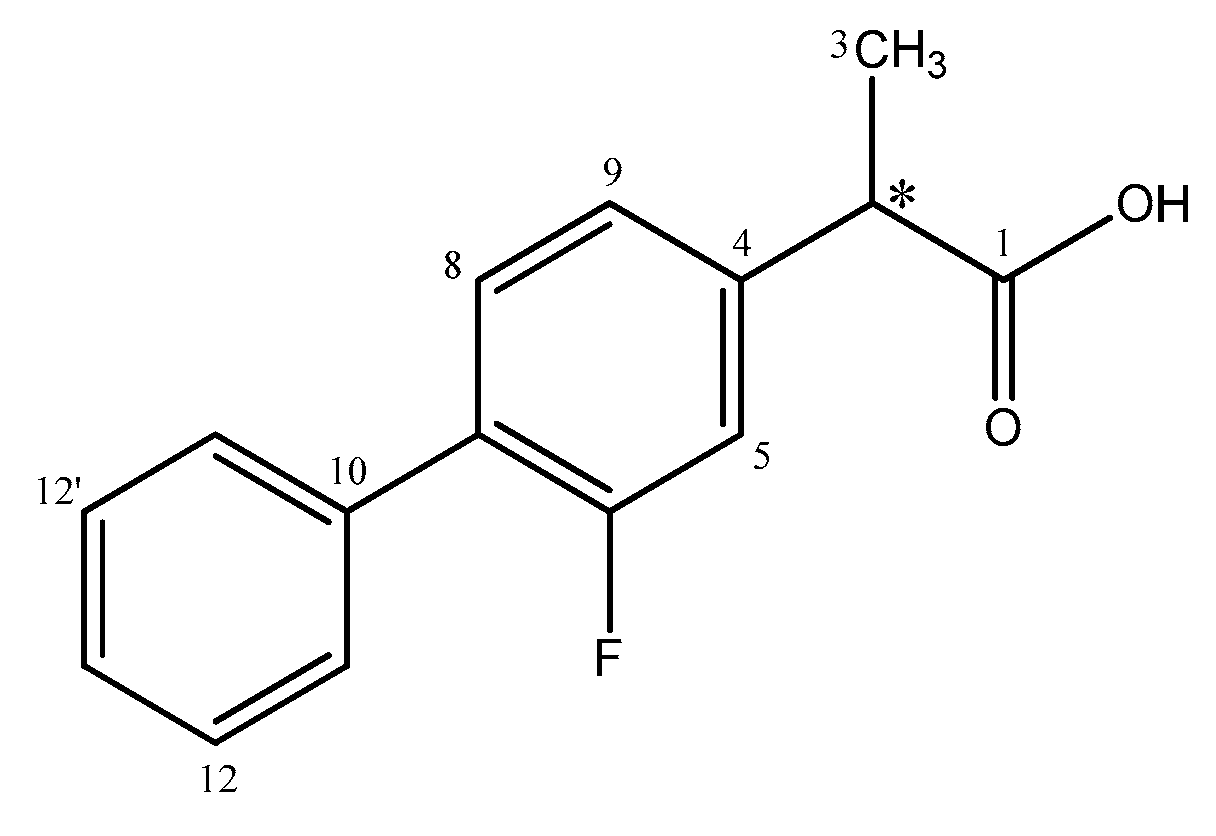
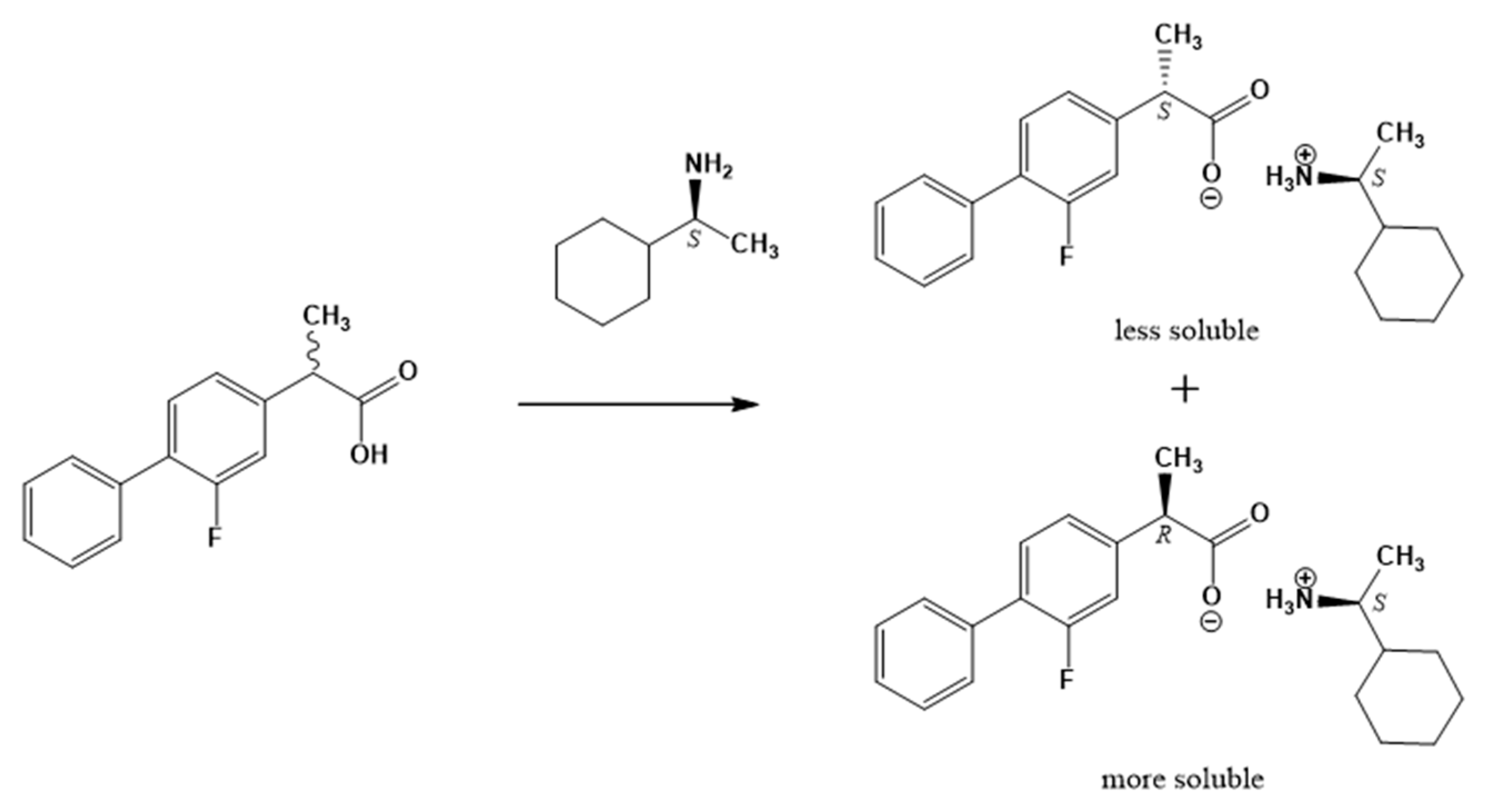
2.1. Resolution of (rac)-Flurbiprofen
2.2. Solid-State Properties of Flurbiprofen
2.3. SDEvS
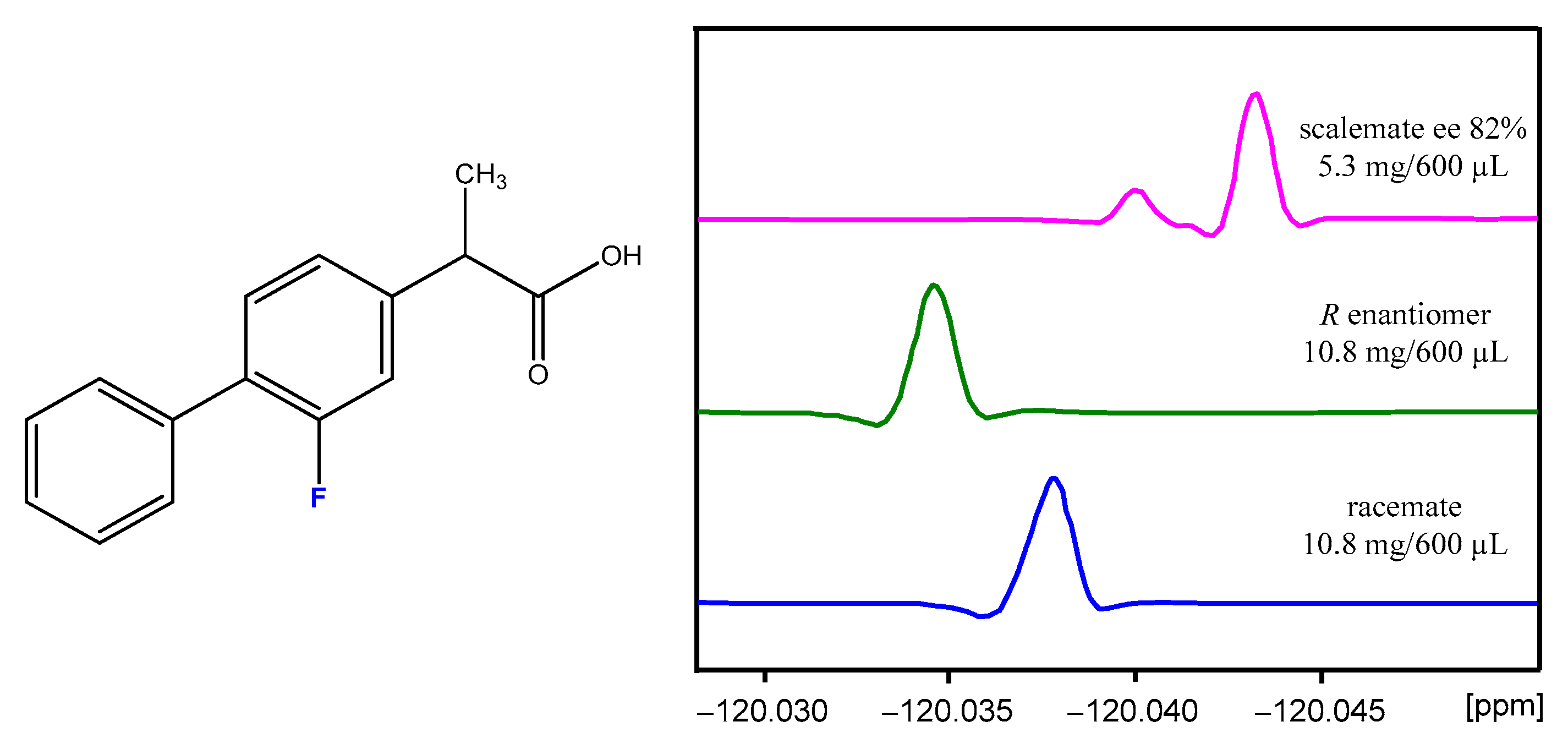
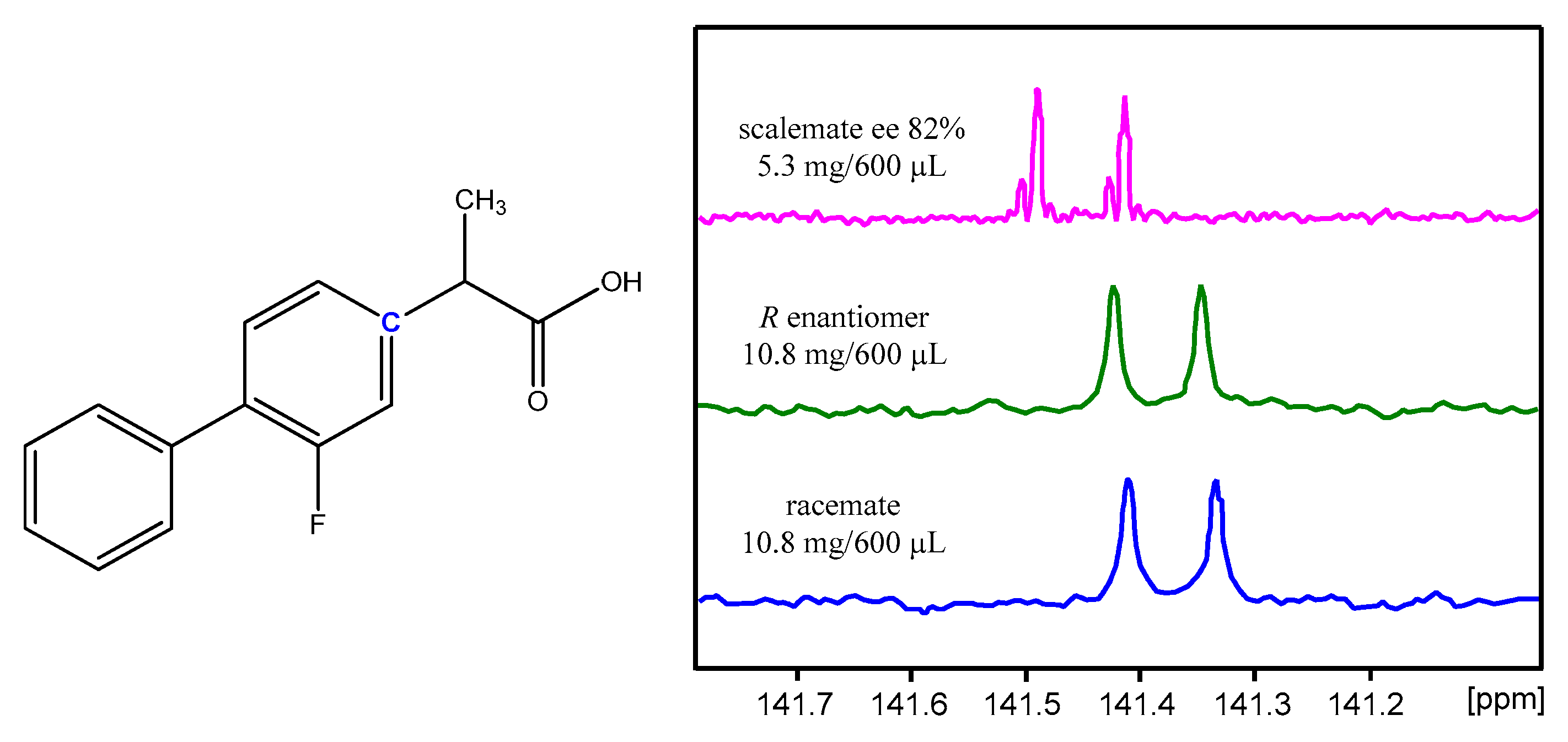
2.4. NMR
2.5. SDEvC
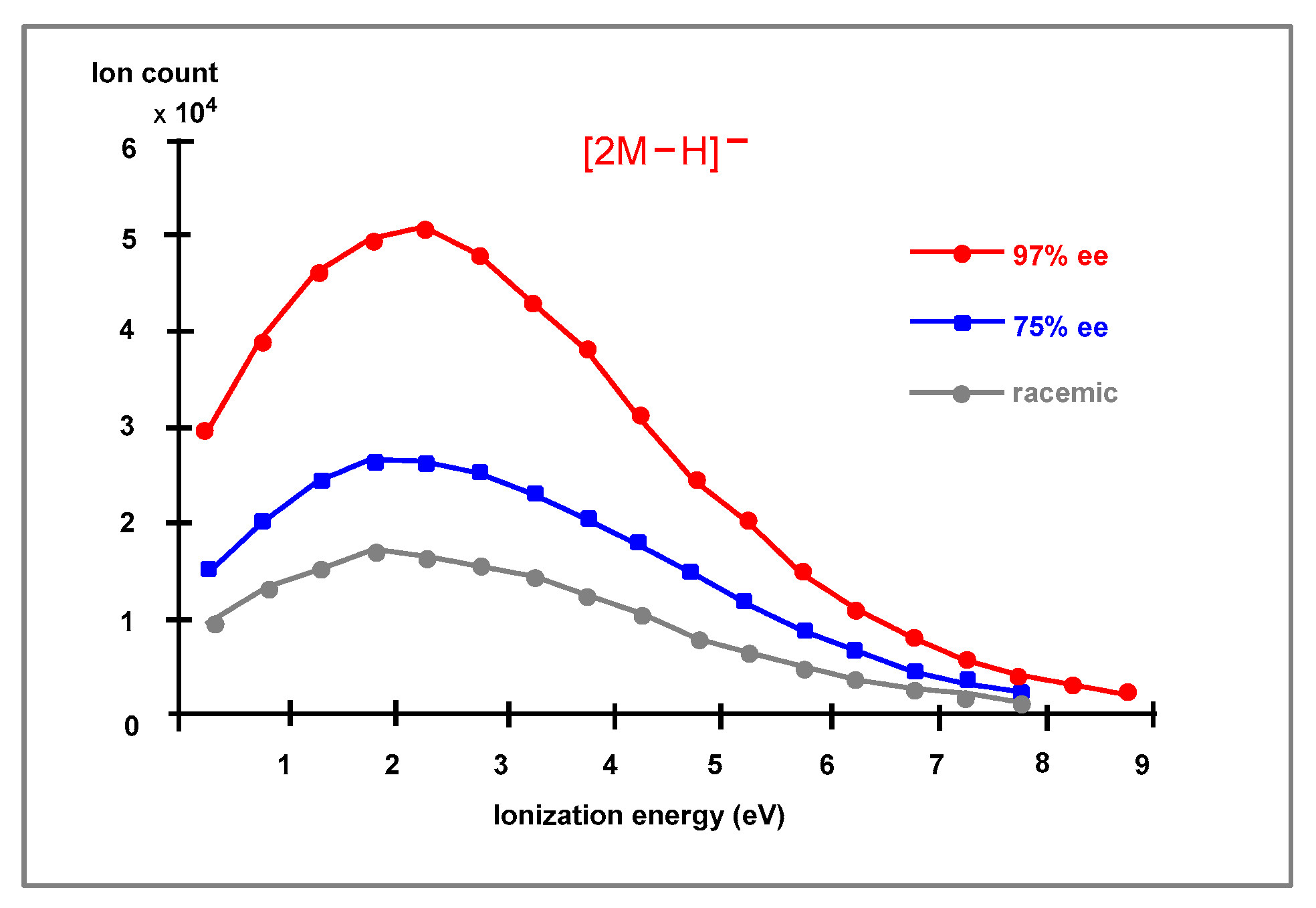
2.6. ESI-MS
3. Conclusions
4. Experimental
4.1. General
4.2. Preparation of Enantioenriched Flurbiprofen
4.3. NMR
4.4. IR
4.5. Chromatography
4.6. ESI-MS
4.7. Sublimation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szakács, Z.; Sánta, Z.; Lomoschitz, A.; Szántay, C., Jr. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. Trends Anal. Chem. 2018, 109, 180–197. [Google Scholar] [CrossRef]
- Nieminen, V.; Murzin, D.Y.; Klika, K.D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1’-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. [Google Scholar] [CrossRef]
- Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106. [Google Scholar] [CrossRef]
- Storch, G.; Haas, M.; Trapp, O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5414–5418. [Google Scholar] [CrossRef] [PubMed]
- Klika, K.D.; Budovská, M.; Kutschy, P. Enantiodifferentiation of phytoalexin spirobrassinin derivatives using the chiral solvating agent (R)-(+)-1,1’-bi-2-naphthol in conjunction with molecular modeling. Tetrahedron Asymmetry 2010, 21, 647–658. [Google Scholar] [CrossRef]
- Klika, K.D.; Budovská, M.; Kutschy, P. NMR spectral enantioresolution of spirobrassinin and 1-methoxy-spirobrassinin enantiomers using (S)-(−)-ethyl lactate and modeling of spirobrassinin self-association for rationalization of its self-induced diastereomeric anisochronism (SIDA) and enantiomer self-disproportionation on achiral-phase chromatography (ESDAC) phenomena. J. Fluor. Chem. 2010, 131, 467–476. [Google Scholar]
- Ouryupin, A.B.; Kadyko, M.I.; Petrovskii, P.V.; Fedin, E.I.; Okruszek, A.; Kinas, R.; Stec, W.J. Enantiomeric 2-Anilino-2-oxo-l,3,2-oxazaphosphorinanes: Synthesis and NMR-Investigation of Their Non-racemic Mixtures. Tetrahedron Asymmetry 1995, 6, 1813–1824. [Google Scholar] [CrossRef]
- Jursic, B.S.; Goldberg, S.I. Enantiomer Discrimination Arising from Solute-Solute Interactions in Partially Resolved Chloroform Solutions of Chiral Carboxamides. J. Org. Chem. 1992, 57, 7172–7174. [Google Scholar] [CrossRef]
- Cung, M.T.; Marraud, M.; Neel, J. Experimental Study on Aggregation of Model Dipeptide Molecules. V. Stereoselective Association of Leucine Dipeptides. Biopolymers 1978, 17, 1149–1173. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Mastryukova, T.A.; Fedin, E.I.; Vaisberg, M.S.; Morozov, L.L.; Petrovsky, P.V.; Shipov, A.E. An NMR Study of Optical Isomers in Solution. Tetrahedron 1976, 32, 1719–1728. [Google Scholar] [CrossRef]
- Fedin, E.I.; Davankov, V.A. NMR Investigations of the Enantiomeric Excess Effects in Solutions with Weak Intermolecular Association. Chirality 1995, 7, 326–330. [Google Scholar] [CrossRef]
- Horeau, A.; Guetté, J.P. Interactions Diastereoisomeres d’Antipodes en Phase Liquide. Tetrahedron 1974, 30, 1923–1931. [Google Scholar] [CrossRef]
- Ouryupin, A.B.; Kadyko, M.I.; Petrovskii, P.V.; Fedin, E.I.; Sanshe, M.; Wolf, R.; Mastryukova, T.A.; Kabachnik, M.I. NMR determination of enantiomeric composition in the case of statistically controlled associate-diastereomer anisochrony. Zh. Obshch. Khim. 1995, 65, 18–22. [Google Scholar]
- Ajisaka, K.; Kainosho, M. Diastereomeric Interaction of Partially Resolved Amines Facilitated by Lanthanide Chelates. Evidence for Dynamic Equilibrium between Seven-Coordinate and Eight-Coordinate Alkylamine-Lanthanide Chelate Adducts. J. Am. Chem. Soc. 1975, 97, 1761–1765. [Google Scholar] [CrossRef]
- Reuben, J. Aqueous Lanthanide Shift Reagents. 8. Chiral Interactions and Stereochemical Assignments of Chemically and Isotopically Chiral Ligands. J. Am. Chem. Soc. 1980, 102, 2232–2237. [Google Scholar] [CrossRef]
- Ouryupin, A.B.; Kadyko, M.I.; Petrovskii, P.V.; Fedin, E.I. Chiral Discrimination in Nonracemic Mixtures of Methanephosphonic Acid, N, N’-Bis(1-phenylethyl) diamides. Chirality 1994, 6, 1–4. [Google Scholar] [CrossRef]
- Fedin, E.I.; Morozov, L.L.; Petrovskii, P.V.; Vaisberg, M.S.; Shipov, A.E.; Mastryukova, T.A.; Kabachnik, M.I. Statistically controlled associate-diastereomeric anisochronism. Temperature dependence. Dokl. Akad. Nauk. 1974, 219, 1181–1184. [Google Scholar]
- Kabachnik, M.I.; Mastryukova, T.A.; Shipov, A.E.; Vaisberg, M.S.; Petrovskii, P.V.; Morozov, L.L.; Fedin, E.I. Associate-diastereomerism. Statistically controlled nonequivalence of the magnetic screening of nuclei in solutions of optical antipodes in achiral solvents. Dokl. Akad. Nauk. 1974, 215, 1153–1156. [Google Scholar]
- Kabachnik, M.I.; Mastryukova, T.A.; Fedin, E.I.; Vaisberg, M.S.; Morozov, L.L.; Petrovskii, P.V.; Shipov, A.E. Optical Isomers in Solution Investigated by Nuclear Magnetic Resonance. Russ. Chem. Rev. 1978, 47, 821–834. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Fedin, E.I.; Morozov, L.L.; Petrovskii, P.V.; Vaisberg, M.S.; Shipov, A.E.; Mastryukova, T.A. Theoretical analysis of NMR spectra of associate-diastereomers. Dokl. Akad. Nauk. 1974, 215, 1400–1403. [Google Scholar]
- Klika, K.D. Use of sub-stoichiometric amounts of chiral auxiliaries for enantiodifferentiation by NMR; caveats and potential utility. Tetrahedron Asymmetry 2009, 20, 1099–1102. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-disproportionation of enantiomers via achiral chromatography: A warning and an extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology related to the phenomenon ’self-disproportionation of enantiomers’ (SDE). Helv. Chem. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Wzorek, A.; Klika, K.D.; Drabowicz, J.; Sato, A.; Aceña, J.L.; Soloshonok, V.A. The self-disproportionation of the enantiomers (SDE) of methyl n-pentyl sulfoxide via achiral, gravity-driven column chromatography: A case study. Org. Biomol. Chem. 2014, 12, 4738–4746. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. RSC Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019, 51, 865–889. [Google Scholar] [CrossRef] [PubMed]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Enantiomeric Enrichments via the Self-Disproportionation of Enantiomers (SDE) by Achiral, Gravity-Driven Column Chromatography: A Case Study Using N-(1-Phenylethyl) acetamide for Optimizing the Enantiomerically Pure Yield and Magnitude of the SDE. Helv. Chim. Acta 2015, 98, 1147–1159. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Marcinkowska, M.; Wzorek, A.; Pajkert, R.; Han, J.; Klika, K.D.; Soloshonok, V.A.; Röschenthaler, G.-V. The self-disproportionation of enantiomers (SDE) via column chromatography of β-amino-α,α-difluorophosphonic acid derivatives. Amino Acids 2019, 51, 1377–1385. [Google Scholar] [CrossRef]
- Davankov, V.A. Can Enantiomers Be Separated in Achiral Chromatographic Systems? Russ. J. Phys. Chem. A 2016, 90, 2119–2121. [Google Scholar] [CrossRef]
- Ueki, H.; Yasumoto, M.; Soloshonok, V.A. Rational application of self-disproportionation of enantiomers via sublimation—A novel methodological dimension for enantiomeric purifications. Tetrahedron Asymmetry 2010, 21, 1396–1400. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Katagiri, T.; Soloshonok, V.A. Unconventional preparation of racemic crystals of isopropyl 3,3,3-trifluoro-2-hydroxypropanoate and their unusual crystallographic structure: The ultimate preference for homochiral intermolecular interactions. Chem. Commun. 2013, 49, 373–375. [Google Scholar] [CrossRef]
- Kurdi, R.; Táborosi, A.; Zucchi, C.; Pályi, G. Autosolvation: Architecture and Selection of Chiral Conformers in Alkylcobalt Carbonyl Molecular Clocks. Symmetry 2014, 6, 551–565. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of enantiomers via achiral gravity-driven column chromatography: A case study of N-acyl-α-phenylethylamines. J. Chromatogr. A 2016, 1467, 270–278. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-disproportionation of enantiomers of chiral, non-racemic fluoroorganic compounds: Role of fluorine as enabling element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Berbasov, D.O. Self-Disproportionation of Enantiomers on Achiral Phase Chromatography. One More Example of Fluorine’s Magic Powers. Chim. Oggi Chem. Today 2006, 24, 44–47. [Google Scholar]
- Soloshonok, V.A.; Berbasov, D.O. Self-Disproportionation of Enantiomers of (R)-Ethyl 3-(3,5-Dinitrobenzamido)-4,4,4-trifluorobutanoate on Achiral Silica Gel Stationary Phase. J. Fluor. Chem. 2006, 127, 597–603. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical purifications via self-disproportionation of enantiomers by achiral chromatography: Case study of a series of α-CF3-containing secondary alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of Optical Self-Purification of Chiral Non-Racemic Compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Soloshonok, V.A. Self-disproportionation of enantiomers of 3,3,3-trifluorolactic acid amides via sublimation. J. Fluor. Chem. 2010, 131, 266–269. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Soloshonok, V.A. Self-disproportionation of enantiomers of α-trifluoromethyl lactic acid amides via sublimation. J. Fluor. Chem. 2010, 131, 540–544. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Orita, H.; Ueki, H.; Soloshonok, V.A. First principle lattice energy calculations for enantiopure and racemic crystals of α-(trifluoromethyl)lactic acid: Is self-disproportionation of enantiomers controlled by thermodynamic stability of crystals? J. Fluor. Chem. 2010, 131, 461–466. [Google Scholar] [CrossRef]
- Albrecht, M.; Soloshonok, V.A.; Schrader, L.; Yasumoto, M.; Suhm, M.A. Chirality-dependent sublimation of α-(trifluoromethyl)-lactic acid: Relative vapor pressures of racemic, eutectic, and enantiomerically pure forms, and vibrational spectroscopy of isolated (S, S) and (S, R) dimers. J. Fluor. Chem. 2010, 131, 495–504. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Ono, T.; Katagiri, T.; Soloshonok, V.A. Self-disproportionation of enantiomers of isopropyl 3,3,3-(trifluoro)lactate via sublimation: Sublimation rates vs. enantiomeric composition. J. Fluor. Chem. 2010, 131, 535–539. [Google Scholar] [CrossRef]
- Katagiri, T.; Takahashi, S.; Tsuboi, A.; Suzaki, M.; Uneyama, K. Discrimination of enantiomeric excess of optically active trifluorolactate by distillation: Evidence for a multi-center hydrogen bonding network in the liquid state. J. Fluor. Chem. 2010, 131, 517–520. [Google Scholar] [CrossRef]
- Katagiri, T.; Uneyama, K. Chiral Recognition by Multicenter Single Proton Hydrogen Bonding of Trifluorolactates. Chem. Lett. 2001, 30, 1330–1331. [Google Scholar] [CrossRef]
- Katagiri, T.; Yoda, C.; Furuhashi, K.; Ueki, K.; Kubota, T. Separation of an Enantiomorph and Its Racemate by Distillation: Strong Chiral Recognizing Ability of Trifluorolactates. Chem. Lett. 1996, 25, 115–116. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-disproportionation of enantiomers via sublimation; new and truly green dimension in optical purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef] [PubMed]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; J. Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Zhang, H.Y.; Wang, X.; Ching, C.B.; Wu, J.C. Experimental optimization of enzymic kinetic resolution of racemic flurbiprofen. Biotechnol. Appl. Biochem. 2005, 42, 67–71. [Google Scholar] [PubMed]
- Chelius, E.C.; Reutzel-Edens, S.M.; Van den Berghe Snorek, S. Crystalline of N-[4-[2-(2-amino-4,7-dihydro-4oxo-3H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic Acid and Process Therefor. U.S. Patent 7,138,521 B2, 21 November 2006. [Google Scholar]
- Han, J.; Takeda, R.; Sato, T.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. Optical Resolution of Rimantadine. Molecules 2019, 24, 1828. [Google Scholar] [CrossRef] [PubMed]
- Flippen, J.L.; Gilardi, R.D. (±)-2-(2-Fluoro-4-biphenyl)propionic Acid (Flurbiprofen). Acta Cryst. 1975, B31, 926–928. [Google Scholar] [CrossRef]
- Habibi-Yangjeh, A.; Pourbasheer, E.; Danandeh-Jenagharad, M. Prediction of Melting Point for Drug-like Compounds Using Principal Component-Genetic Algorithm-Artificial Neural Network. Bull. Korean Chem. Soc. 2008, 29, 833–841. [Google Scholar]
- Swart, H.; Breytenbach, J.C.; Hadgraft, J.; du Plessis, J. Synthesis and transdermal penetration of NSAID glycoside esters. Int. J. Pharm. 2005, 301, 71–79. [Google Scholar] [CrossRef]
- Pajula, K.; Taskinen, M.; Lehto, V.-P.; Ketolainen, J.; Korhonen, O. Predicting the Formation and Stability of Amorphous Small Molecule Binary Mixtures from Computationally Determined Flory-Huggins Interaction Parameter and Phase Diagram. Mol. Pharmaceutics 2010, 7, 795–804. [Google Scholar] [CrossRef]
- Henck, J.-O.; Kuhnert-Brandstätter, M. Demonstration of the Terms Enantiotropy and Monotropy in Polymorphism Research Exemplified by Flurbiprofen. J. Pharm. Sci. 1999, 88, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Kawai, N.; Kato, N.; Hamada, Y.; Shioiri, T. New Methods and Reagents in Organic Synthesis. 35. A New Synthesis of Some Non-Steroidal Anti-Inflammatory Agents with the 2-Arylpropionic Acid Skeleton by the Use of Diphenyl Phosphorazidate (DPPA) as a 1,3-Dipole. Chem. Pharm. Bull. 1983, 31, 3139–3148. [Google Scholar] [CrossRef][Green Version]
- Smith, C.R.; RajanBabu, T.V. Catalytic Asymmetric Synthesis Using Feedstocks: An Enantioselective Route to 2-Arylpropionic Acids and 1-Arylethyl Amines via Hydrovinylation of Vinyl Arenes. J. Org. Chem. 2009, 74, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Tarasevych, A.V.; Sorochinsky, A.E.; Kukhar, V.P.; Chollet, A.; Daniellou, R.; Guillemin, J.-C. Partial Sublimation of Enantioenriched Amino Acids at Low Temperature. Is it Coming from the Formation of a Euatmotic Composition of the Gaseous Phase? J. Org. Chem. 2013, 78, 10530–10533. [Google Scholar] [CrossRef]
- Clark, J.B.; Hastie, J.W.; Kihlborg, L.H.E.; Metselaar, R.; Thackeray, M.M. Definitions of terms relating to phase transitions of the solid state. Pure Appl. Chem. 1994, 66, 577–594. [Google Scholar] [CrossRef]
- Gamsjäger, H.; Lorimer, J.W.; Scharlin, P.; Shaw, D.G. Glossary of terms related to solubility. Pure Appl. Chem. 2008, 80, 233–276. [Google Scholar] [CrossRef]
- Bellec, A.; Guillemin, J.-C. A simple explanation of the enhancement or depletion of the enantiomeric excess in the partial sublimation of enantiomerically enriched amino acids. Chem. Commun. 2010, 46, 1482–1484. [Google Scholar] [CrossRef]
- Bellec, A.; Guillemin, J.-C. Attempts to explain the self-disproportionation observed in the partial sublimation of enantiomerically enriched carboxylic acids. J. Fluor. Chem. 2010, 131, 545–548. [Google Scholar] [CrossRef]
- Carman, R.M.; Klika, K.D. Partially racemic compounds as brushtail possum urinary metabolites. Aust. J. Chem. 1992, 45, 651–657. [Google Scholar] [CrossRef]
- Flynn, A.J.; Ford, A.; Maguire, A.R. Localized Partitioning of Enantiomers in Solid Samples of Sulfoxides: Importance of Sampling Method in Determination of Enantiopurity. J. Org. Chem. 2020, 85, 10216–10221. [Google Scholar] [CrossRef]
- Doucet, H.; Fernandez, E.; Layzell, T.P.; Brown, J.M. The scope of catalytic asymmetric hydroboration/oxidation with rhodium complexes of 1,1’-(2-diarylphosphino-1-naphthyl)isoquinolines. Chem. Eur. J. 1999, 5, 1320–1330. [Google Scholar] [CrossRef]
- Abás, S.; Arróniz, C.; Molins, E.; Escolano, C. Access to the enantiopure pyrrolobenzodiazepine (PBD) dilactam nucleus via self-disproportionation of enantiomers. Tetrahedron 2018, 74, 867–871. [Google Scholar] [CrossRef]
- Wzorek, A.; Kamizela, A.; Sato, A.; Soloshonok, V.A. Self-disproportionation of Enantiomers (SDE) via achiral gravity-driven column chromatography of N-fluoroacyl-1-phenylethylamines. J. Fluor. Chem. 2017, 196, 37–43. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE): The effect of scaling down, potential problems versus prospective applications, possible new occurrences, and unrealized opportunities? Electrophoresis 2019, 40, 1869–1880. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Wzorek, A.; Klika, K.D. A question of policy: Should tests for the self-disproportionation of enantiomers (SDE) be mandatory for reports involving scalemates? Tetrahedron Asymmetry 2017, 28, 1430–1434. [Google Scholar] [CrossRef]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-Made Amino Acids and Fluorinated Motifs as Prominent Traits in the Modern Pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of Fluorine-Containing Amino Acids for Drug Design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, E.; Yamamoto, T.; Ito, E.; Shibata, N. Understanding the Thalidomide Chirality in Biological Processes by the Self-disproportionation of Enantiomers. Sci. Rep. 2018, 8, 17131. [Google Scholar] [CrossRef]
- Wu, D.H.; Chen, A.D.; Johnson, C.S., Jr. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- McDonald, G.G.; Leigh, J.S., Jr. A New Method for Measuring Longitudinal Relaxation Times. J. Magn. Reson. 1973, 9, 358–362. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR Solvent: | Toluene-d8 | c-Hexane a-d12–MTBE a, 4:1 | CDCl3 | 1,4-Dioxane-d8 | Acetonitrile-d3 | ESI-MS Using Acetonitrile |
| preferred association | heterochiral b | homochiral c | homochiral d | undetermined | indeterminate | homochiral |
| ΔD e × 10−10, m2s−1 | 0.13 | 0.26 | ~0 | not examined | ~0 | – |
| SIDA magnitude | seen on 1H, 19F, and 13C nuclei | not observed but aSIDA a evident | weak | very, very weak | not observed but aSIDA a evident | – |
| Chromatographic Method: | MPLC a | PTLC a | Column a | Column a | SEC a,f | SEC a,g |
| eluent | n-hexane–ethyl acetate, 4:1 | n-hexane–ethyl acetate, 1:5 | n/c-hexane a–ethyl acetate, 2:1 | toluene–MTBE a, 20:1 | CHCl3 | CHCl3 |
| 1st eluting portion | enantiomer | enantiomer | enantiomer | racemate | enantiomer | racemic |
| Δee, % | 6.8 | 3.2 | 0.2/0.4, erratic | −12.6, erratic | 4.2 | −7.8 |
| eluent | c-hexane a–MTBE a, 1:7 | c-hexane a–MTBE a, 4:1 | n/c-hexane a–MTBE a, 1:1 | The separation of the enantiomers/enantiomeric excess and racemic portions is based on: | ||
| 1st eluting portion | racemate | racemate | racemate | chiral interaction | size exclusion | |
| Δee, % | −1.2 | −5.0 | −3.4/−6.6 | – | – | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiatkowska, M.; Wzorek, A.; Kolbus, A.; Urbaniak, M.; Han, J.; Soloshonok, V.A.; Klika, K.D. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry 2021, 13, 543. https://doi.org/10.3390/sym13040543
Kwiatkowska M, Wzorek A, Kolbus A, Urbaniak M, Han J, Soloshonok VA, Klika KD. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry. 2021; 13(4):543. https://doi.org/10.3390/sym13040543
Chicago/Turabian StyleKwiatkowska, Magdalena, Alicja Wzorek, Anna Kolbus, Mariusz Urbaniak, Jianlin Han, Vadim A. Soloshonok, and Karel D. Klika. 2021. "Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR" Symmetry 13, no. 4: 543. https://doi.org/10.3390/sym13040543
APA StyleKwiatkowska, M., Wzorek, A., Kolbus, A., Urbaniak, M., Han, J., Soloshonok, V. A., & Klika, K. D. (2021). Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry, 13(4), 543. https://doi.org/10.3390/sym13040543