Model Reduction for Kinetic Models of Biological Systems


Abstract
1. Introduction
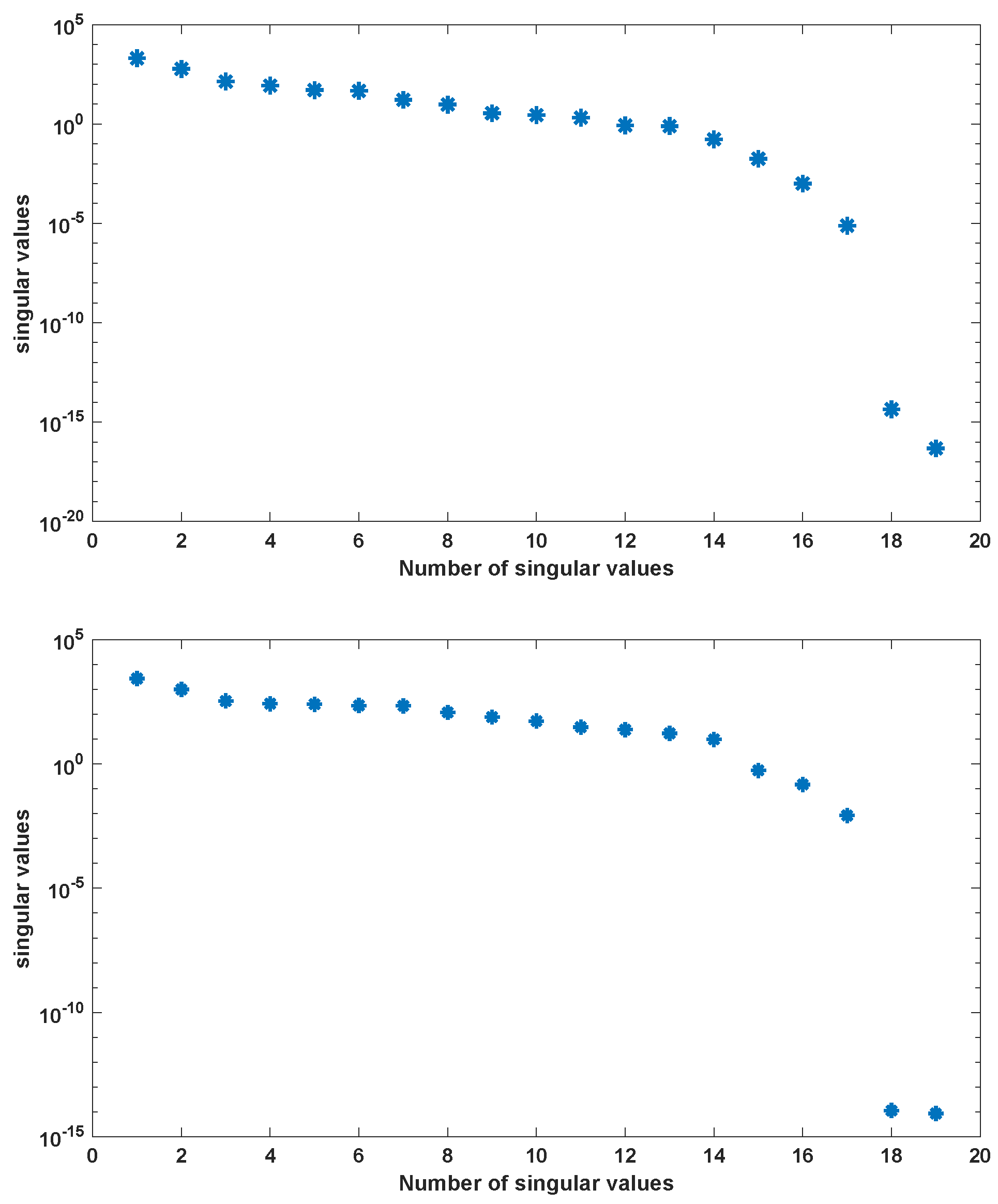
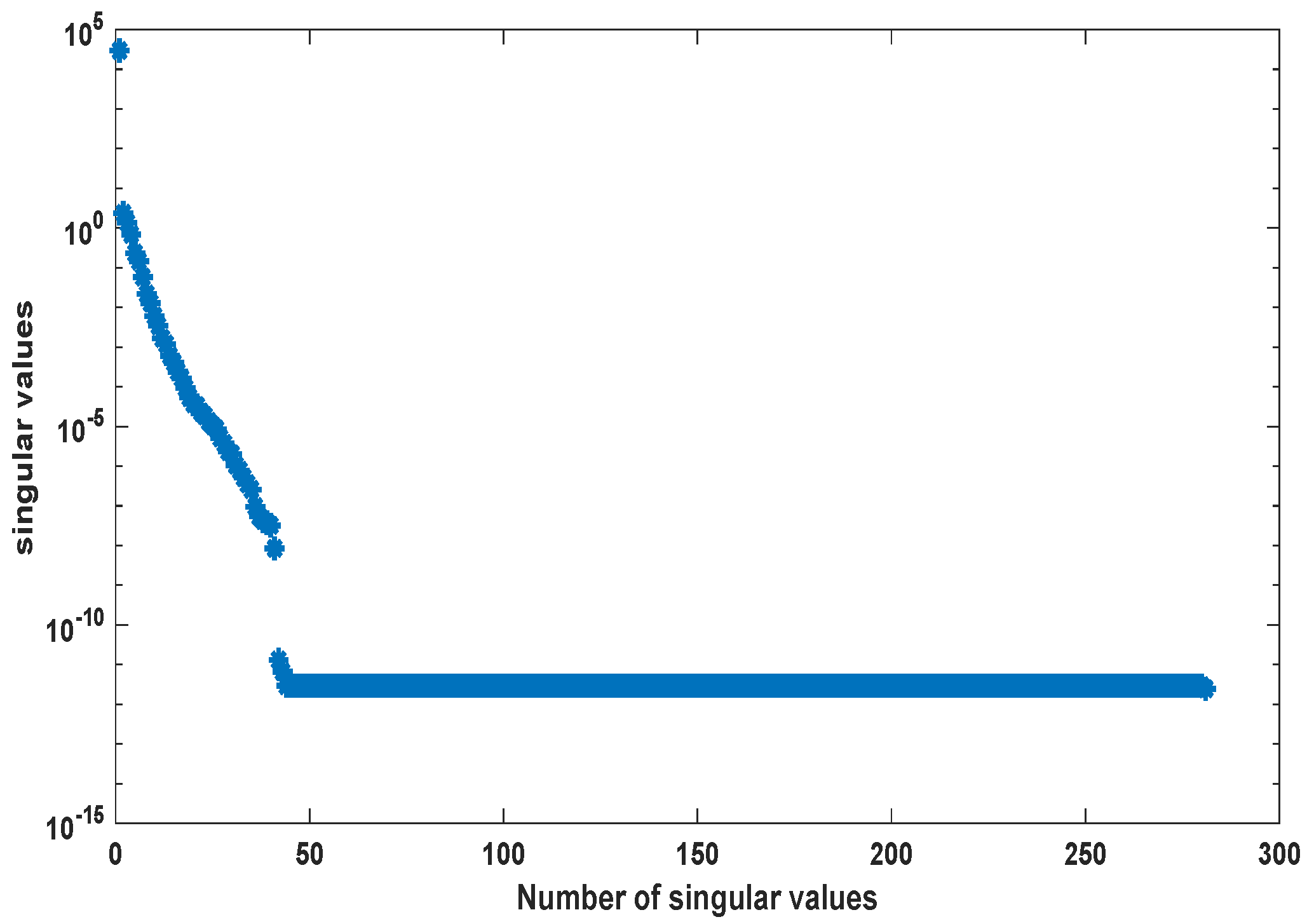
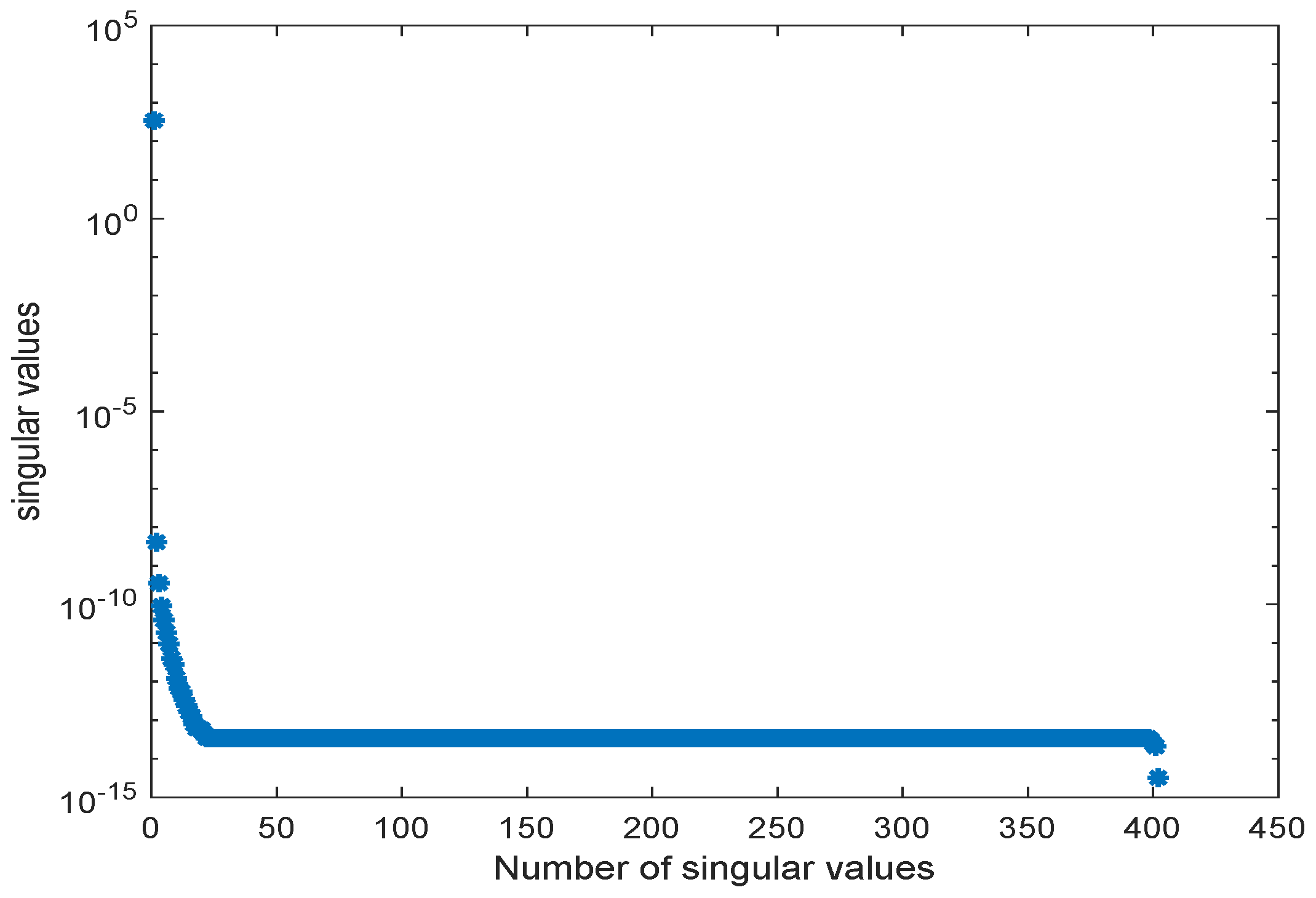
2. Proper Orthogonal Decomposition for Differential Equations
3. Application of the POD-DEIM Approach to Kinetic Model Examples
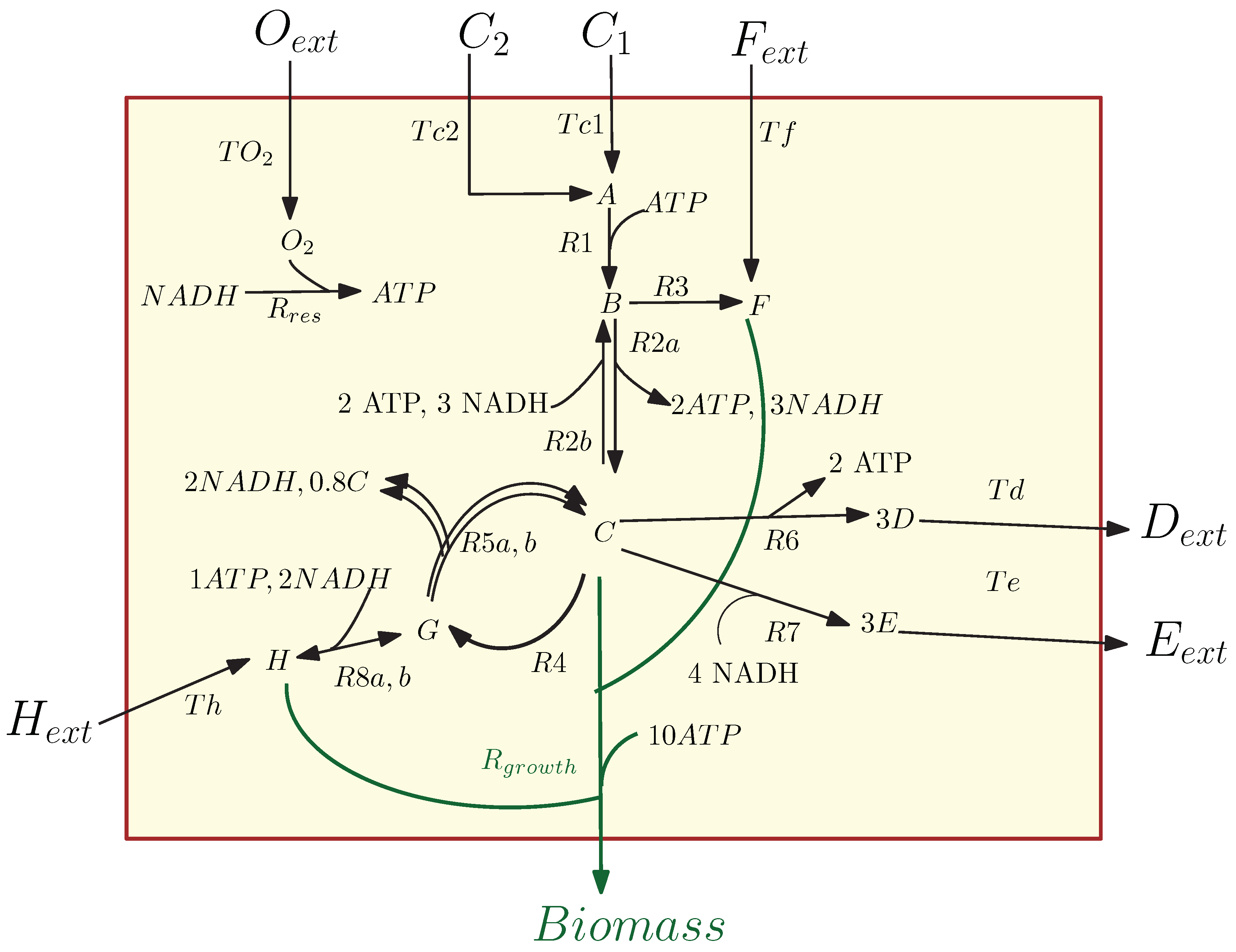
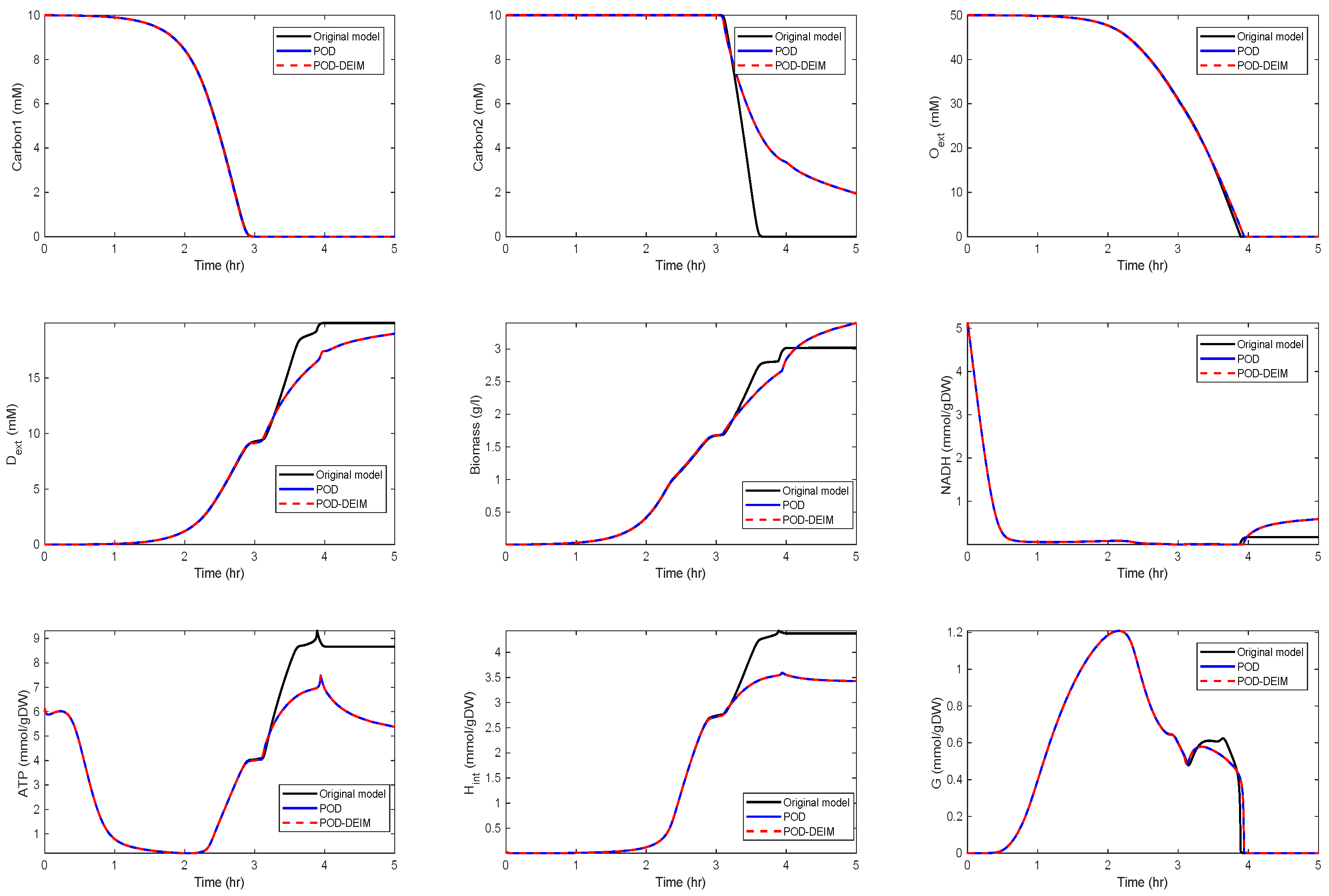
3.1. Kinetic Model of the Metabolic-Genetic Network
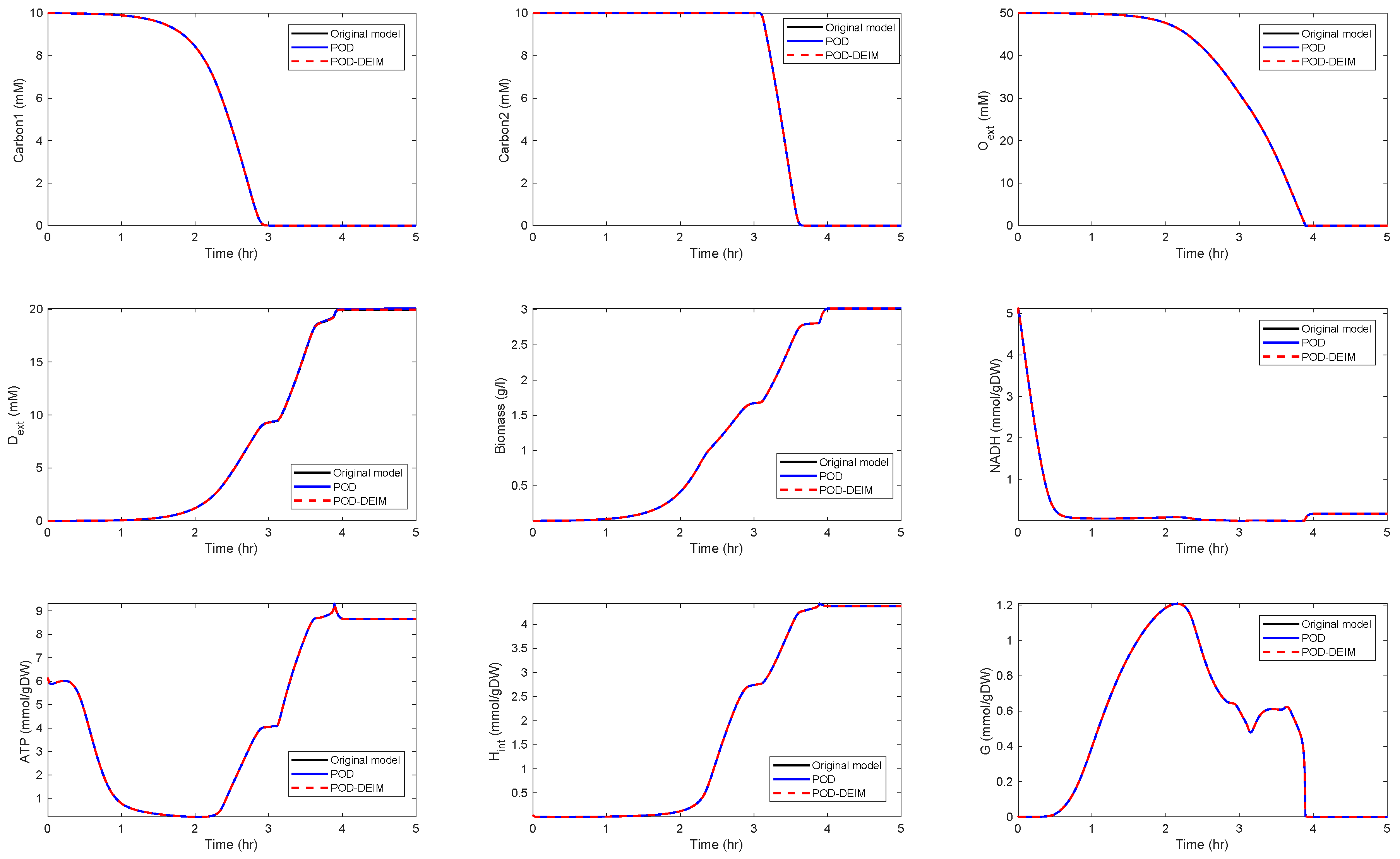
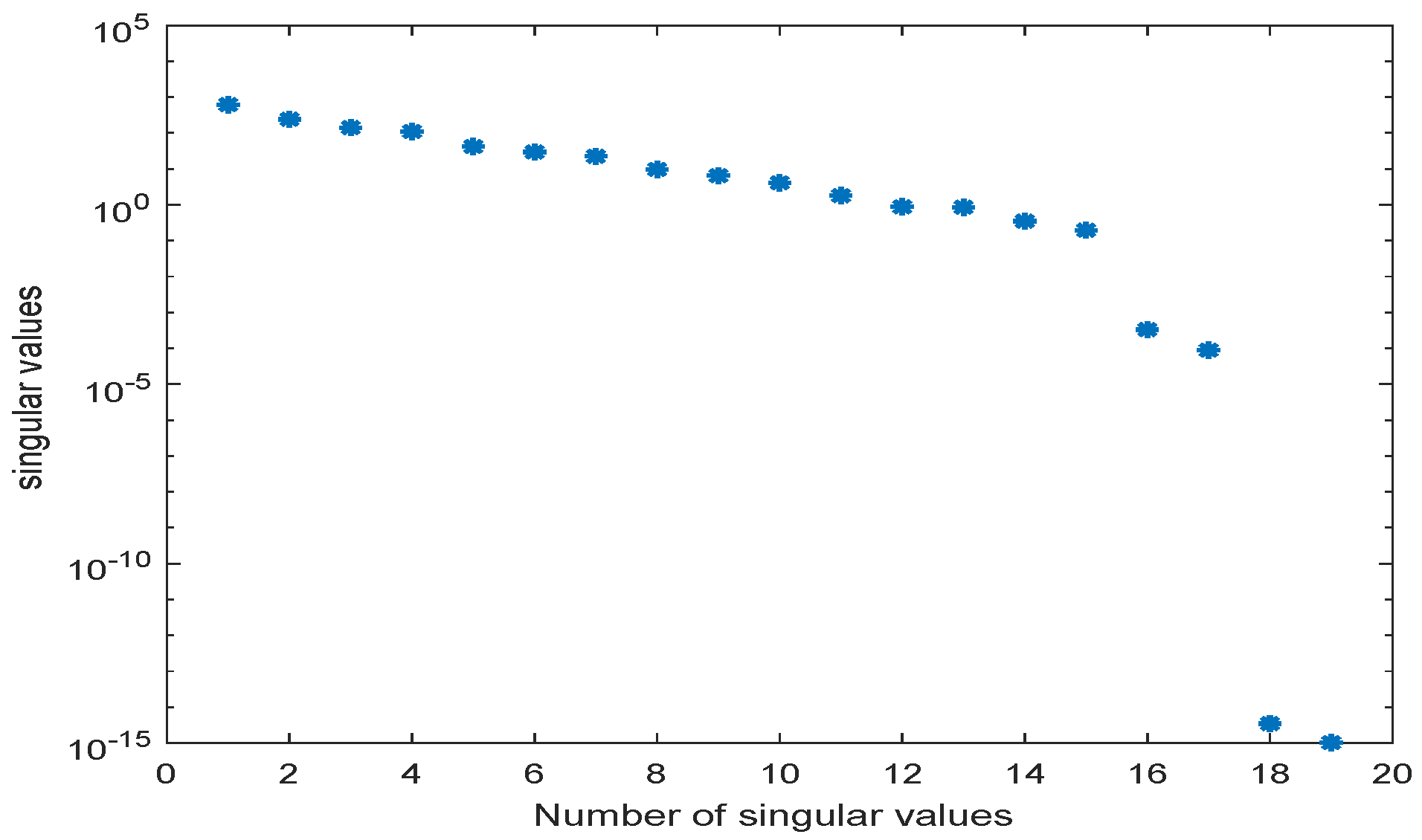
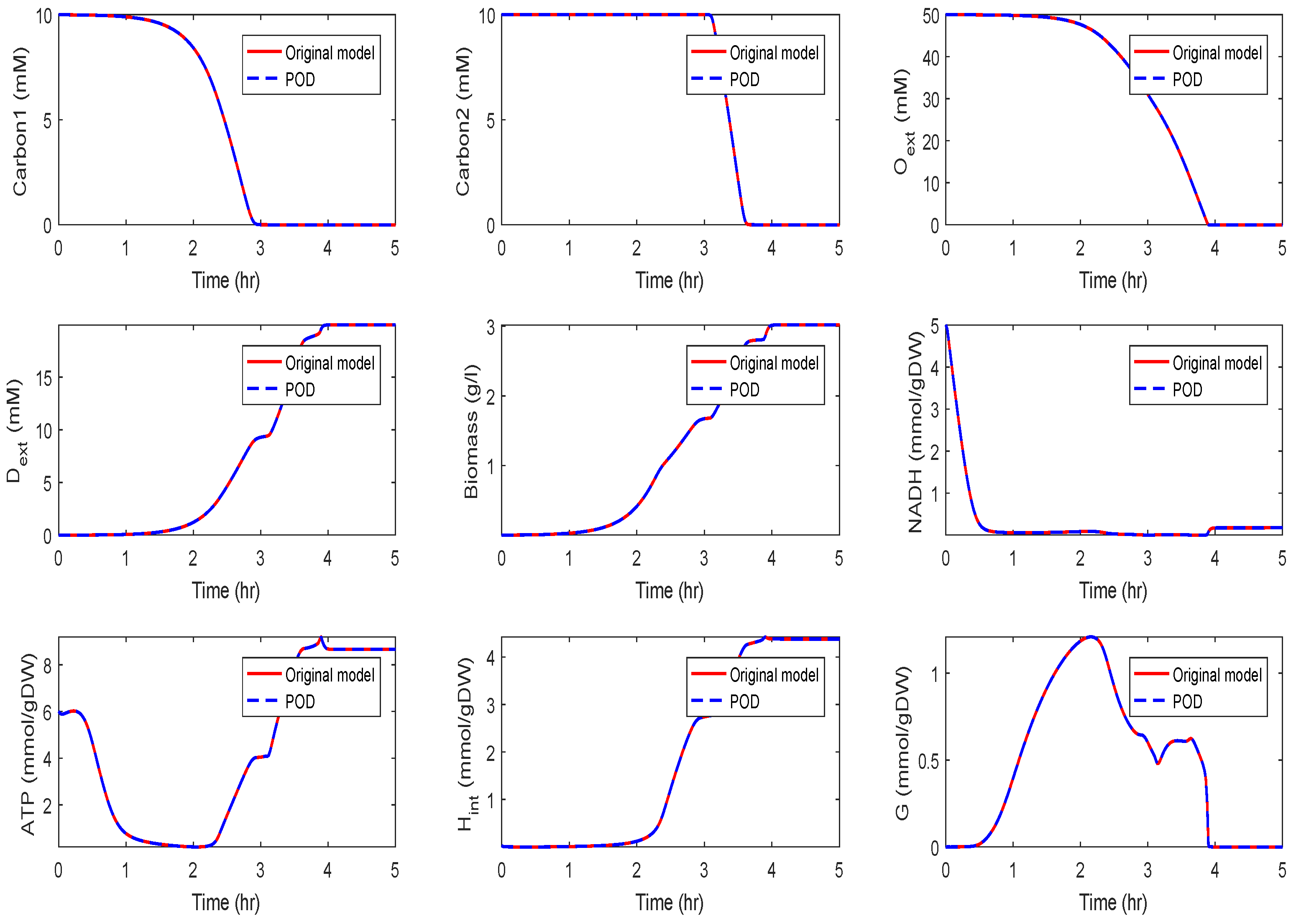
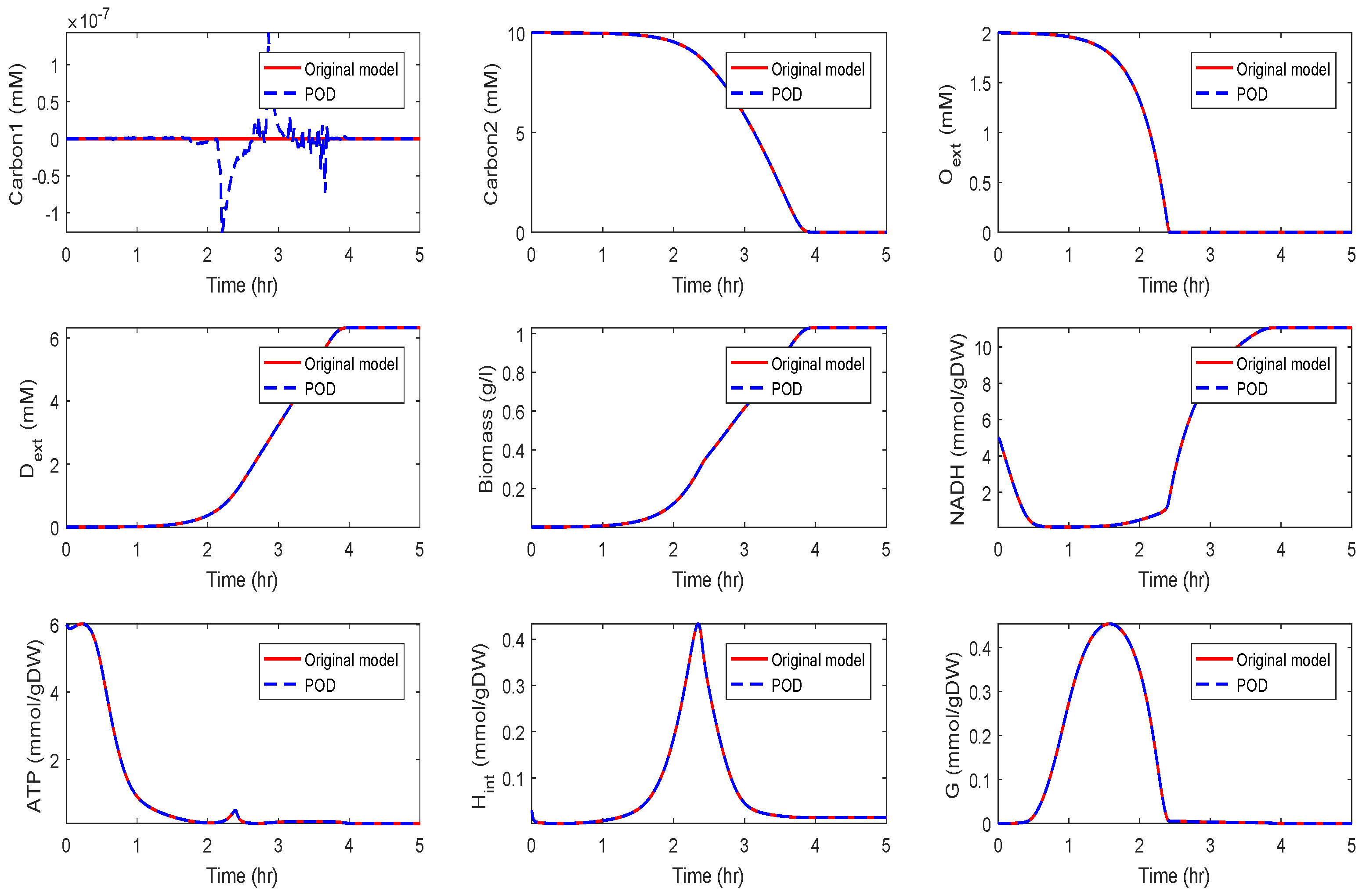
Application of POD-DEIM to the Diauxic-Switch Scenario
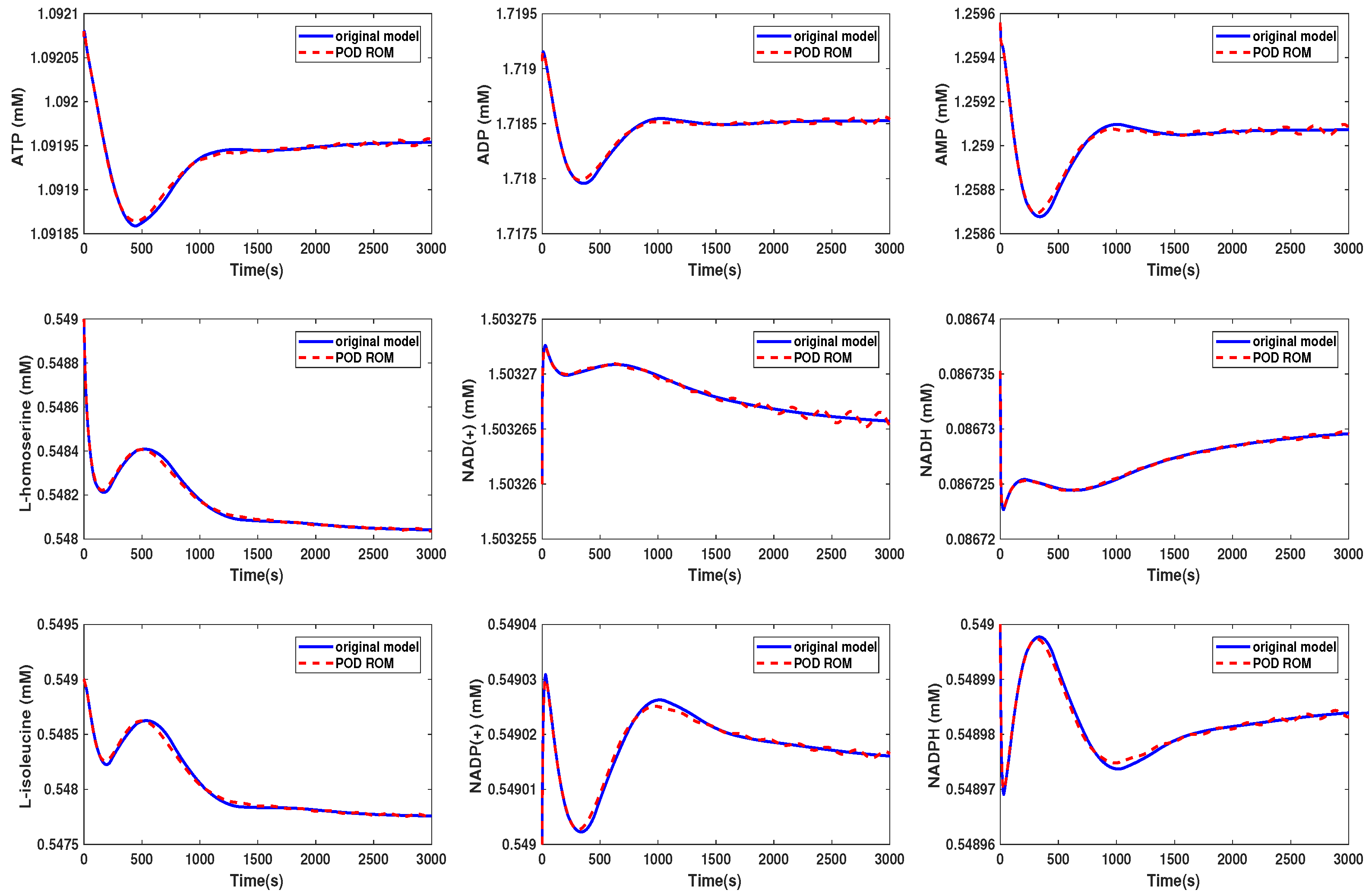
3.2. Kinetic Model of the Yeast Metabolic Network
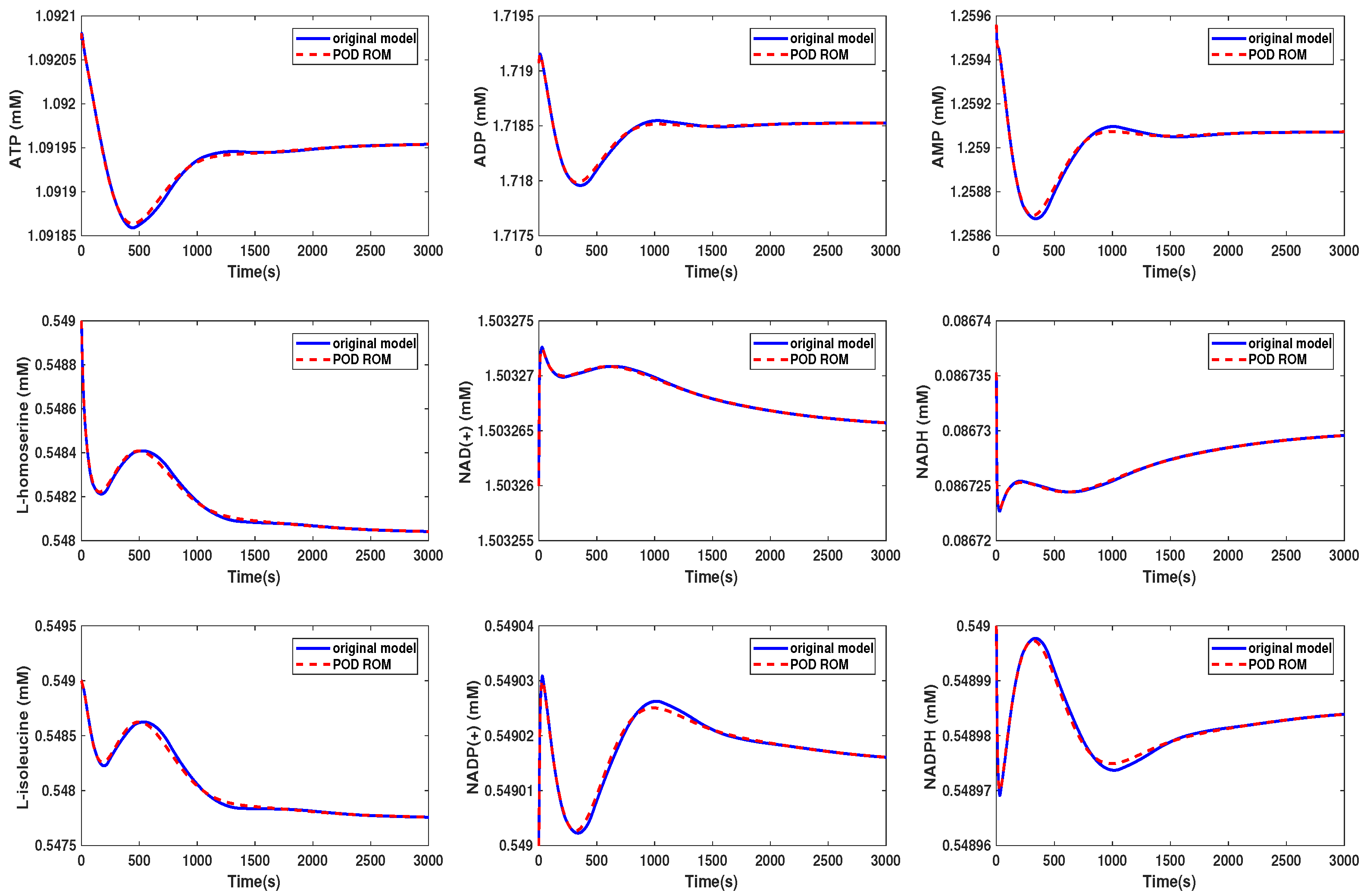
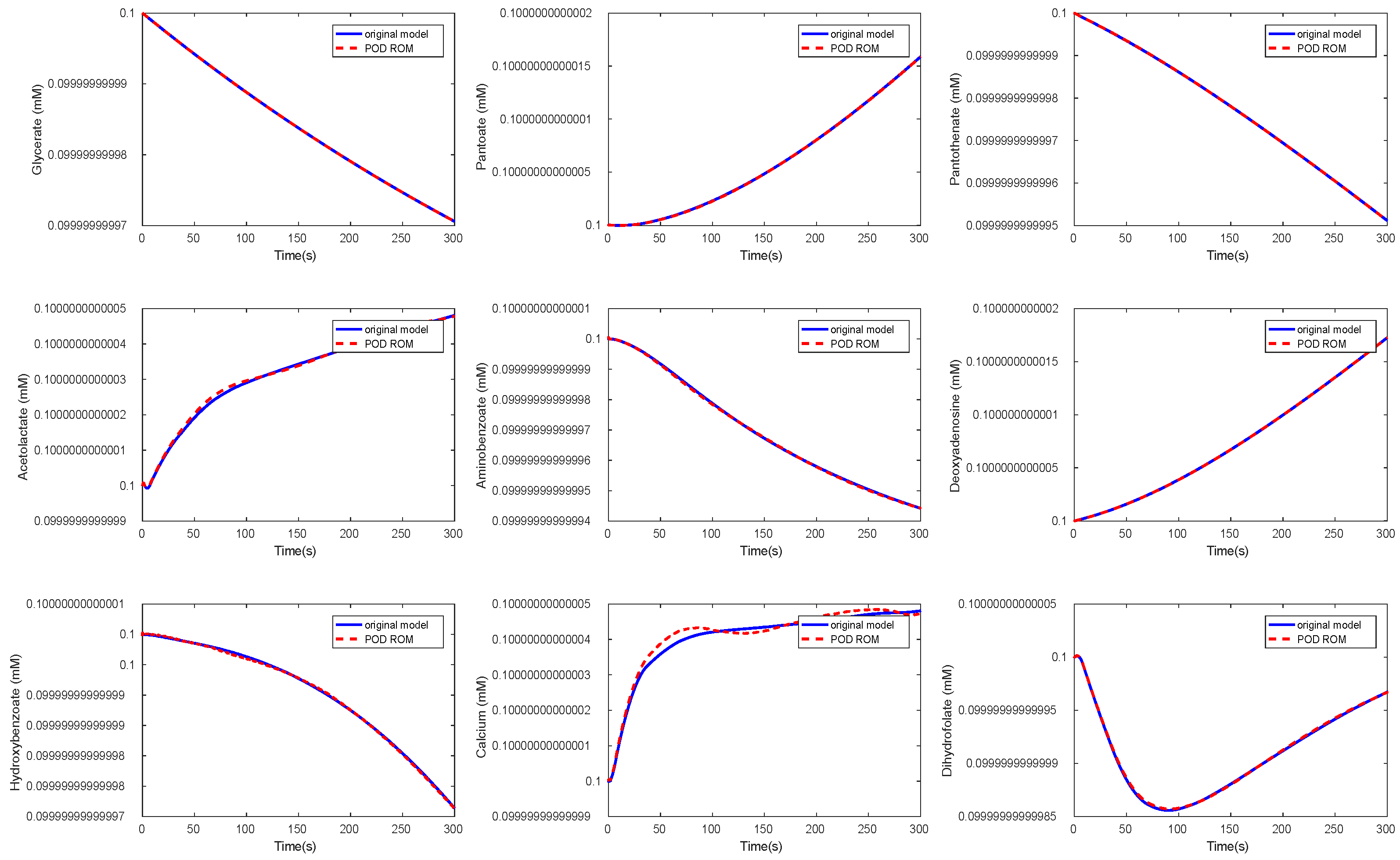
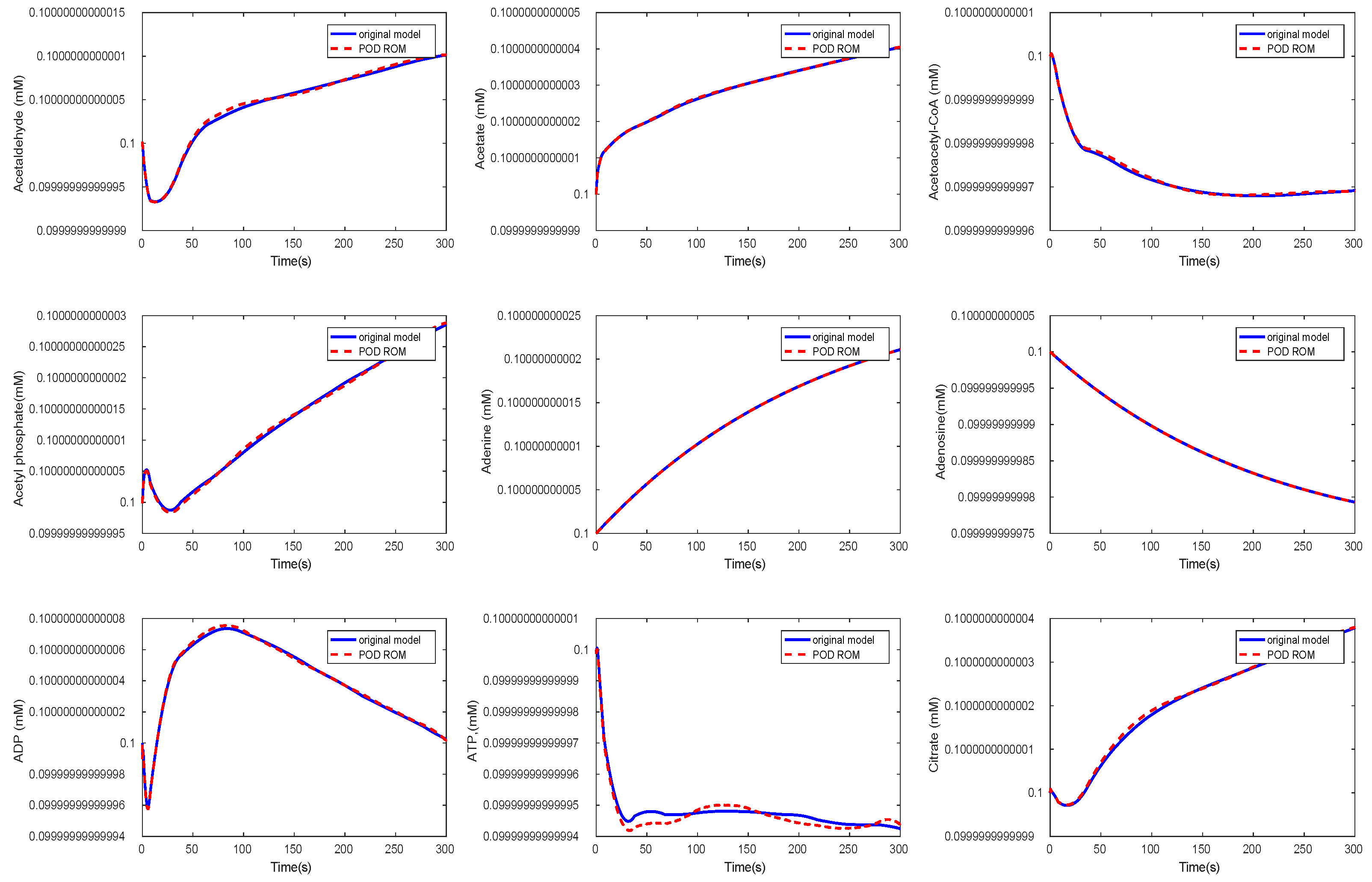
3.3. Kinetic Model of the E. coli Metabolic Network
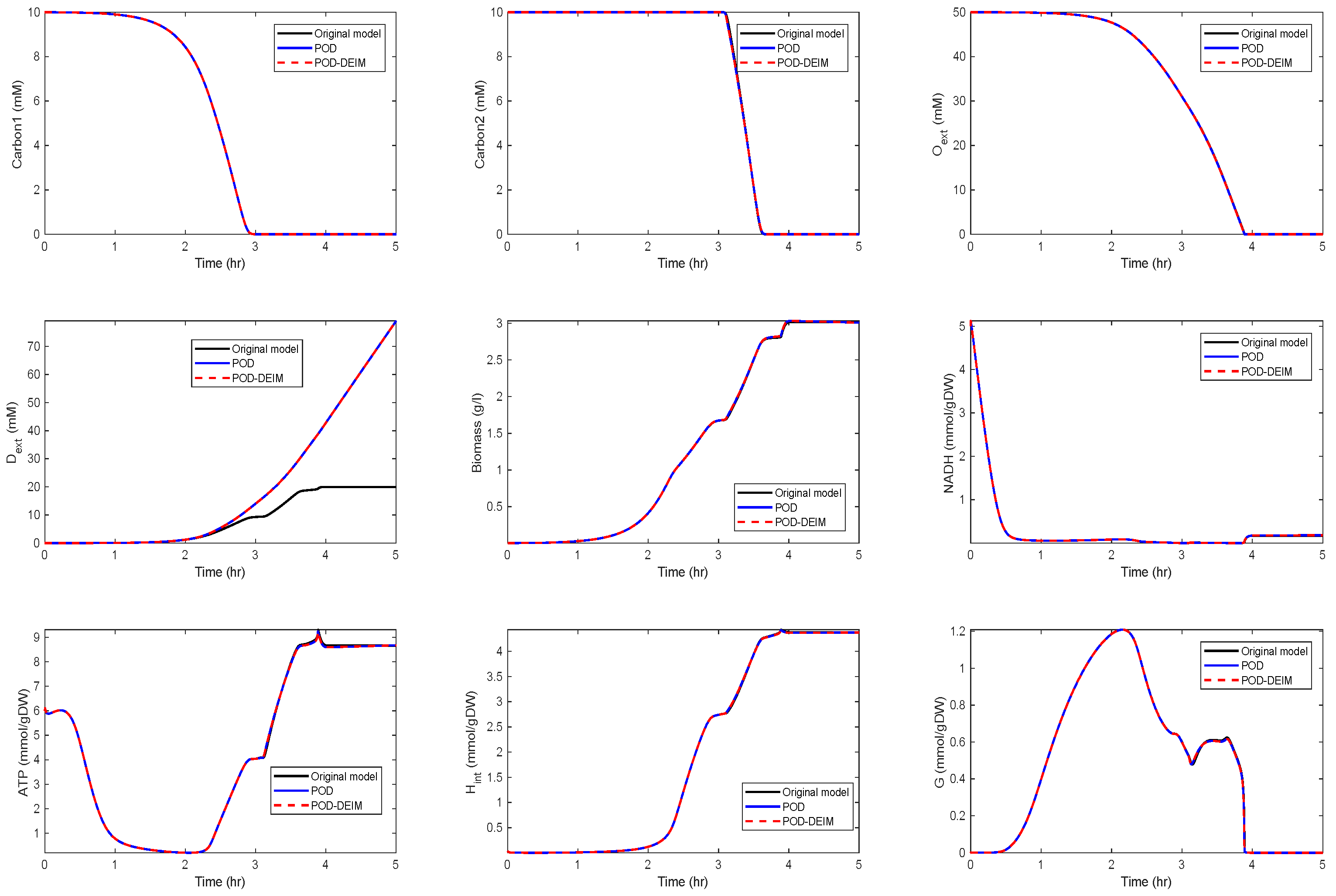
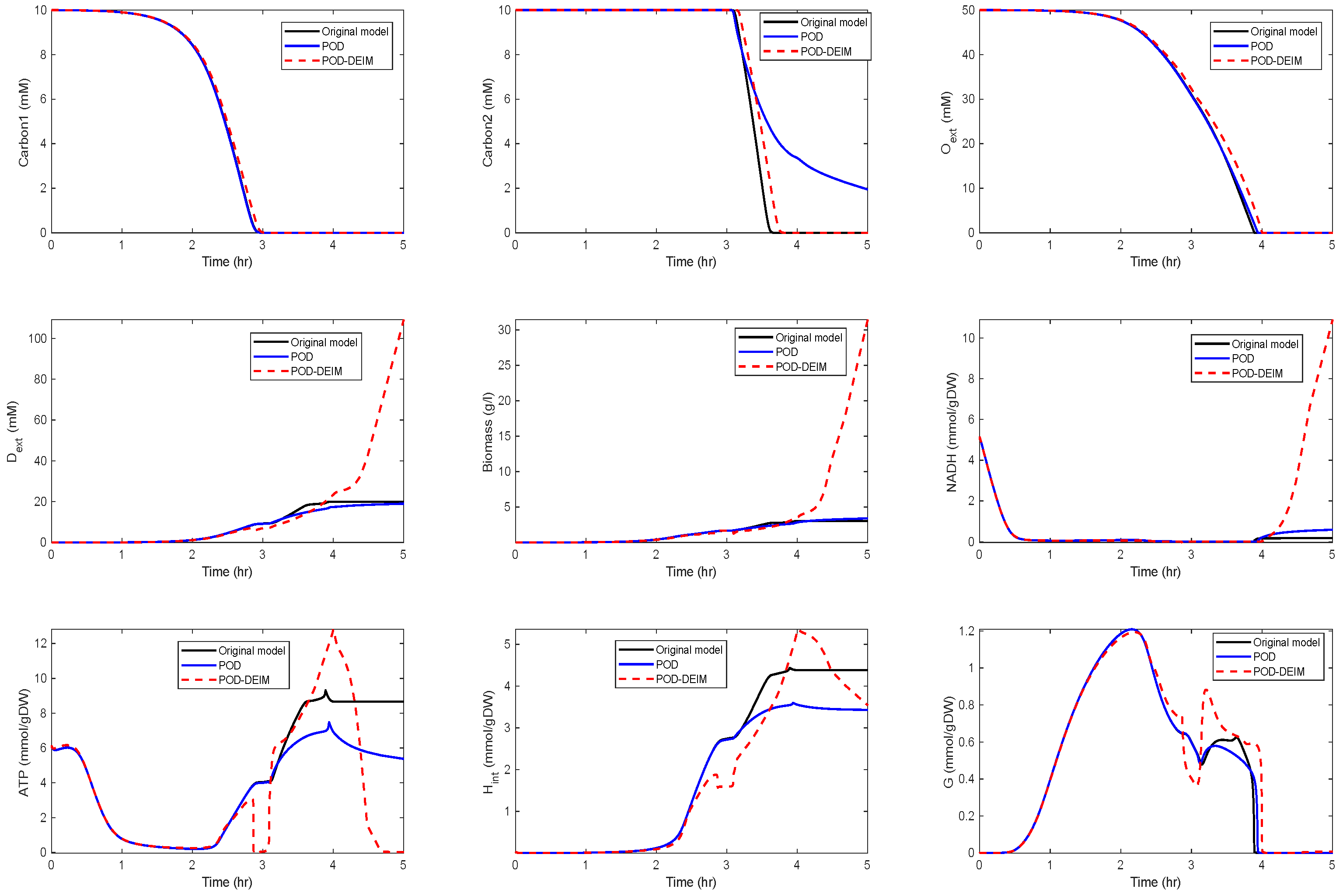
4. POD for Kinetic Models With Different Initial Conditions
Application to the Kinetic Model of a Metabolic-Genetic Network
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Derivation of Kinetic Model Equations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulatory Proteins | Transcriptional Regulation | ||
|---|---|---|---|
| IF () | IF NOT ( ) | ||
| IF () | IF NOT ( ) | ||
| IF () | IF NOT ( ) | ||
| IF Not () | IF NOT () | ||
Appendix A.2. Model Fitting
| Constant Rates | Value | Unit | |
|---|---|---|---|
| mM | estimated | ||
| mM | estimated | ||
| mM | estimated | ||
| mM | estimated | ||
| mmol/gDW | estimated | ||
| mmol/gDW | estimated | ||
| 41 | mM | estimated | |
| 980 | mmol3/gDW3 · h−1 | estimated | |
| 1000 | mmol2/gDW2 · h−1 | estimated | |
| 300 | mmol/gDW · h−1 | estimated | |
| 140 | mmol3/gDW3 · h−1 | estimated | |
| 23 | mmol/gDW · h−1 | estimated | |
| 25 | mmol/gDW · h−1 | estimated | |
| mmol/gDW· h−1 | estimated | ||
| 1000 | mmol/gDW· h−1 | estimated | |
| 13 | mmol/gDW· h−1 | estimated | |
| mmol2/gDW2· h−1 | estimated | ||
| 150 | mmol3/gDW3 · h−1 | estimated | |
| mmol/gDW· h−1 | estimated | ||
| 170 | mmol2/gDW2 · h−1 | estimated | |
| mM | estimated | ||
| mmol/gDW | estimated | ||
| mM | estimated | ||
| 290 | mmol/gDW | estimated | |
| 1 | gDW/mmol | assumed |
References
- Gerdtzen, Z.P.; Daoutidis, P.; Hu, W.-S. Non-linear reduction for kinetic models of metabolic reaction networks. Metab. Eng. 2004, 6, 140–154. [Google Scholar] [CrossRef]
- Briggs, G.E.; Sanderson Haldane, J.B. A note on the kinetics of enzyme action. Biochem. J. 1925, 19, 338. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M.L. Die Kinetik der Invertinwirkung. Biochem Z 1913, 49, 333–369. [Google Scholar]
- Nelson, D.L.; Cox, M.M. Hormonal regulation of food metabolism. In Lehninger Principles of Biochemistry, 4th ed.; WH Freeman: New York, NY, USA, 2005; pp. 881–992. [Google Scholar]
- Sontag, E.D. Lecture Notes on Mathematical Systems Biology; Northeastern University: Boston, MA, USA, 2005. [Google Scholar]
- Snowden, T.J.; van der Graaf, P.H.; Tindall, M.J. Methods of model reduction for large-scale biological systems: A survey of current methods and trends. Bull. Math. Biol. 2017, 79, 1449–1486. [Google Scholar] [CrossRef]
- Costa, R.S.; Rocha, I.; Ferreira, E.C. Model Reduction Based on Dynamic Sensitivity Analysis: A Systems Biology Case of Study. 2008. Available online: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.619.3225 (accessed on 8 May 2020).
- Zi, Z. Sensitivity analysis approaches applied to systems biology models. IET Syst. Biol. 2011, 5, 336–346. [Google Scholar] [CrossRef]
- Kuo, J.C.W.; Wei, J. Lumping analysis in monomolecular reaction systems. analysis of approximately lumpable system. Ind. Eng. Chem. Fund. 1969, 8, 124–133. [Google Scholar] [CrossRef]
- Wei, J.; Kuo, J.C.W. Lumping analysis in monomolecular reaction systems. analysis of the exactly lumpable system. Ind. Eng. Chem. Fund. 1969, 8, 114–123. [Google Scholar] [CrossRef]
- Okeke, B.E. Lumping Methods for Model Reduction. Ph.D. Thesis, University of Lethbridge, Lethbridge, AB, Canada, 2013. [Google Scholar]
- Pepiot-Desjardins, P.; Pitsch, H. An automatic chemical lumping method for the reduction of large chemical kinetic mechanisms. Combust. Theory Model. 2008, 12, 1089–1108. [Google Scholar] [CrossRef]
- Okino, M.S.; Mavrovouniotis, M.L. Simplification of mathematical models of chemical reaction systems. Chem. Rev. 1998, 98, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Flach, E.; Schnell, S. Use and abuse of the quasi-steady-state approximation. IEE Proc.-Syst. Biol. 2006, 153, 187–191. [Google Scholar] [CrossRef]
- Khalil, H.K.; Grizzle, J.W. Nonlinear Systems; Prentice Hall: Upper Saddle River, NJ, USA, 2002; p. 3. [Google Scholar]
- Tikhonov, A.N. Systems of differential equations containing small parameters in the derivatives. Matematicheskii Sbornik 1952, 73, 575–586. [Google Scholar]
- Kuntz, J.; Oyarzún, D.; Stan, G.-B. Model reduction of genetic-metabolic networks via time scale separation. In A Systems Theoretic Approach to Systems and Synthetic Biology I: Models and System Characterizations; Springer: Berlin, Germany, 2014; pp. 181–210. [Google Scholar]
- Duan, Z.; Cruz Bournazou, M.N.; Kravaris, C. Dynamic model reduction for two-stage anaerobic digestion processes. Chem. Eng. J. 2017, 327, 1102–1116. [Google Scholar] [CrossRef]
- Belgacem, I.; Casagranda, S.; Grac, E.; Ropers, D.; Gouzé, J.-L. Reduction and stability analysis of a transcription–translation model of RNA polymerase. Bull. Math. Biol. 2018, 80, 294–318. [Google Scholar] [CrossRef] [PubMed]
- Karhunen, K. Über Lineare Methoden in der Wahrscheinlichkeitsrechnung. Sana 1947, 37. [Google Scholar]
- Loeve, M. Elementary probability theory. In Probability Theory I; Springer: Berlin, Germany, 1977; pp. 1–52. [Google Scholar]
- Brunton, S.L.; Kutz, J.N. Data-Driven Science and Engineering: Machine Learning, Dynamical Systems, and Control; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Golub, G.H.; Reinsch, C. Singular value decomposition and least squares solutions. In Linear Algebra; Springer: Berlin, Germany, 1971; pp. 134–151. [Google Scholar]
- Strang, G. Introduction to Linear Algebra; Wellesley-Cambridge Press: Wellesley, MA, USA, 1993; Volume 3. [Google Scholar]
- Chaturantabut, S.; Sorensen, D. Nonlinear model reduction via discrete empirical interpolation. SIAM J. Sci. Comput. 2010, 32, 2737–2764. [Google Scholar] [CrossRef]
- Covert, M.W.; Schilling, C.H.; Palsson, B. Regulation of gene expression in flux balance models of metabolism. J. Theor. Biol. 2001, 213, 73–88. [Google Scholar] [CrossRef]
- Beattieand, C.; Gugercin, S.; Mehrmann, V. Model reduction for systems with inhomogeneous initial conditions. Syst. Control Lett. 2017, 99, 99–166. [Google Scholar] [CrossRef]
- Chaturantabut, S.; Sorensen, D. A state space error estimate for POD-DEIM nonlinear model reduction. SIAM J. Numer. Anal. 2012, 50, 46–63. [Google Scholar] [CrossRef]
- Afanasiev, K.; Hinze, M. Adaptive control of a wake flow using proper orthogonal decomposition1. Lect. Notes Pure Appl. Math. 2001, 1, 216. [Google Scholar] [CrossRef]
- Stanford, N.J.; Lubitz, T.; Smallbone, K.; Klipp, E.; Mendes, P.; Liebermeister, W. Systematic Construction of Kinetic Models from Genome-Scale Metabolic Networks. PLoS ONE 2013, 8, e79195. [Google Scholar] [CrossRef]
- Smallbone, K.; Mendes, P. Large-scale metabolic models: From reconstruction to differential equations. Ind. Biotechnol. 2013, 9, 179–184. [Google Scholar] [CrossRef]
- Badreddine, A.-A.; Henrik, P. Simulation of switching phenomena in biological systems. In Biochemical Engineering for 2001; Springer: Berlin, Germany, 1992; pp. 701–704. [Google Scholar]
- Kremling, A.; Kremling, S.; Bettenbrock, K. Catabolite repression in escherichia coli—a comparison of modelling approaches. FEBS J. 2009, 276, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Rosa, C.S.; Boris, L.; Rudy, E. Stability Preservation in Projection-based Model Order Reduction of Large Scale Systems. Eur. J. Control 2012, 18, 122–132. [Google Scholar]
- Roland, P. Stability preservation in Galerkin-type projection-based model order reduction. arXiv 2017, arXiv:1711.02912. [Google Scholar]
- Gesztelyi, R.; Zsuga, J.; Kemeny-Beke, A.; Varga, B.; Juhasz, B.; Tosaki, A. The Hill Equation and the Origin of Quantitative Pharmacology; Springer: Berlin, Germany, 2012; Volume 66, pp. 427–438. [Google Scholar]
- Hill, A.V. The possible effects of the aggregation of the molecules of hæmoglobin on its dissociation curves. J. Physiol. 1910, 40, i–vii. [Google Scholar]
- Guldberg, C.M.; Waage, P. Etudes sur Les Affinités Chimiques; Brøgger & Christie: Oslo, Norway, 1867. [Google Scholar]
- Raue, A.; Steiert, B.; Schelker, M.; Kreutz, C.; Maiwald, T.; Hass, H.; Vanlier, J.; Tönsing, C.; Adlung, L.; Engesser, R.; et al. Data2Dynamics: A modeling environment tailored to parameter estimation in dynamical systems. Bioinformatics 2015, 31, 3558–3560. [Google Scholar] [CrossRef]
- Raue, A.; Schilling, M.; Bachmann, J.; Matteson, A.; Schelke, M.; Kaschek, D.; Hug, S.; Kreutz, C.; Harms, B.D.; Theis, F.J.; et al. Lessons Learned from Quantitative Dynamical Modeling in Systems Biology. PLoS ONE 2013, 8, 1932–6203. [Google Scholar] [CrossRef]















| Original Model | POD ROM | POD-DEIM ROM | |
|---|---|---|---|
| Scenario 1 (, ) | 0.3 s | 1.4 s | 0.7 s |
| Scenario 2 (, ) | 0.3 s | 1.4 s | 0.6 s |
| Scenario 3 (, ) | 0.3 s | 1.9 s | 0.9 s |
| Scenario 4 (, ) | 0.3 s | 1.9 s | 0.9 s |
| Original Model | POD ROM | |
|---|---|---|
| Scenario 1 () | 0.28 s | 0.20 s |
| Scenario 2 () | 0.28 s | 0.46 s |
| Original Model | POD ROM | |
|---|---|---|
| Scenario () | 0.10 s | 0.014 s |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali Eshtewy, N.; Scholz, L. Model Reduction for Kinetic Models of Biological Systems. Symmetry 2020, 12, 863. https://doi.org/10.3390/sym12050863
Ali Eshtewy N, Scholz L. Model Reduction for Kinetic Models of Biological Systems. Symmetry. 2020; 12(5):863. https://doi.org/10.3390/sym12050863
Chicago/Turabian StyleAli Eshtewy, Neveen, and Lena Scholz. 2020. "Model Reduction for Kinetic Models of Biological Systems" Symmetry 12, no. 5: 863. https://doi.org/10.3390/sym12050863
APA StyleAli Eshtewy, N., & Scholz, L. (2020). Model Reduction for Kinetic Models of Biological Systems. Symmetry, 12(5), 863. https://doi.org/10.3390/sym12050863