Simultaneous Analysis of d,l-Amino Acids in Human Urine Using a Chirality-Switchable Biaryl Axial Tag and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Human Urine Samples
2.3. Instrumentation
2.4. Standard Solution Preparation
2.5. Internal Standard Solution Preparation
2.6. Urine Sample Preparation and Derivatization
2.7. LC-MS/MS Analysis
3. Results and Discussion
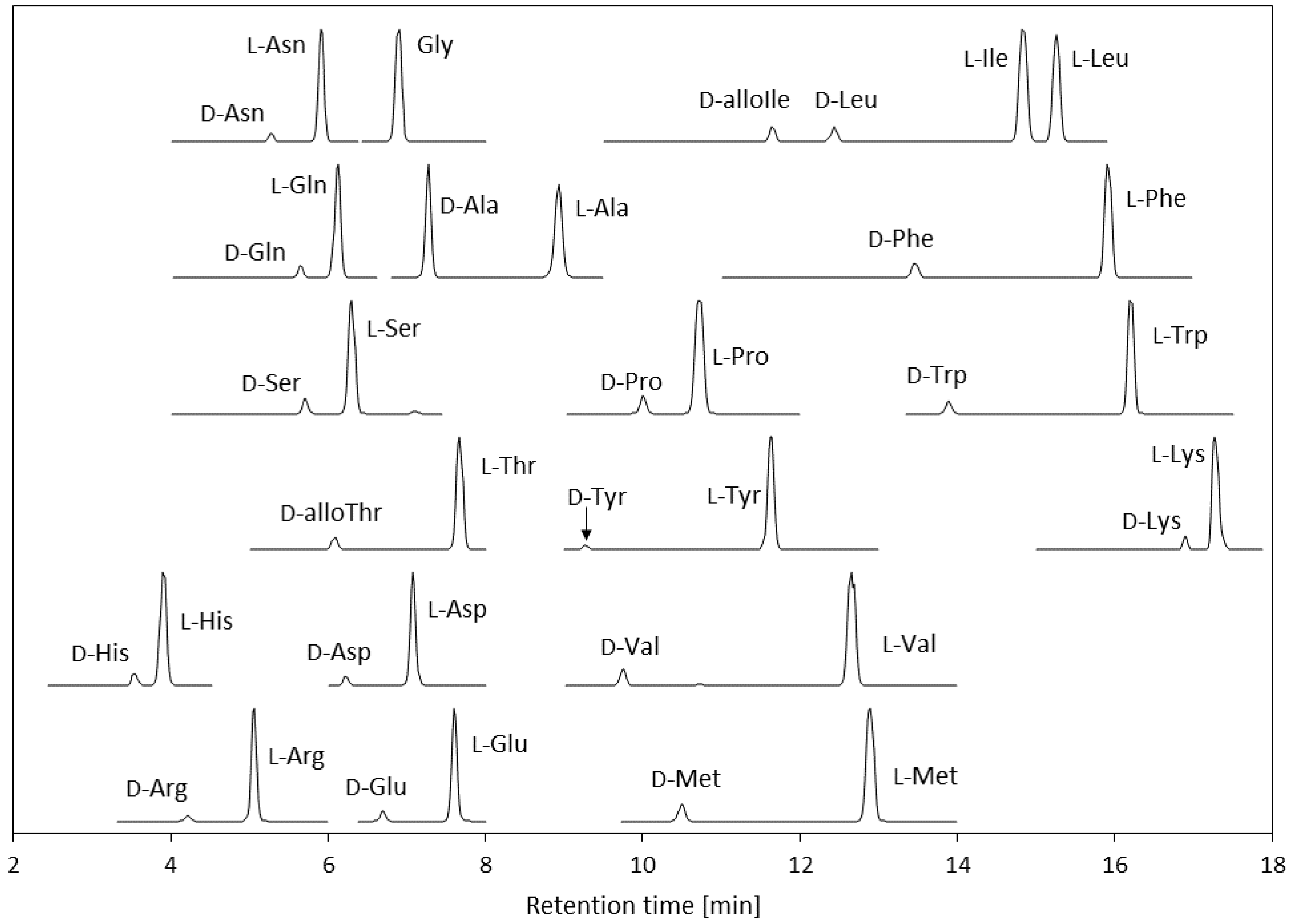
3.1. Optimization of the LC-MS/MS Conditions for Amino Acids Derivatized Using (R)-BiAC
3.2. Method Validation
3.2.1. Linearity and Quantification Ranges
3.2.2. Precision Results, Accuracies, and Recoveries
3.3. Verifying the Analytical Results Using a Chirality-Switching Method Using (S)-BiAC
3.4. Human Urine Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor Ion [m/z] | DP | CE | CXP | ||
|---|---|---|---|---|---|---|
| Analyte (d-isomer) | Analyte (l-isomer) | IS 4 | ||||
| Ala | 385.1 1 | 385.1 1 | 388.1 | 136 | 29 | 10 |
| Arg | 235.2 | 235.7 1 | 240.0 | 46 | 17 | 10 |
| Asn | 427.1 | 428.0 1 | 433.0 | 136 | 29 | 10 |
| Asp | 428.2 | 429.2 1 | 433.1 | 66 | 35 | 10 |
| Gln | 441.0 | 443.0 2 | 448.1 | 136 | 29 | 10 |
| Glu | 442.1 | 443.0 1 | 448.0 | 66 | 35 | 10 |
| His | 450.1 | 451.1 1 | 459.1 | 66 | 23 | 16 |
| Ile | 426.03 | 427.0 1 | 433.1 | 136 | 29 | 10 |
| Leu | 426.0 | 427.0 1 | 433.1 | 136 | 29 | 10 |
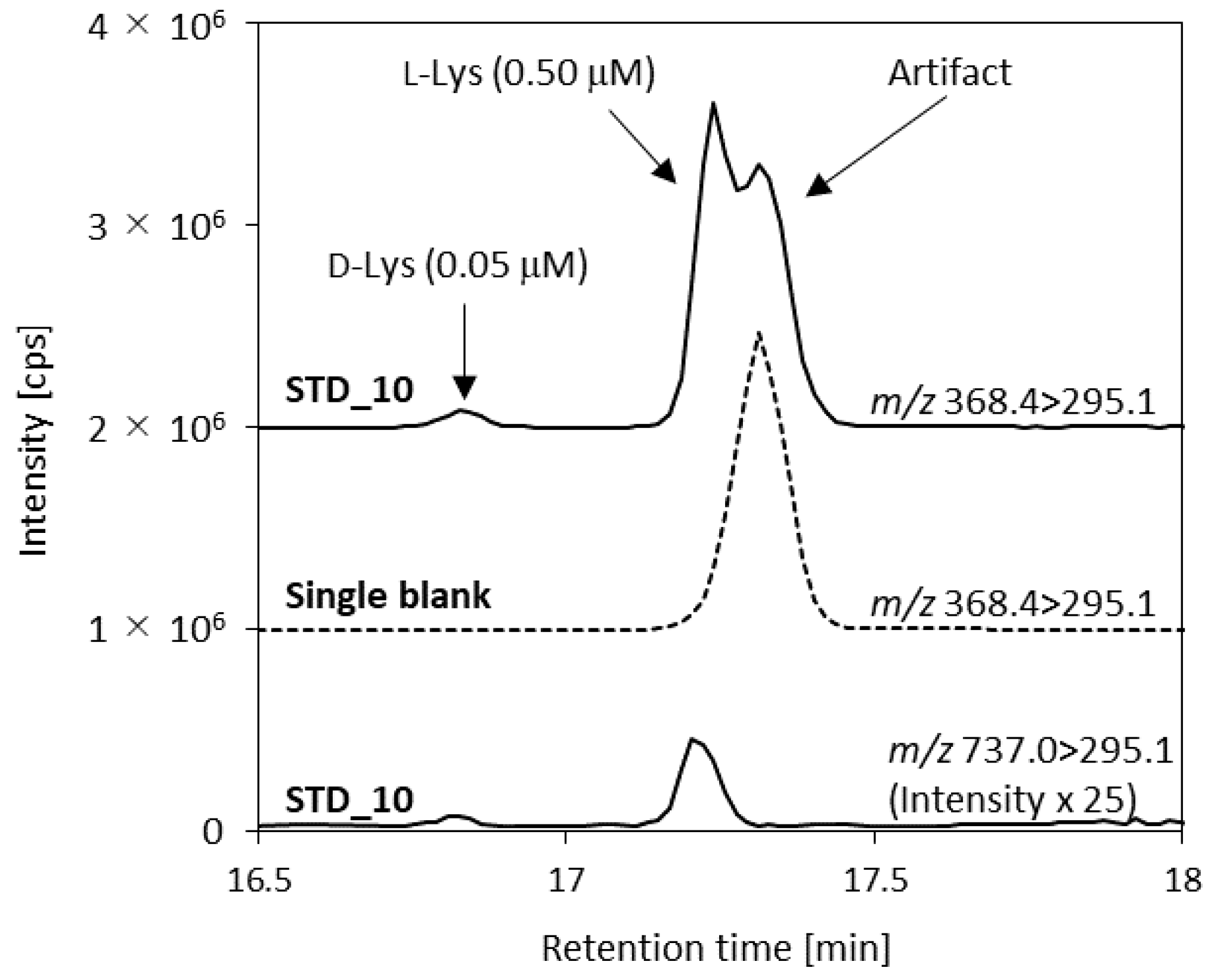
| Lys (2+) | 368.4 | – | 372.4 | 46 | 29 | 36 |
| Lys (1+) | – | 737.0 2 | 743.0 | 106 | 39 | 18 |
| Met | 444.0 | 445.0 1 | 450.0 | 136 | 29 | 10 |
| Phe | 460.1 | 461.1 1 | 470.0 | 26 | 33 | 8 |
| Pro | 410.2 | 411.2 1 | 416.0 | 46 | 31 | 8 |
| Ser | 401.0 1 | 401.0 1 | 404.0 | 136 | 29 | 10 |
| Thr | 414.03 | 415.0 1 | 419.0 | 136 | 29 | 10 |
| Trp | 499.0 | 500.0 1 | 512.0 | 26 | 33 | 8 |
| Tyr | 476.0 | 477.0 1 | 486.0 | 26 | 33 | 8 |
| Val | 412.0 | 413.0 1 | 418.0 | 136 | 29 | 10 |
| Gly | 371.0 1 | 373.0 | 136 | 29 | 10 | |
References
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef]
- Hashimoto, A.; Oka, T.; Nishikawa, T. Anatomical distribution and postnatal changes in endogenous free D-aspartate and D-serine in rat brain and periphery. Eur. J. Neurosci. 1995, 7, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Molliver, M.E.; Snyder, S.H. D-serine, an endogenous synaptic modulator: Localization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. USA 1995, 92, 3948–3952. [Google Scholar] [CrossRef]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef]
- D’Aniello, A. D-Aspartic acid: An endogenous amino acid with an important neuroendocrine role. Brain Res. Rev. 2007, 53, 215–234. [Google Scholar] [CrossRef]
- Homma, H. Biochemistry of D-aspartate in mammalian cells. Amino Acids 2007, 32, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Billard, J.M. D-Amino acids in brain neurotransmission and synaptic plasticity. Amino Acids 2012, 43, 1851–1860. [Google Scholar] [CrossRef]
- Martineau, M.; Shi, T.; Puyal, J.; Knolhoff, A.M.; Dulong, J.; Gasnier, B.; Klingauf, J.; Sweedler, J.V.; Jahn, R.; Mothet, J.P. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J. Neurosci. 2013, 33, 3413–3423. [Google Scholar] [CrossRef]
- Kimura, R.; Tsujimura, H.; Tsuchiya, M.; Soga, S.; Ota, N.; Tanaka, A.; Kim, H. Development of a cognitive function marker based on D-amino acid proportions using new chiral tandem LC-MS/MS systems. Sci. Rep. 2020, 10, 804. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Duncan, J.D.; Lee, S.H. Evaluation of D-amino acid levels in human urine and in commercial L-amino acid samples. Amino Acids 1991, 1, 97–106. [Google Scholar] [CrossRef]
- Bruckner, H.; Haasmann, S.; Friedrich, A. Quantification of D-amino acids in human urine using GC-MS and HPLC. Amino Acids 1994, 6, 205–211. [Google Scholar] [CrossRef]
- Bruckner, H.; Schieber, A. Determination of amino acid enantiomers in human urine and blood serum by gas chromatography-mass spectrometry. Biomed. Chromatogr. 2001, 15, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Hamase, K.; Morikawa, A.; Ohgusu, T.; Lindner, W.; Zaitsu, K. Comprehensive analysis of branched aliphatic D-amino acids in mammals using an integrated multi-loop two-dimensional column-switching high-performance liquid chromatographic system combining reversed-phase and enantioselective columns. J. Chromatogr. A 2007, 1143, 105–111. [Google Scholar] [CrossRef]
- Sasabe, J.; Suzuki, M.; Miyoshi, Y.; Tojo, Y.; Okamura, C.; Ito, S.; Konno, R.; Mita, M.; Hamase, K.; Aiso, S. Ischemic acute kidney injury perturbs homeostasis of serine enantiomers in the body fluid in mice: Early detection of renal dysfunction using the ratio of serine enantiomers. PLoS ONE 2014, 9, e86504. [Google Scholar] [CrossRef]
- Kimura, T.; Hamase, K.; Miyoshi, Y.; Yamamoto, R.; Yasuda, K.; Mita, M.; Rakugi, H.; Hayashi, T.; Isaka, Y. Chiral amino acid metabolomics for novel biomarker screening in the prognosis of chronic kidney disease. Sci. Rep. 2016, 6, 26137. [Google Scholar] [CrossRef]
- Furusho, A.; Koga, R.; Akita, T.; Mita, M.; Kimura, T.; Hamase, K. Three-Dimensional High-Performance Liquid Chromatographic Determination of Asn, Ser, Ala, and Pro Enantiomers in the Plasma of Patients with Chronic Kidney Disease. Anal. Chem. 2019, 91, 11569–11575. [Google Scholar] [CrossRef]
- Hesaka, A.; Sakai, S.; Hamase, K.; Ikeda, T.; Matsui, R.; Mita, M.; Horio, M.; Isaka, Y.; Kimura, T. D-Serine reflects kidney function and diseases. Sci. Rep. 2019, 9, 5104. [Google Scholar] [CrossRef]
- Morikawa, A.; Fukuoka, H.; Uezono, K.; Mita, M.; Koyanagi, S.; Ohdo, S.; Zaitsu, K.; Hamase, K. Sleep-Awake Profile Related Circadian D-Alanine Rhythm in Human Serum and Urine. Chromatography 2017, 38, 53–58. [Google Scholar] [CrossRef]
- Szoko, E.; Vincze, I.; Tabi, T. Chiral separations for d-amino acid analysis in biological samples. J. Pharm. Biomed. Anal. 2016, 130, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Koga, R.; Oyama, T.; Han, H.; Ueno, K.; Masuyama, K.; Itoh, Y.; Hamase, K. HPLC analysis of naturally occurring free D-amino acids in mammals. J. Pharm. Biomed. Anal. 2012, 69, 42–49. [Google Scholar] [CrossRef]
- Ishii, C.; Akita, T.; Mita, M.; Ide, T.; Hamase, K. Development of an online two-dimensional high-performance liquid chromatographic system in combination with tandem mass spectrometric detection for enantiomeric analysis of free amino acids in human physiological fluid. J. Chromatogr. A 2018, 1570, 91–98. [Google Scholar] [CrossRef]
- Karakawa, S.; Shimbo, K.; Yamada, N.; Mizukoshi, T.; Miyano, H.; Mita, M.; Lindner, W.; Hamase, K. Simultaneous analysis of D-alanine, D-aspartic acid, and D-serine using chiral high-performance liquid chromatography-tandem mass spectrometry and its application to the rat plasma and tissues. J. Pharm. Biomed. Anal. 2015, 115, 123–129. [Google Scholar] [CrossRef]
- Umakoshi, Y.; Nakano, Y.; Fukuda, K.; Watanabe, K.; Miyawaki, I.; Fukusaki, E. Automatic switching valve system to minimize variation of liquid chromatography-tandem mass spectrometry-based chiral amino acid profiling. J. Biosci. Bioeng. 2019, 128, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Kunisawa, A.; Hattori, T.; Kawana, S.; Kitada, Y.; Tamada, H.; Kawano, S.; Hayakawa, Y.; Iida, J.; Fukusaki, E. Free D-amino acids produced by commensal bacteria in the colonic lumen. Sci. Rep. 2018, 8, 17915. [Google Scholar] [CrossRef] [PubMed]
- Ilisz, I.; Aranyi, A.; Peter, A. Chiral derivatizations applied for the separation of unusual amino acid enantiomers by liquid chromatography and related techniques. J. Chromatogr. A 2013, 1296, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Karakawa, S.; Yamada, N.; Miyano, H.; Shimbo, K. Biaryl axially chiral derivatizing agent for simultaneous separation and sensitive detection of proteinogenic amino acid enantiomers using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2019, 1593, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-L.; Lin, P.-Y.; Akita, T.; Mita, M.; Ide, T.; Lee, J.-A.; Hamase, K. Development of a Three-Dimensional HPLC System for the Simultaneous Determination of Lactate and 3-Hydroxybutyrate Enantiomers in Mammalian Urine. Chromatography 2019, 40, 25–32. [Google Scholar] [CrossRef]
- Harada, M.; Karakawa, S. Method for Producing Stereoisomers from Various Amino Compounds. Japan Patent JP 2019055921, 11 April 2019. [Google Scholar]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef]
- Jung, J.; Jeong, K.; Choi, Y.; Kim, S.A.; Kim, H.; Lee, J.W.; Kim, V.N.; Kim, K.P.; Kim, J.S. Deuterium-Free, Three-Plexed Peptide Diethylation for Highly Accurate Quantitative Proteomics. J. Proteome Res. 2019, 18, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Karakawa, S.; Nishimoto, R.; Harada, M.; Arashida, N.; Nakayama, A. Simultaneous Analysis of Tryptophan and Its Metabolites in Human Plasma Using Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry. Chromatography 2019, 40, 127–133. [Google Scholar] [CrossRef]
- Takayama, T.; Mochizuki, T.; Todoroki, K.; Min, J.Z.; Mizuno, H.; Inoue, K.; Akatsu, H.; Noge, I.; Toyo’oka, T. A novel approach for LC-MS/MS-based chiral metabolomics fingerprinting and chiral metabolomics extraction using a pair of enantiomers of chiral derivatization reagents. Anal. Chim. Acta 2015, 898, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.P.; Dudzik, D.; Varas, E.; Gibellini, M.; Skotnicki, M.; Zorawski, M.; Zarzycki, W.; Pellati, F.; Garcia, A. Optimization and validation of a chiral GC-MS method for the determination of free D-amino acids ratio in human urine: Application to a gestational diabetes mellitus study. J. Pharm. Biomed. Anal. 2015, 107, 480–487. [Google Scholar] [CrossRef] [PubMed]





| Analyte | Range [µM] | Pooled Urine 1 [µM] | Spiked Concentration [µM] | Recovery (n = 5) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RSD [%] | Accuracy [%] | ||||||||||
| LQC | MQC | HQC | LQC | MQC | HQC | LQC | MQC | HQC | |||
| d-Ala | 0.025–50.0 | 3.85 | 1.25 | 5.00 | 12.5 | 3.2 | 1.2 | 1.5 | 93 | 92 | 93 |
| d-Arg | 0.025–50.0 | 0.510 | 0.250 | 1.25 | 5.00 | 4.6 | 2.8 | 2.3 | 92 | 97 | 101 |
| d-Asn | 0.010–10.0 | 1.32 | 0.250 | 1.25 | 5.00 | 1.2 | 3.6 | 2.1 | 107 | 103 | 96 |
| d-Asp | 0.025–25.0 | BLOQ 2 | 0.250 | 1.25 | 5.00 | 5.6 | 3.1 | 2.2 | 106 | 104 | 104 |
| d-Gln | 0.025–10.0 | 0.306 | 0.250 | 1.25 | 5.00 | 2.0 | 3.7 | 2.7 | 109 | 102 | 103 |
| d-Glu | 0.025–25.0 | 0.197 | 0.250 | 1.25 | 5.00 | 5.9 | 1.7 | 1.8 | 125 | 108 | 106 |
| d-His | 0.010–10.0 | 0.319 | 0.250 | 1.25 | 5.00 | 2.2 | 1.7 | 3.7 | 95 | 98 | 99 |
| d-alloIle | 0.010–5.00 | 0.064 | 0.250 | 1.25 | 5.00 | 1.7 | 2.8 | —3 | 101 | 100 | —3 |
| d-Leu | 0.010–5.00 | 0.072 | 0.250 | 1.25 | 5.00 | 0.6 | 1.4 | —3 | 105 | 103 | —3 |
| d-Lys | 0.010–5.00 | 0.484 | 0.250 | 1.25 | 5.00 | 1.6 | 2.2 | —3 | 110 | 104 | —3 |
| d-Met | 0.010–5.00 | 0.148 | 0.250 | 1.25 | 5.00 | 3.9 | 2.6 | —3 | 97 | 98 | —3 |
| d-Phe | 0.010–5.00 | 0.071 | 0.250 | 1.25 | 5.00 | 4.6 | 1.6 | —3 | 104 | 102 | —3 |
| d-Pro | 0.010–5.00 | BLOQ 2 | 0.250 | 1.25 | 5.00 | 2.5 | 3.2 | —3 | 108 | 106 | —3 |
| d-Ser | 0.100–50.0 | 9.62 | 1.25 | 5.00 | 12.5 | 2.8 | 0.6 | 4.5 | 140 | 105 | 110 |
| d-alloThr | 0.010–10.0 | 0.778 | 0.250 | 1.25 | 5.00 | 3.3 | 1.6 | 2.2 | 101 | 101 | 97 |
| d-Trp | 0.010–10.0 | BLOQ 2 | 0.250 | 1.25 | 5.00 | 2.5 | 1.2 | 3.6 | 98 | 103 | 107 |
| d-Tyr | 0.010–10.0 | 0.024 | 0.250 | 1.25 | 5.00 | 2.8 | 4.7 | 3.4 | 99 | 94 | 95 |
| d-Val | 0.010–5.00 | 0.051 | 0.250 | 1.25 | 5.00 | 1.0 | 2.6 | —3 | 103 | 101 | —3 |
| Gly | 0.250–500 | 132 | 12.5 | 50.0 | 125 | 3.6 4 | 2.2 | 1.4 | 111 4 | 101 | 102 |
| l-Ala | 0.250–500 | 25.5 | 12.5 | 50.0 | 125 | 1.6 4 | 1.7 | 2.5 | 112 | 98 4 | 103 |
| l-Arg | 0.250–500 | 1.21 | 2.50 | 12.5 | 50.0 | 4.0 | 9.6 | 1.9 | 102 | 107 | 102 |
| l-Asn | 0.250–500 | 7.55 | 2.50 | 12.5 | 50.0 | 2.5 | 4.3 | 1.5 | 99 | 104 | 102 |
| l-Asp | 0.500–500 | BLOQ 2 | 2.50 | 12.5 | 50.0 | 3.0 | 0.7 4 | 2.9 | 111 | 110 4 | 103 |
| l-Gln | 0.500–500 | 43.0 | 12.5 | 50.0 | 125 | 3.0 | 2.2 | 3.5 | 84 | 80 | 83 |
| l-Glu | 0.500–500 | 0.816 | 2.50 | 12.5 | 50.0 | 1.9 | 1.6 4 | 1.5 | 106 | 104 4 | 103 |
| l-His | 0.250–500 | 74.4 | 12.5 | 50.0 | 125 | 2.7 | 1.5 | 1.1 | 98 | 96 | 97 |
| l-Ile | 0.250–100 | 0.964 | 2.50 | 12.5 | 50.0 | 5.2 | 4.5 | 1.4 | 104 | 106 | 96 |
| l-Leu | 0.250–100 | 2.50 | 2.50 | 12.5 | 50.0 | 1.6 | 5.4 | 0.7 | 103 | 111 | 101 |
| l-Lys | 0.500–500 | 27.5 | 12.5 | 50.0 | 125 | 9.2 | 4.9 | 2.9 | 109 | 96 | 95 |
| l-Met | 0.250–100 | 0.641 | 2.50 | 12.5 | 50.0 | 1.9 | 2.4 | 1.0 | 99 | 101 | 101 |
| l-Phe | 0.250–50.0 | 3.96 | 2.50 | 12.5 | 50.0 | 1.6 | 3.8 | —3 | 98 | 100 | —3 |
| l-Pro | 0.250–100 | 0.501 | 2.50 | 12.5 | 50.0 | 2.2 | 7.9 | 2.5 | 107 | 112 | 100 |
| l-Ser | 0.250–500 | 20.8 | 12.5 | 50.0 | 125 | 1.8 4 | 2.0 | 1.0 | 101 4 | 99 | 101 |
| l-Thr | 0.250–250 | 14.4 | 12.5 | 50.0 | 125 | 6.6 | 2.2 | 0.9 | 108 | 95 | 93 |
| l-Trp | 0.250–100 | 6.94 | 2.50 | 12.5 | 50.0 | 1.4 | 2.1 | 2.2 | 97 | 109 | 106 |
| l-Tyr | 0.250–250 | 8.42 | 2.50 | 12.5 | 50.0 | 1.4 | 3.2 | 2.3 | 98 | 108 | 105 |
| l-Val | 0.250–100 | 2.92 | 2.50 | 12.5 | 50.0 | 1.5 | 6.5 | 2.5 | 101 | 106 | 99 |
| Analyte | Concentration of Pooled Urine [µM] | Ratio (S)-BiAC/(R)-BiAC | |
|---|---|---|---|
| (R)-BiAC 1 | (S)-BiAC 2 | ||
| d-Ala | 3.85 | 4.17 | 108% |
| d-Arg | 0.510 | 0.497 | 97% |
| d-Asn | 1.32 | 1.38 | 105% |
| d-Asp | BLOQ 3 | 0.025 | — |
| d-Gln | 0.306 | 0.751 | 246% |
| d-Glu | 0.197 | 0.435 | 221% |
| d-His | 0.319 | 0.276 | 87% |
| d-alloIle | 0.064 | 0.043 | 67% |
| d-Leu | 0.072 | 0.791 | 1104% |
| d-Lys | 0.484 | 0.539 | 111% |
| d-Met | 0.148 | 0.149 | 101% |
| d-Phe | 0.071 | 0.077 | 109% |
| d-Pro | BLOQ 3 | BLOQ 3 | — |
| d-Ser | 9.62 | 9.74 | 101% |
| d-alloThr | 0.778 | 0.766 | 99% |
| d-Trp | BLOQ 3 | BLOQ 3 | — |
| d-Tyr | 0.024 | 0.036 | 150% |
| d-Val | 0.051 | 0.048 | 94% |
| Gly | 132 | 129 | 97% |
| l-Ala | 25.5 | 25.2 | 99% |
| l-Arg | 1.21 | 1.13 | 94% |
| l-Asn | 7.55 | 7.84 | 104% |
| l-Asp | BLOQ 3 | BLOQ 3 | — |
| l-Gln | 43.0 | 46.3 | 108% |
| l-Glu | 0.816 | 0.809 | 99% |
| l-His | 74.4 | 74.8 | 100% |
| l-Ile | 0.964 | 0.813 | 84% |
| l-Leu | 2.50 | 2.46 | 98% |
| l-Lys | 27.5 | 26.3 | 96% |
| l-Met | 0.641 | 0.666 | 104% |
| l-Phe | 3.96 | 3.84 | 97% |
| l-Pro | 0.501 | 0.516 | 103% |
| l-Ser | 20.8 | 19.8 | 95% |
| l-Thr | 14.4 | 14.2 | 99% |
| l-Trp | 6.94 | 6.84 | 98% |
| l-Tyr | 8.42 | 8.56 | 102% |
| l-Val | 2.92 | 2.90 | 99% |
| Analyte | Concentration of Individual Urine Sample 1 [µM] | Average (n = 5) [µM] | % D 5 | ||||
|---|---|---|---|---|---|---|---|
| #1 | #2 | #3 | #4 | #5 | |||
| d-Ala | 19.2 | 59.4 | 21.9 | 17.4 | 57.7 | 35.1 | 12 |
| d-Arg | 6.28 | 5.83 | 4.27 | 3.51 | 5.42 | 5.06 | 29 |
| d-Asn | 24.0 | 13.3 | 11.1 | 12.9 | 14.3 | 15.1 | 17 |
| d-Asp | 0.548 | BLOQ 2 | 0.257 | BLOQ 2 | BLOQ 2 | (0.403) 3 | – |
| d-Gln | 5.13 | 2.74 | 2.51 | 3.35 | 3.59 | 3.46 | 0.8 |
| d-Glu | 3.16 | 2.53 | 1.43 | 1.59 | 2.56 | 2.25 | 22 |
| d-His | 4.64 | 2.50 | 4.46 | 3.43 | 2.40 | 3.49 | 0.5 |
| d-alloIle | 0.681 | 0.307 | 0.310 | 0.976 | 0.847 | 0.624 4 | 5.3 4 |
| d-Leu | 0.524 | 0.451 | 0.326 | 0.987 | 0.864 | 0.630 | 2.3 |
| d-Lys | 9.77 | 3.76 | 3.89 | 5.27 | 5.87 | 5.71 | 2.2 |
| d-Met | 0.383 | 6.517 | 0.191 | 0.273 | 0.338 | 1.54 | 16 |
| d-Phe | 0.848 | 0.538 | 0.400 | 0.706 | 1.03 | 0.705 | 1.5 |
| d-Pro | BLOQ 2 | BLOQ 2 | BLOQ 2 | BLOQ 2 | BLOQ 2 | – | – |
| d-Ser | 246 | 94.2 | 93.5 | 105 | 89.1 | 126 | 37 |
| d-alloThr | 16.8 | 9.52 | 7.19 | 7.54 | 8.00 | 9.80 | 9.5 |
| d-Trp | 0.106 | BLOQ 2 | BLOQ 2 | 0.124 | BLOQ 2 | (0.115) 3 | 0.2 |
| d-Tyr | 0.298 | 0.225 | 0.109 | 0.191 | 0.338 | 0.232 | 0.2 |
| d-Val | 0.829 | 0.514 | 0.366 | 0.459 | 0.638 | 0.561 | 1.6 |
| Gly | 2,120 | 761 | 2,470 | 1,350 | 685 | 1,480 | – |
| l-Ala | 300 | 166 | 234 | 278 | 320 | 260 | – |
| l-Arg | 13.9 | 13.2 | 8.98 | 15.0 | 9.44 | 12.1 | – |
| l-Asn | 61.8 | 56.9 | 87.5 | 91.7 | 63.2 | 72.2 | – |
| l-Asp | BLOQ 2 | BLOQ 2 | BLOQ 2 | BLOQ 2 | BLOQ 2 | – | – |
| l-Gln | 553 | 290 | 475 | 511 | 371 | 440 | – |
| l-Glu | 12.1 | 5.45 | 6.68 | 11.3 | 5.62 | 8.23 | – |
| l-His | 966 | 584 | 763 | 806 | 695 | 763 | – |
| l-Ile | 17.5 | 7.51 | 8.09 | 10.8 | 11.4 | 11.1 | – |
| l-Leu | 40.7 | 19.2 | 21.0 | 29.3 | 26.2 | 27.3 | – |
| l-Lys | 154 | 201 | 184 | 338 | 407 | 257 | – |
| l-Met | 14.0 | 5.49 | 7.53 | 9.28 | 4.96 | 8.25 | – |
| l-Phe | 74.4 | 37.1 | 34.2 | 43.2 | 39.9 | 45.7 | – |
| l-Pro | 6.14 | 3.69 | 4.93 | 6.67 | 4.14 | 5.11 | – |
| l-Ser | 227 | 111 | 314 | 287 | 110 | 210 | – |
| l-Thr | 173 | 94.6 | 176 | 196 | 96.3 | 147 | – |
| l-Trp | 91.4 | 51.1 | 69.5 | 69.2 | 78.1 | 71.9 | – |
| l-Tyr | 126 | 82.1 | 73.5 | 89.6 | 95.2 | 93.4 | – |
| l-Val | 53.4 | 23.7 | 29.9 | 28.5 | 31.8 | 33.5 | – |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harada, M.; Karakawa, S.; Miyano, H.; Shimbo, K. Simultaneous Analysis of d,l-Amino Acids in Human Urine Using a Chirality-Switchable Biaryl Axial Tag and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Symmetry 2020, 12, 913. https://doi.org/10.3390/sym12060913
Harada M, Karakawa S, Miyano H, Shimbo K. Simultaneous Analysis of d,l-Amino Acids in Human Urine Using a Chirality-Switchable Biaryl Axial Tag and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Symmetry. 2020; 12(6):913. https://doi.org/10.3390/sym12060913
Chicago/Turabian StyleHarada, Masashi, Sachise Karakawa, Hiroshi Miyano, and Kazutaka Shimbo. 2020. "Simultaneous Analysis of d,l-Amino Acids in Human Urine Using a Chirality-Switchable Biaryl Axial Tag and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry" Symmetry 12, no. 6: 913. https://doi.org/10.3390/sym12060913
APA StyleHarada, M., Karakawa, S., Miyano, H., & Shimbo, K. (2020). Simultaneous Analysis of d,l-Amino Acids in Human Urine Using a Chirality-Switchable Biaryl Axial Tag and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Symmetry, 12(6), 913. https://doi.org/10.3390/sym12060913