Equilibrium Geometries, Adiabatic Excitation Energies and Intrinsic C=C/C–H Bond Strengths of Ethylene in Lowest Singlet Excited States Described by TDDFT




Abstract
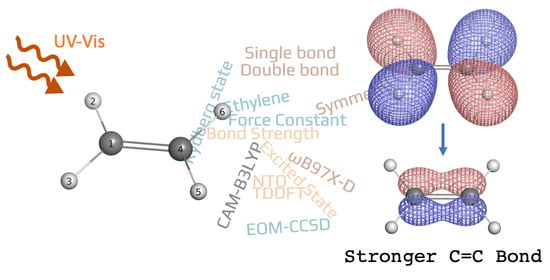
1. Introduction
- Does bond strengthening exist for the excited states of ethylene?
- To what extent can the bond strengthening/weakening be for the excited states of ethylene?
- How can we rationalize and interpret the result of bond strength changes determined with the local mode analysis?
2. Computational Details
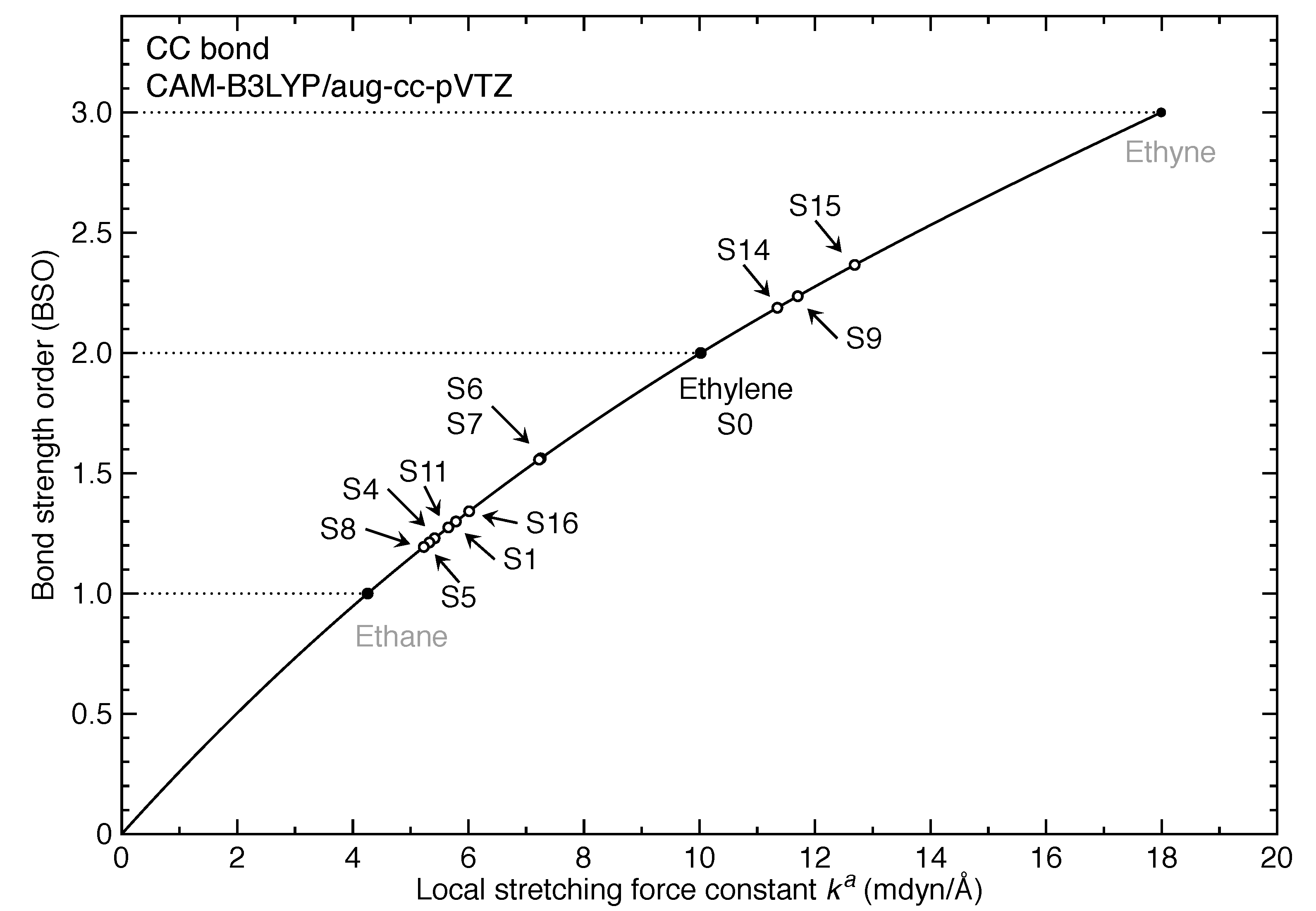
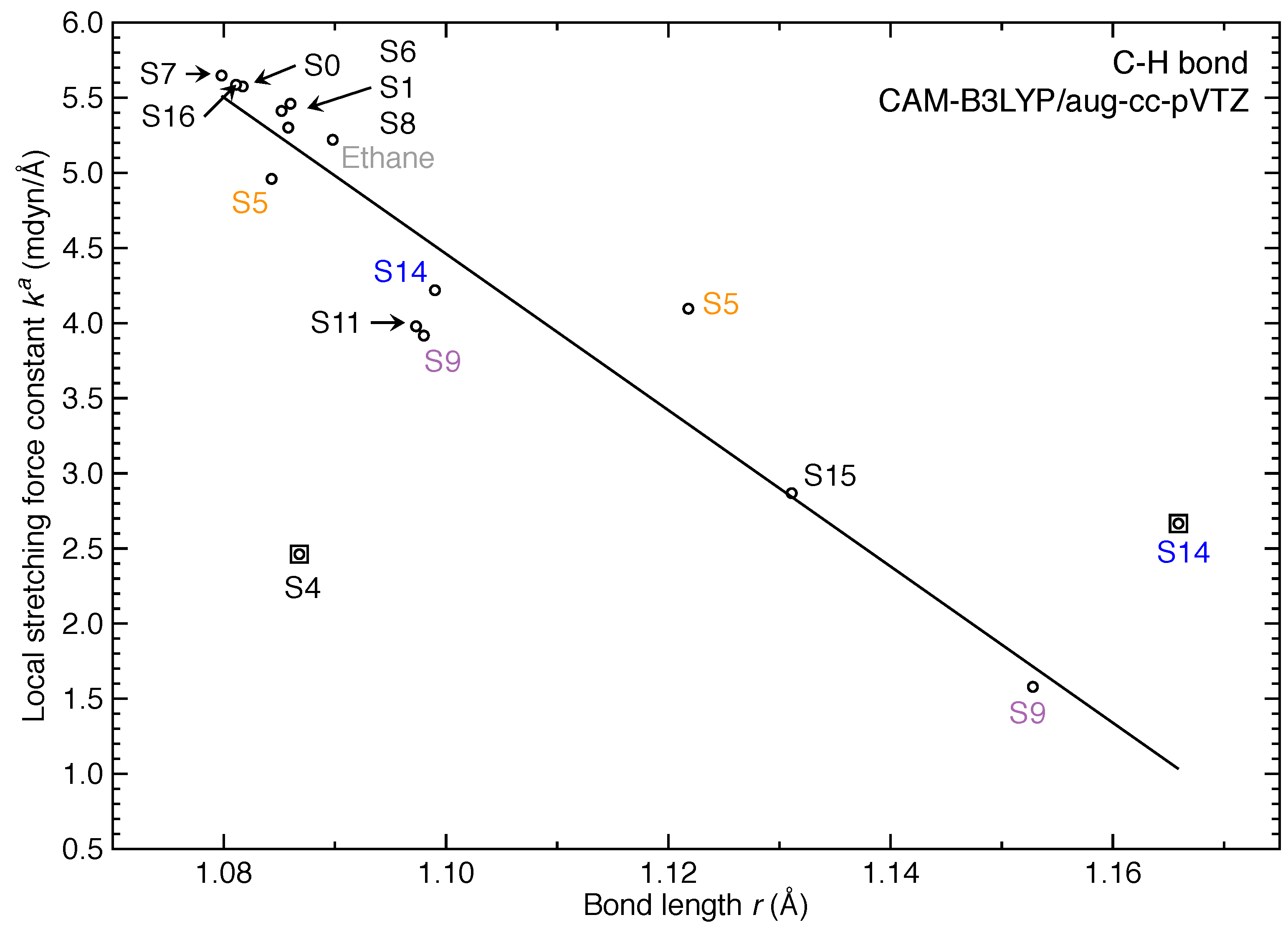
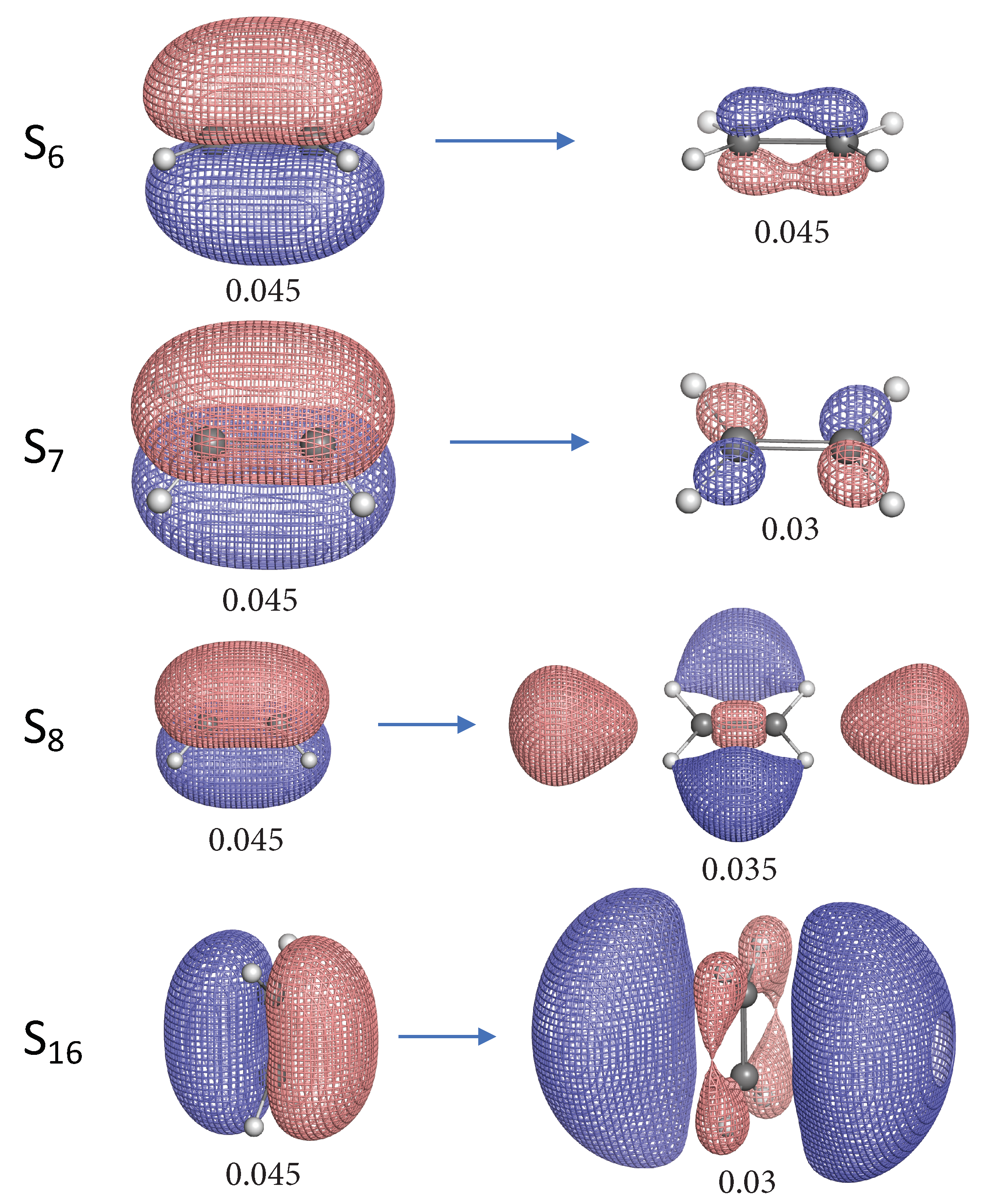
3. Results and Discussion
4. Conclusions
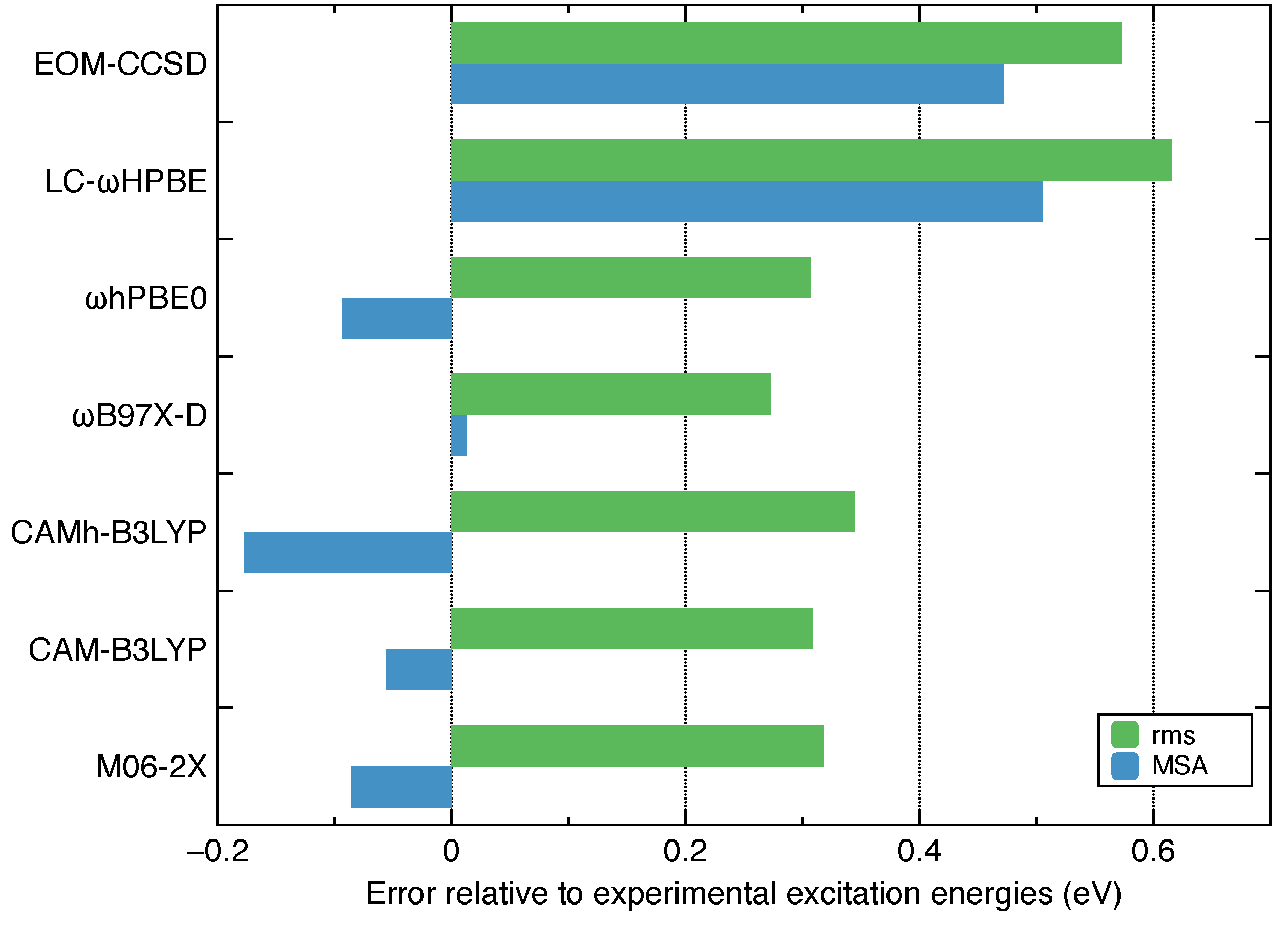
- Among the several commonly used density functionals for describing the Rydberg states, B97X-D, CAM-B3LYP and hPBE0 give relatively low rms deviations (∼0.3 eV) against the experimental excitation energies for ethylene.
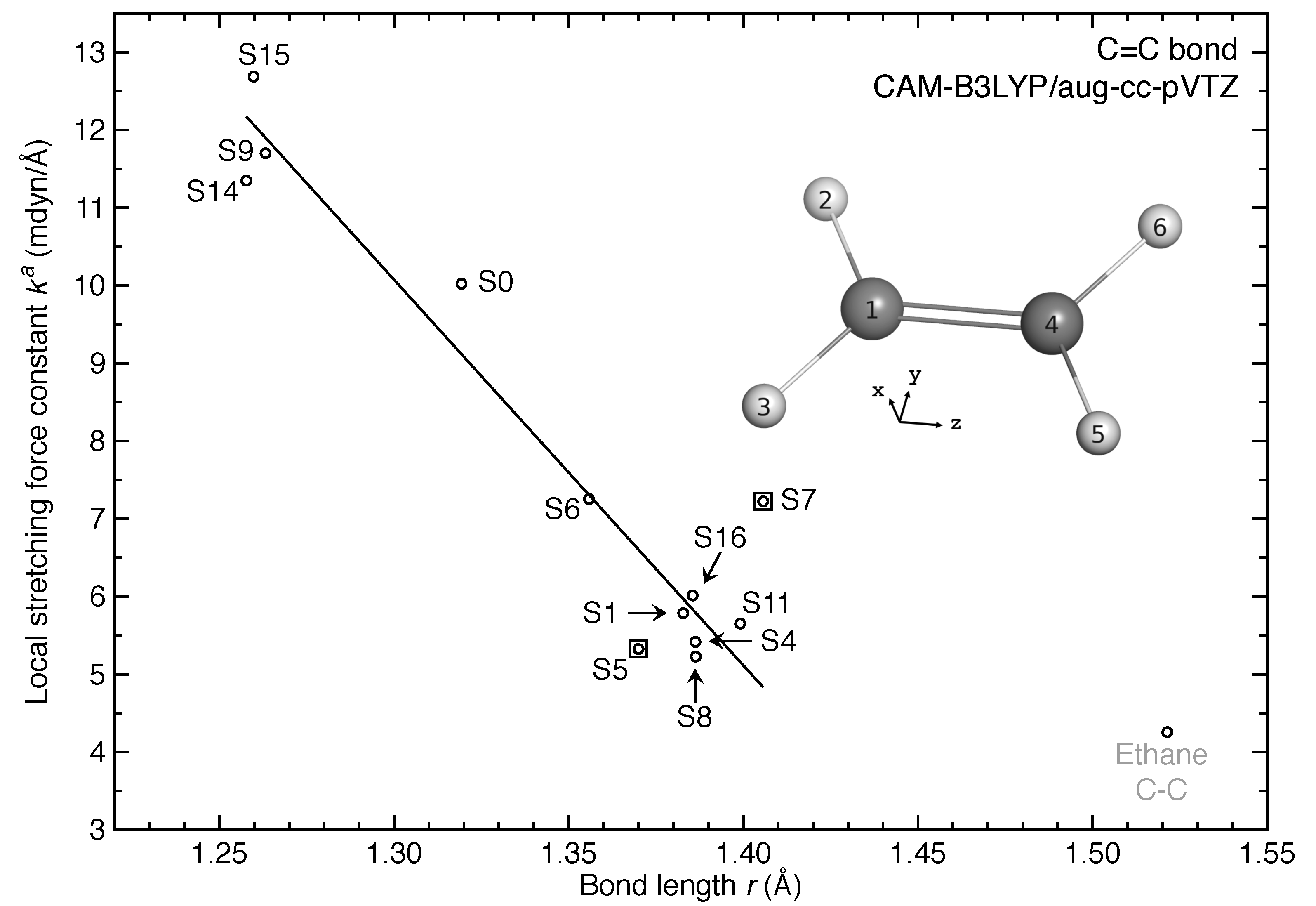
- The equilibrium geometries for 11 out of 17 excited states of ethylene were successfully modeled at the CAM-B3LYP/aug-cc-pVTZ level. Six of them are twisted in contrast to the planar ground-state structure.
- For the first time, the local vibrational mode theory has been applied to molecules in excited states to characterize the intrinsic bond strength which can be directly compared with the ground-state counterpart. In this study on ethylene, the majority of the 11 excited states with optimized geometries exhibit weakened C=C/C–H bonds as characterized by the local stretching force constant , which is consistent with the assumption that a molecule in its excited states with higher electronic energy has in general smaller bond (dissociation) energies. However, the results from the local mode analysis show that 3 excited states have stronger C=C bonds with BSO values of 2.4, which almost reaches the midpoint between ethylene C=C (BSO = 2.0) and ethyne C≡C (BSO = 3.0). Meanwhile, 2 excited states show only marginally stronger C–H bond strength the ground state. Overall, the local stretching force constants for these 11 excited states of ethylene show a larger range of variation for the C=C bond compared to the ground state (variation of −48 % to +27 %) than for C–H bonds (variation of −72 % to +1 %). This leads to the important conclusion that the C=C bond of ethylene is more tunable by electronic excitation, while this does not hold for the C–H bond.
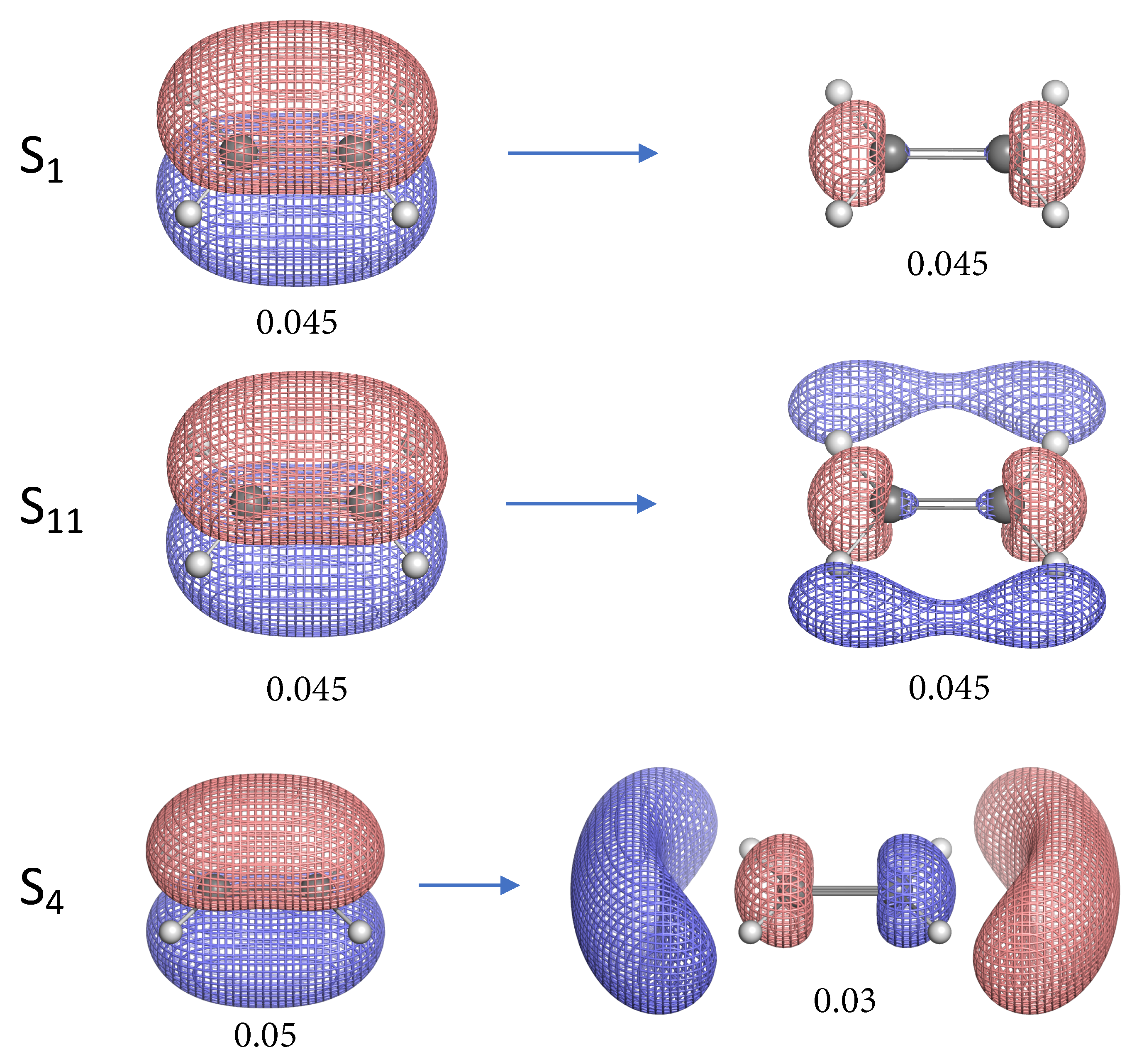
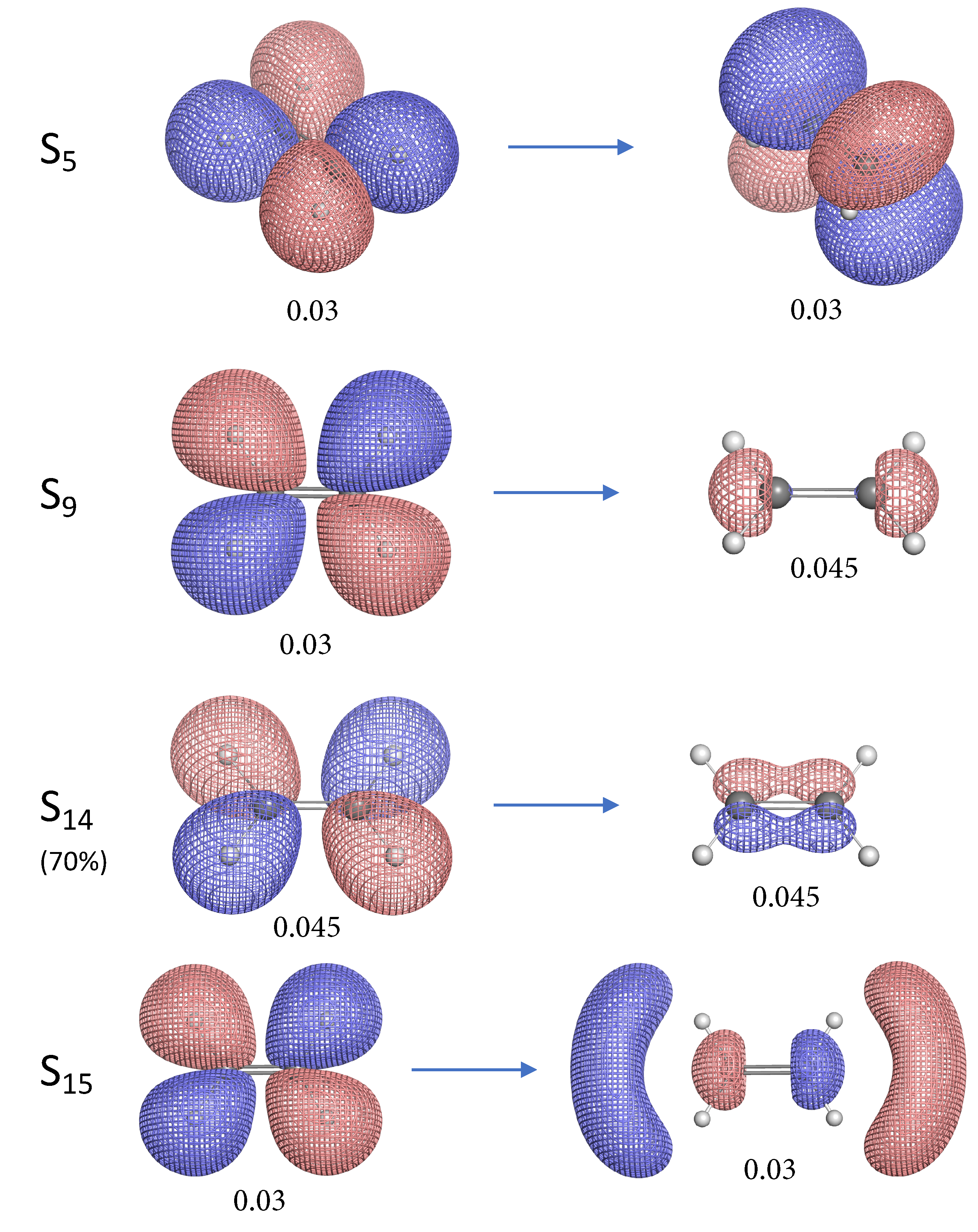
- The NTO analysis plays an important role in helping interpret the result of local stretching force constants for C=C/C–H bonds of ethylenes in excited states.
- When the particle NTO is the bonding orbital, the C=C bond is always weakened in terms of local stretching force constant.
- When the particle NTO is the orbital arising from the -backbone of ethylene, the C=C bond is in general strengthened except when the hole NTO is the anti-bonding orbital. Surprisingly, when the hole NTO orbital is 3p the C=C bond is the strongest and this hole NTO has (C=C) anti-bonding character.
- The excited state leading to slightly increased C–H bond strength has its hole NTO as 4d which consists of 4 C–H bonding orbitals.
- When the hole NTO has significant C–H anti-bonding character, the C–H bonds are greatly weakened.
- Computational modeling of molecular excited states is generally more challenging than for the ground state. Therefore, we used ethylene as a simple theoretical model system to provide the proof of concept that fine-tuning chemical bond strength via electronic excitation is feasible, although the calculations performed in this work can be further improved by more advanced wavefunction-based correlation methods other than TDDFT [5,27]. It is also possible to find the equilibrium geometries of more excited states with dedicated algorithms [43], which we will test in the future.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TDDFT | Time-Dependent Density-Functional Theory |
| NTO | Natural Transition Orbitals |
| BSO | Bond Strength Order |
| BO | Bond Order |
| VEE | Vertical Excitation Energy |
| AEE | Adiabatic Excitation Energy |
| rms | root mean square |
| MSA | Mean Signed Average |
References
- Mai, S.; González, L. Molecular Photochemistry: Recent Developments in Theory. Angew. Chem. Int. Ed. 2020, 59, 16832–16846. [Google Scholar] [CrossRef]
- Hait, D.; Head-Gordon, M. Excited State Orbital Optimization via Minimizing the Square of the Gradient: General Approach and Application to Singly and Doubly Excited States via Density Functional Theory. J. Chem. Theory Comput. 2020, 16, 1699–1710. [Google Scholar] [CrossRef]
- Mewes, S.A.; Dreuw, A. Density-based descriptors and exciton analyses for visualizing and understanding the electronic structure of excited states. Phys. Chem. Chem. Phys. 2019, 21, 2843–2856. [Google Scholar] [CrossRef]
- Liu, Y.; Kilby, P.; Frankcombe, T.J.; Schmidt, T.W. Electronic transitions of molecules: Vibrating Lewis structures. Chem. Sci. 2019, 10, 6809–6814. [Google Scholar] [CrossRef] [PubMed]
- Loos, P.F.; Scemama, A.; Blondel, A.; Garniron, Y.; Caffarel, M.; Jacquemin, D. A Mountaineering Strategy to Excited States: Highly Accurate Reference Energies and Benchmarks. J. Chem. Theory Comput. 2018, 14, 4360–4379. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Escudero, D.; Serrano-Andrés, L. Progress and Challenges in the Calculation of Electronic Excited States. ChemPhysChem 2012, 13, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Dick, B.; Freund, H.J. Analysis of Bonding Properties in Molecular Ground and Excited States by a Cohen-Type Bond Order. Int. J. Quantum Chem. 1983, 24, 747–765. [Google Scholar] [CrossRef]
- Wiberg, K. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Burnus, T.; Marques, M.A.L.; Gross, E.K.U. Time-Dependent Electron Localization Function. Phys. Rev. A 2005, 71, 010501. [Google Scholar] [CrossRef]
- Kadantsev, E.S.; Schmider, H.L. Analysis of Chemical Bonding in Electronic Excited States Using Parity Function. Int. J. Quantum Chem. 2007, 108, 1–14. [Google Scholar] [CrossRef]
- Ferro-Costas, D.; Pendás, Á.M.; González, L.; Mosquera, R.A. Beyond the Molecular Orbital Conception of Electronically Excited States Through the Quantum Theory of Atoms in Molecules. Phys. Chem. Chem. Phys. 2014, 16, 9249–9258. [Google Scholar] [CrossRef] [PubMed]
- Jara-Cortés, J.; Guevara-Vela, J.M.; Pendás, Á.M.; Hernández-Trujillo, J. Chemical Bonding in Excited States: Energy Transfer and Charge Redistribution From a Real Space Perspective. J. Comput. Chem. 2017, 38, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Vannay, L.; Brémond, E.; de Silva, P.; Corminboeuf, C. Visualizing and Quantifying Interactions in the Excited State. Chem. Eur. J. 2016, 22, 18442–18449. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, N.V.; Boldyrev, A.I. Chemical Bonding Analysis of Excited States Using the Adaptive Natural Density Partitioning Method. Phys. Chem. Chem. Phys. 2019, 21, 9590–9596. [Google Scholar] [CrossRef]
- Birks, J.B. Excimers. Rep. Prog. Phys. 1975, 38, 903–974. [Google Scholar] [CrossRef]
- Zimmerman, H.E.; Hixson, S.S.; McBride, E.F. Excited State Bond Weakening in Photochemical Rearrangements of Cyclopropyl Ketones. Exploratory and Mechanistic Organic Photochemistry. XLVIII. J. Am. Chem. Soc. 1970, 92, 2000–2015. [Google Scholar] [CrossRef]
- Badenhoop, J.K.; Scheiner, S. Characterization of Ground and Excited Electronic State Deprotonation Energies of Systems Containing Double Bonds Using Natural Bond Orbital Analysis. J. Chem. Phys. 1996, 105, 4675–4691. [Google Scholar] [CrossRef][Green Version]
- Zink, J.I. Photo-Induced Metal-Ligand Bond Weakening, Potential Surfaces, and Spectra. Coord. Chem. Rev. 2001, 211, 69–96. [Google Scholar] [CrossRef]
- Loose, F.; Wang, D.; Tian, L.; Scholes, G.D.; Knowles, R.R.; Chirik, P.J. Evaluation of Excited State Bond Weakening for Ammonia Synthesis from a Manganese Nitride: Stepwise Proton Coupled Electron Transfer is Preferred over Hydrogen Atom Transfer. Chem. Commun. 2019, 55, 5595–5598. [Google Scholar] [CrossRef]
- Zhao, G.J.; Han, K.L. Hydrogen Bonding in the Electronic Excited State. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef]
- Chu, T.S.; Liu, B.T. Establishing New Mechanisms with Triplet and Singlet Excited-State Hydrogen Bonding Roles in Photoinduced Liquid Dynamics. Int. Rev. Phys. Chem. 2016, 35, 187–208. [Google Scholar] [CrossRef]
- Chu, T.S.; Xu, J. Photoinduced Hydrogen-Bonding Dynamics. J. Mol. Model. 2016, 22, 200. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Zou, W.; Tao, Y. Decoding Chemical Information from Vibrational Spectroscopy Data—Local Vibrational Mode Theory. WIREs Comput. Mol. Sci. 2020, 10, e1480. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Hadad, C.M.; Foresman, J.B.; Chupka, W.A. Electronically Excited States of Ethylene. J. Phys. Chem. 1992, 96, 10756–10768. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Rico, R.J.; Oumi, M.; Lee, T.J. A Doubles Correction to Electronic Excited States from Configuration Interaction in the Space of Single Substitutions. Chem. Phys. Lett. 1994, 219, 21–29. [Google Scholar] [CrossRef]
- Feller, D.; Peterson, K.A.; Davidson, E.R. A Systematic Approach to Vertically Excited States of Ethylene Using Configuration Interaction and Coupled Cluster Techniques. J. Chem. Phys. 2014, 141, 104302. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F. An Efficient Reformulation of the Closed-Shell Coupled Cluster Single and Double Excitation (CCSD) Equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Stanton, J.F.; Bartlett, R.J. The Equation of Motion Coupled-Cluster Method. A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition Probabilities, and Excited State Properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Liu, J.; Liang, W. Analytical Hessian of Electronic Excited states in Time-Dependent Density Functional Theory with Tamm-Dancoff Approximation. J. Chem. Phys. 2011, 135, 014113. [Google Scholar] [CrossRef]
- Liu, J.; Liang, W. Analytical Approach for the Excited-State Hessian in Time-Dependent Density Functional Theory: Formalism, Implementation, and Performance. J. Chem. Phys. 2011, 135, 184111. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zou, W.; Tao, Y.; Freindorf, M.; Makos, M.; Verma, N.; Kraka, E. LMODEA2020; Southern Methodist University: Dallas, TX, USA, 2020. [Google Scholar]
- Henderson, T.M.; Izmaylov, A.F.; Scalmani, G.; Scuseria, G.E. Can Short-Range Hybrids Describe Long-Range-Dependent Properties? J. Chem. Phys. 2009, 131, 044108. [Google Scholar] [CrossRef]
- Shao, Y.; Mei, Y.; Sundholm, D.; Kaila, V.R.I. Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores. J. Chem. Theory Comput. 2019, 16, 587–600. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- García, J.S.; Boggio-Pasqua, M.; Ciofini, I.; Campetella, M. Excited State Tracking During the Relaxation of Coordination Compounds. J. Comput. Chem. 2019, 40, 1420–1428. [Google Scholar] [CrossRef]
- Bersuker, I. The Jahn-Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Leang, S.S.; Zahariev, F.; Gordon, M.S. Benchmarking the Performance of Time-Dependent Density Functional Methods. J. Chem. Phys. 2012, 136, 104101. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | State | Assignment | VEE [exp.] | f | Optimized Geometry | |
|---|---|---|---|---|---|---|
| Symmetry | AEE | |||||
| 0 | 1 A | Ground State | 0.00 | - | D | – |
| 1 | 1 B | → 3s | 6.86 [7.11] [25] | 0.0695 | D | 6.80 |
| 2 | 1 B | → | 7.34 [7.63] [25,48] | 0.3421 | n/a | |
| 3 | 1 B | → 3p | 7.47 [7.80] [25] | 0.0000 | n/a | |
| 4 | 1 B | → 3p | 7.53 [8.00] [48] | 0.0000 | D | 7.45 |
| 5 | 2 B | → 3d () | 8.01 | 0.0000 | C | 7.80 |
| 6 | 2 A | → 3p | 8.36 [8.29] [25] | 0.0000 | D | 7.96 |
| 7 | 1 A | → 4d | 8.58 | 0.0000 | D | 8.52 |
| 8 | 2 B | → 3d | 8.72 [8.62] [25] | 0.0043 | D | 8.65 |
| 9 | 1 B | → 3s | 9.18 | 0.0000 | C | 8.69 |
| 10 | 2 B | → 3d () | 9.39 | 0.0000 | n/a | |
| 11 | 3 B | → 4s | 9.56 | 0.0188 | D | 9.49 |
| 12 | 3 B | → 4p | 9.68 [9.34] [25] | 0.0000 | n/a | |
| 13 | 3 B | → 4p | 9.68 | 0.0000 | n/a | |
| 14 | 2 B | → 4d (30%), → 3p (70%) | 9.72 [9.33] [25] | 0.0617 | C | 9.33 |
| 15 | 1 B | → 3p | 9.80 | 0.0839 | D | 9.18 |
| 16 | 4 B | → 3d | 9.95 | 0.2225 | D | 9.85 |
| 17 | 3 B | → 4d (70%), → 3p (30%) | 9.99 | 0.0086 | n/a | |
| No. | rCC | BSO | rCH | ||
|---|---|---|---|---|---|
| 0 | 1.3193 | 10.022 | 2.0 | 1.0817 | 5.575 |
| 1 | 1.3828 | 5.785 | 1.3 | 1.0852 | 5.412 |
| 4 | 1.3863 | 5.416 | 1.2 | 1.0868 | 2.462 |
| 5 | 1.3700 | 5.325 | 1.2 | 1.1218 (C1H2,C4H5) | 4.096 |
| 1.0843 (C1H3,C4H6) | 4.960 | ||||
| 6 | 1.3558 | 7.254 | 1.6 | 1.0860 | 5.461 |
| 7 | 1.4057 | 7.223 | 1.6 | 1.0798 | 5.648 |
| 8 | 1.3864 | 5.228 | 1.2 | 1.0858 | 5.301 |
| 9 | 1.2632 | 11.701 | 2.2 | 1.0980 (C1H2,C1H3) | 3.917 |
| 1.1528 (C4H5,C4H6) | 1.579 | ||||
| 11 | 1.3991 | 5.653 | 1.3 | 1.0973 | 3.979 |
| 14 | 1.2577 | 11.348 | 2.2 | 1.0990 (C1H2,C4H5) | 4.219 |
| 1.1659 (C1H3,C4H6) | 2.666 | ||||
| 15 | 1.2598 | 12.686 | 2.4 | 1.1313 | 2.864 |
| 16 | 1.3855 | 6.014 | 1.3 | 1.0812 | 5.583 |
| Ethane | 1.5214 | 4.256 | 1.0 | 1.0898 | 5.220 |
| Ethyne | 1.1911 | 17.992 | 3.0 | 1.0619 | 6.416 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, Y.; Zhang, L.; Zou, W.; Kraka, E. Equilibrium Geometries, Adiabatic Excitation Energies and Intrinsic C=C/C–H Bond Strengths of Ethylene in Lowest Singlet Excited States Described by TDDFT. Symmetry 2020, 12, 1545. https://doi.org/10.3390/sym12091545
Tao Y, Zhang L, Zou W, Kraka E. Equilibrium Geometries, Adiabatic Excitation Energies and Intrinsic C=C/C–H Bond Strengths of Ethylene in Lowest Singlet Excited States Described by TDDFT. Symmetry. 2020; 12(9):1545. https://doi.org/10.3390/sym12091545
Chicago/Turabian StyleTao, Yunwen, Linyao Zhang, Wenli Zou, and Elfi Kraka. 2020. "Equilibrium Geometries, Adiabatic Excitation Energies and Intrinsic C=C/C–H Bond Strengths of Ethylene in Lowest Singlet Excited States Described by TDDFT" Symmetry 12, no. 9: 1545. https://doi.org/10.3390/sym12091545
APA StyleTao, Y., Zhang, L., Zou, W., & Kraka, E. (2020). Equilibrium Geometries, Adiabatic Excitation Energies and Intrinsic C=C/C–H Bond Strengths of Ethylene in Lowest Singlet Excited States Described by TDDFT. Symmetry, 12(9), 1545. https://doi.org/10.3390/sym12091545