Genetic Similarity Analysis Based on Positive and Negative Sequence Patterns of DNA



Abstract
1. Introduction
2. Related Work
2.1. Pattern Similarity Analysis of Biological Sequences
2.2. Biological Sequential Pattern Mining Technique
3. Basic Principles
3.1. Definition
3.2. Data Sets of DNA Sequence
3.3. Similarity Distances
3.4. Output Data
4. F-NSP Algorithm Based on Biological Sequences
4.1. Preprocessing
4.2. The Main Idea and Data Structure of f-NSP
- (1)
- the GSP algorithm is used in order to obtain all positive frequent sequences, and the bitmap corresponding to each sequence is stored in a hash table;
- (2)
- corresponding NSCs based on all the positive sequences are generated; and,
- (3)
- support of NSCs can be calculated by bit operation. If the support of a NSC is greater than min_sup, then it is a frequent sequential pattern;
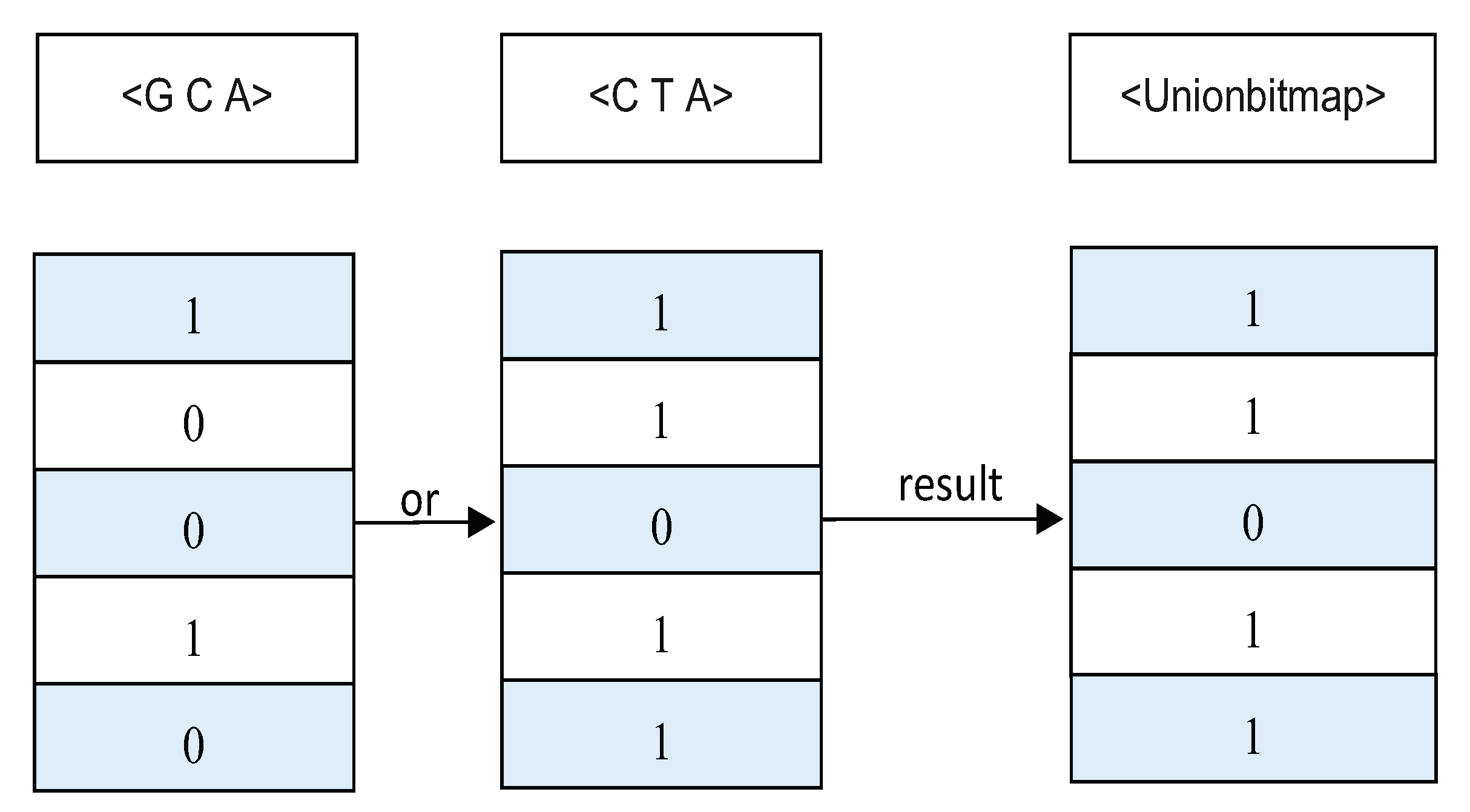
4.3. Calculating the Supports of Negative Sequences in f-NSP
4.4. The f-NSP Algorithm
| Algorithm 1:f-NSP Algorithm. |
|
5. Similarity Analysis of Negative DNA Sequences
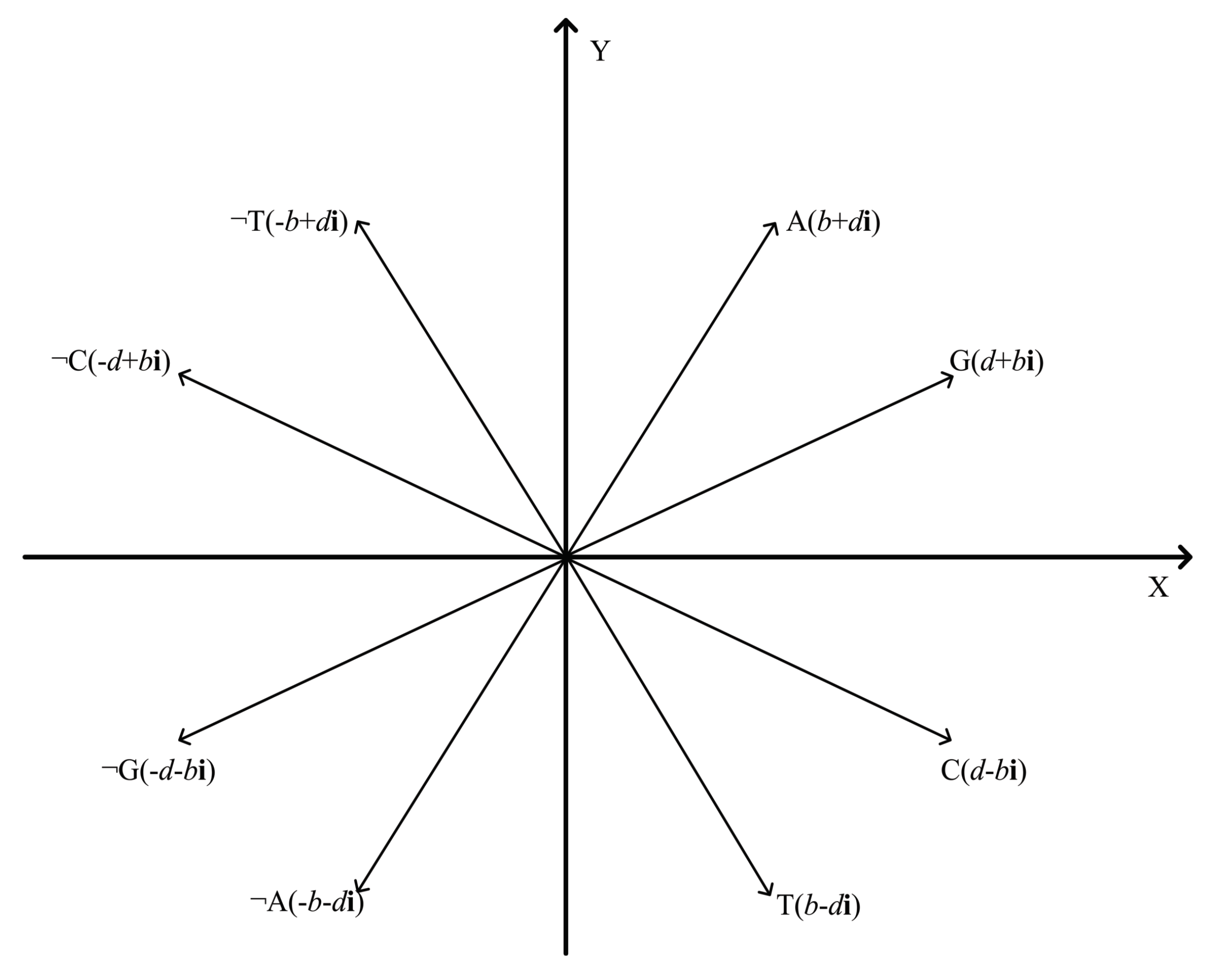
5.1. 2-D Representation of Negative DNA Sequential Patterns
5.2. Algorithm Principle of DTW Distance
5.3. Similarity Analysis of Negative DNA Sequences
6. Experiment Results
6.1. Experiment Data Set
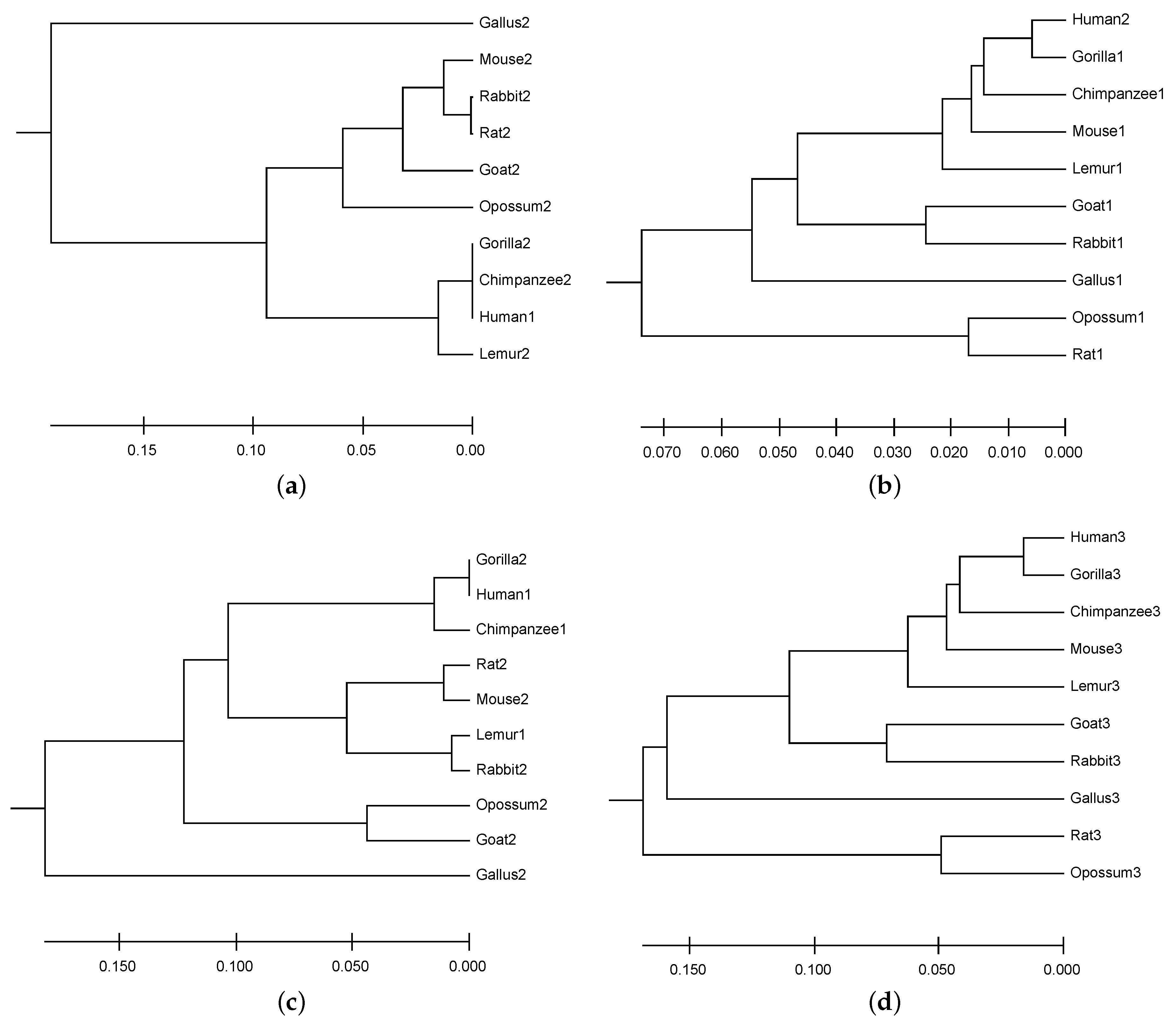
6.2. Result of Mining Patterns
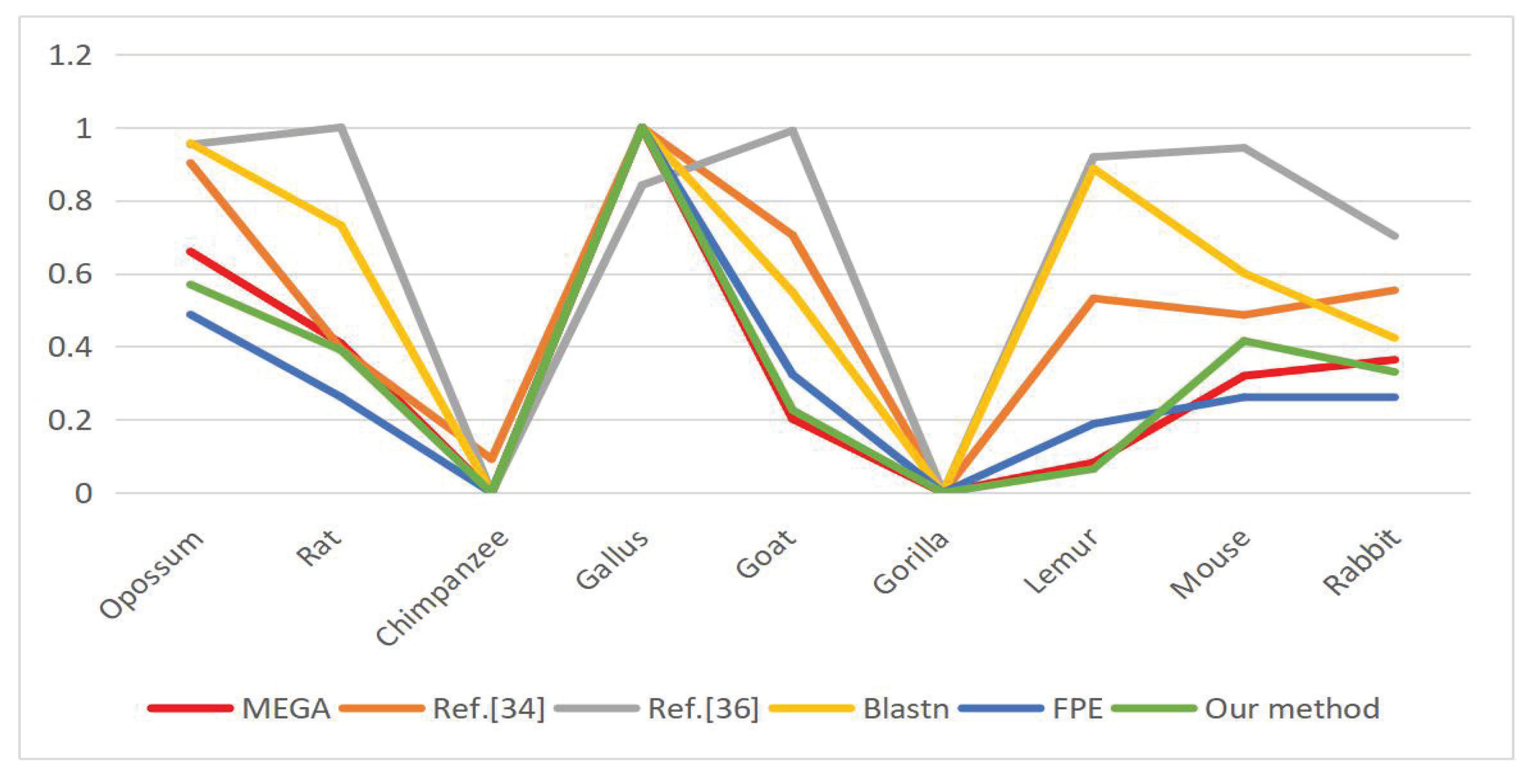
6.3. DNA Sequence Similarity Analysis
7. Conclusions Future Work
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PSPs | Positive Sequential Patterns |
| NSPs | Negative Sequential Patterns |
| NSC | (Negative Sequential Candidate) |
References
- Zhang, W.; Wang, X.; Huang, Z. A System of Mining Semantic Trajectory Patterns from GPS Data of Real Users. Sysmmetry 2019, 11, 889. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhang, C.; Shi, Y. Mining Contiguous Sequential Generators in Biological Sequences. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Matloob, I.; Khan, S.; Rehman, H. Sequence Mining and Prediction-Based Healthcare Fraud Detection Methodology. IEEE Access 2020, 8, 143256–143273. [Google Scholar] [CrossRef]
- Cao, L.; Yu, P.; Kumar, V. Nonoccurring Behavior Analytics: A New Area. IEEE Intell. Syst. 2015, 30, 4–11. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, T.; Dong, X. Campus Data Analysis Based on Positive and Negative Sequential Patterns. Int. J. Pattern Recognit. Artif. Intell. 2019, 33. [Google Scholar] [CrossRef]
- Cao, L.; Dong, X.; Zheng, Z. e-NSP: Efficient negative sequential pattern mining. Artif. Intell. 2016, 235, 156–182. [Google Scholar] [CrossRef]
- Dong, X.; Gong, Y.; Cao, L. F-NSP+: A fast negative sequential patterns mining method with self-adaptive data storage. Pattern Recognit. 2018, 84, 13–27. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Paterson, A.; Freeling, M.; Tang, H.; Wang, X. Insights from the Comparison of Plant Genome Sequences. Annu. Rev. Plant Biol. 2010, 61, 349–372. [Google Scholar] [CrossRef]
- Eugene, H.; John, R. A novel method of representation of nucleotide series especially suited for long DNA sequences. J. Biol. Chem. 1983, 258, 1318–1327. [Google Scholar]
- Liao, B.; Wang, T.; Huang, Z. New 2D graphical representation of DNA sequences. J. Comput. Chem. 2004, 25, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Fan, X. A geometric characterization of DNA sequence. Phys. A Stat. Mech. Its Appl. 2019, 527, 121429. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, T.; Huang, Z. A new method to analyze the similarity of the DNA sequences. Comput. Theor. Chem. 2008, 853, 62–67. [Google Scholar] [CrossRef]
- Ma, T.; Liu, Y.; Dai, Q.; Yao, Y.; He, P. A graphical representation of protein based on a novel iterated function system. Phys. A Stat. Mech. Its Appl. 2014, 403, 21–28. [Google Scholar] [CrossRef]
- Lee, S.; Cha, J.; Theera-Umpon, N.; Kim, K. Analysis of a Similarity Measure for Non-Overlapped Data. Symmetry 2017, 9, 68. [Google Scholar] [CrossRef]
- Xie, G.; Jin, X.; Yang, C.; Pu, J.; Mo, Z. Graphical Representation and Similarity Analysis of DNA Sequences Based on Trigonometric Functions. Acta Biotheor. 2018, 66, 113–133. [Google Scholar] [CrossRef]
- Aboelkhier, M.; Abdelwahaab, M.; Aboelmaaty, M. Measuring Similarity among Protein Sequences Using a New Descriptor. BioMed Res. Int. 2019, 2019, 2796971. [Google Scholar]
- Jafarzadeh, N.; Iranmanesh, A. C-curve: A novel 3D graphical representation of DNA sequence based on codons. Math. Biosci. 2013, 241, 217–224. [Google Scholar] [CrossRef]
- Liao, B.; Xiang, Q.; Cai, L.; Cao, Z. A new graphical coding of DNA sequence and its similarity calculation. Phys. A Stat. Mech. Its Appl. 2013, 392, 4663–4667. [Google Scholar] [CrossRef]
- Olivier, D.; Eric, R. STAR: An algorithm to Search for Tandem Approximate Repeats. Bioinformatics 2004, 20, 2812–2820. [Google Scholar]
- Kurtz, S.; Choudhuri, J.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Chen, X.; Li, S.; Xiong, C. Frequent Patterns Mining in DNA Sequence. IEEE Access 2019, 7, 108400–108410. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Zhang, M.; Yu, X.; Yu, X.; Guo, W.; Zeng, T.; Chen, L. Efficient Mining Multi-mers in a Variety of Biological Sequences. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 17, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, J.; Lin, M.; Chen, C. Mining Negative Sequential Patterns for E-commerce Recommendations. In Proceedings of the 3rd IEEE Asia-Pacific Service Computing Conference, Yilan, Taiwan, 9–12 December 2008; pp. 1213–1218. [Google Scholar]
- Zheng, Z.; Zhao, Y.; Zuo, Y.; Cao, L. Negative-GSP: An efficient method for mining negative sequential patterns. In Proceedings of the 8th Australasian Data Mining Conference, Melbourne, Australia, 1–4 December 2009; pp. 63–67. [Google Scholar]
- Rastogi, V.; Khare, V. Apriori Based: Mining Positive and Negative Frequent Sequential Patterns. Int. J. Latest Trends Eng. Technol. 2012, 1, 24–33. [Google Scholar]
- Khare, V.; Rastogi, V. Mining Positive and Negative Sequential Pattern in Incremental Transaction Databases. Int. J. Comput. Appl. 2013, 71, 18–22. [Google Scholar]
- Lin, N.; Chen, H.; Hao, H.; Wei, H. Mining negative sequential patterns. In Proceedings of the 6th WSEAS International Conference on Applied Computer Science, Corfu, Greece, 16–19 February 2007; pp. 654–658. [Google Scholar]
- Dong, X.; Gong, Y.; Cao, L. e-RNSP: An Efficient Method for Mining Repetition Negative Sequential Patterns. IEEE Trans. Cybern. 2020, 50, 2084–2096. [Google Scholar] [CrossRef]
- Dong, X.; Qiu, P.; Lu, J.; Cao, L.; Xu, T. Mining Top-k Useful Negative Sequential Patterns via Learning. IEEE Trans. Neural Netw. Learn. Syst. 2019, 30, 2764–2778. [Google Scholar] [CrossRef]
- Xie, X.; Guan, J.; Zhou, S. Similarity evaluation of DNA sequences based on frequent patterns and entropy. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef]
- Jin, X.; Jiang, Q.; Chen, Y.; Lee, S.; Nie, R.; Yao, S.; Zhou, D.; He, K. Similarity/dissimilarity calculation methods of DNA sequences: A survey. J. Mol. Graph. Model. 2017, 76, 342–355. [Google Scholar] [CrossRef]
- Bai, F.; Wang, T. A 2-D graphical representation of protein sequences based on nucleotide triplet codons. Chem. Phys. Lett. 2005, 413, 458–462. [Google Scholar] [CrossRef]
- Abd Elwahaab, M.A.; Abo-Elkhier, M.M.; Abo el Maaty, M.I. A Statistical Similarity/Dissimilarity Analysis of Protein Sequences Based on a Novel Group Representative Vector. BioMed Res. Int. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Zhu, W.; Sun, Y.; Xiang, Q.; Zheng, M.; Chen, M.; Li, Z. One novel representation of DNA sequence based on the global and local position information. Sci. Rep. 2018, 8, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.; Madden, T.; Schäffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, D. Graphical representation for DNA sequences via joint diagonalization of matrix pencil. IEEE J. Biomed. Health Inform. 2013, 17, 503–511. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methodsn. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sid | Data Sequence |
|---|---|
| 1 | <ATCTG> |
| 2 | <GGACCT> |
| 3 | <CAGTC> |
| 4 | <AGTCA> |
| 5 | <CCA> |
| Data Set | Code Sequences |
|---|---|
| Human | ATGGTGCACCTGACTCCTGAGGAGAAGTCTGCCGTTACTGCCCTGTGGGGCAAGGTGAACGTGGATTAAGTTGGTGGTGAGGCCCTGGGCAG |
| Opossum | ATGGTGCACTTGACTTCTGAGGAGAAGAACTGCATCACTACCATCTGGTCTAAGGTGCAGGTTGACCAGACTGGTGGTGAGGCCCTTGGCAG |
| Rat | ATGGTGCACCTAACTGATGCTGAGAAGGCTACTGTTAGTGGCCTGTGGGGAAAGGTGAACCCTGATAATGTTGGCGCTGAGGCCCTGGGCAG |
| Chimpanzee | ATGGTGCACCTGACTCCTGAGGAGAAGTCTGCCGTTACTGCCCTGTGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGTTGGTATCAAGG |
| Gallus | ATGGTGCACTGGACTGCTGAGGAGAAGCAGCTCATCACCGGCCTCTGGGGCAAGGTCAATGTGGCCGAATGTGGGGCCGAAGCCCTGGCCAG |
| Goat | ATGCTGACTGCTGAGGAGAAGGCTGCCGTCACCGGCTTCTGGGGCAAGGTGAAAGTGGATGAAGTTGGTGCTGAGGCCCTGGGCAG |
| Gorilla | ATGGTGCACCTGACTCCTGAGGAGAAGTCTGCCGTTACTGCCCTGTGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGG |
| Lemur | ATGACTTTGCTGAGTGCTGAGGAGAATGCTCATGTCACCTCTCTGTGGGGCAAGGTGGATGTAGAGAAAGTTGGTGGCGAGGCCTTGGGCAG |
| Mouse | ATGGTGCACCTGACTGATGCTGAGAAGTCTGCTGTCTCTTGCCTGTGGGCAAAGGTGAACCCCGATGAAGTTGGTGGTGAGGCCCTGGGCAGG |
| Rabbit | ATGGTGCATCTGTCCAGTGAGGAGAAGTCTGCGGTCACTGCCCTGTGGGGCAAGGTGAATGTGGAAGAAGTTGGTGGTGAGGCCCTGGGC |
| No. | Species | GenBank Accession Number | Location | Nucleotide Length |
|---|---|---|---|---|
| 1 | Human | U01317 | 62,187–62,278 | 92 |
| 2 | Opossum | J03643 | 467–558 | 92 |
| 3 | Rat | X06701 | 310–401 | 92 |
| 4 | Chimpanzee | X02345 | 4189–4293 | 104 |
| 5 | Gallus | V00409 | 465–556 | 92 |
| 6 | Goat | M15387 | 279–364 | 86 |
| 7 | Gorilla | X61109 | 4538–4630 | 93 |
| 8 | Lemur | M15734 | 154–245 | 92 |
| 9 | Mouse | V00722 | 259–367 | 93 |
| 10 | Rabbit | V00882 | 277–366 | 90 |
| Data Set | Frequent Pattern | Data Set | Frequent Pattern |
|---|---|---|---|
| Human1 (Hum1) | G T G G A G | Goat (Goa1) | G G T G G T |
| Human2 (Hum2) | G G G G G A | Goat (Goa2) | G C C T G C |
| Human3 (Hum3) | ¬A G T G ¬C G A ¬C G | Goat (Goa3) | C ¬A T G A ¬A G ¬A |
| Opossum1 (Opo1) | G G C G C A | Gorilla (Gor1) | G G G G C A |
| Opossum2 (Opo2) | G G C T T A | Gorilla (Gor2) | G T G G A G |
| Opossum3 (Opo3) | G G C ¬G G C A ¬G | Gorilla (Gor3) | ¬A G G G ¬C G A G |
| Rat1 | G C C T G A | Lemur (Lem1) | G G G G G C |
| Rat2 | G G T G G G | Lemur (Lem2) | G T G G C A |
| Rat3 | G C C ¬A T G A ¬C | Lemur (Lem3) | ¬A G T G ¬C G ¬T C A |
| Chimpanzee1 (Chi1) | G G G G A G | Mouse (Mou1) | T G G G G G |
| Chimpanzee2 (Chi2) | G T G G A G | Mouse (Mou2) | G G C C T G |
| Chimpanzee3 (Chi3) | ¬A G G G ¬C G A G | Mouse (Mou3) | G C C ¬A T G ¬A C |
| Gallus (Gal1) | G G C G C T | Rabbit (Rab1) | G G T G G C |
| Gallus (Gal2) | G G C T T C | Rabbit (Rab2) | C C T G A T |
| Gallus (Gal3) | G G C ¬G G G | Rabbit (Rab3) | ¬A T G A ¬C T G |
| Frequent PSPs | Hum1 | Hum2 | Opo1 | Opo2 | Rat1 | Rat2 | Chi1 | Chi2 | Gal1 | Gal2 | Goa1 | Goa2 | Gor1 | Gor2 | Lem1 | Lem2 | Mou1 | Mou2 | Rab1 | Rab2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hum1 | 0.0000 | X | 0.1451 | 0.2739 | 0.1319 | 0.1876 | 0.0310 | 0.0000 | 0.1021 | 0.4808 | 0.0902 | 0.1086 | 0.0115 | 0.0000 | 0.1288 | 0.0313 | 0.0462 | 0.1998 | 0.0731 | 0.1589 |
| Hum2 | 0.0000 | 0.1390 | 0.8654 | 0.1355 | 0.3845 | 0.0297 | 0.0992 | 0.0978 | 0.9575 | 0.0864 | 0.6786 | 0.0115 | 0.0121 | 0.0311 | 0.5149 | 0.0299 | 0.4719 | 0.0701 | 0.5361 | |
| Opo1 | 0.0000 | X.XXXX | 0.0339 | 0.0354 | 0.1510 | 0.1201 | 0.1690 | 0.0720 | 0.1594 | 0.0829 | 0.1497 | 0.1393 | 0.1560 | 0.1298 | 0.1535 | 0.1289 | 0.1675 | 0.1018 | ||
| Opo2 | 0.0000 | 0.5405 | 0.1163 | 0.2513 | 0.2739 | 0.7460 | 0.2069 | 0.5194 | 0.1653 | 0.8670 | 0.2739 | 0.2411 | 0.3052 | 0.4471 | 0.0741 | 0.3881 | 0.1150 | |||
| Rat1 | 0.0000 | X | 0.1374 | 0.1274 | 0.1527 | 0.0446 | 0.1399 | 0.1074 | 0.1360 | 0.1295 | 0.1387 | 0.1147 | 0.1385 | 0.1419 | 0.1429 | 0.0970 | ||||
| Rat2 | 0.0000 | 0.2081 | 0.1576 | 0.8291 | 0.3232 | 0.5142 | 0.0503 | 0.3871 | 0.1576 | 0.1557 | 0.1889 | 0.2025 | 0.0422 | 0.3168 | 0.0013 | |||||
| Chi1 | 0.0000 | X | 0.1090 | 0.1137 | 0.0774 | 0.0808 | 0.0276 | 0.0288 | 0.0443 | 0.0462 | 0.0411 | 0.0523 | 0.0764 | 0.0798 | ||||||
| Chi2 | 0.0000 | 0.9591 | 0.4808 | 0.7322 | 0.1086 | 0.0937 | 0.0000 | 0.5479 | 0.0313 | 0.5200 | 0.1998 | 0.5720 | 0.1589 | |||||||
| Gal1 | 0.0000 | X | 0.1388 | 0.1449 | 0.0979 | 0.1021 | 0.0829 | 0.0865 | 0.1296 | 0.1353 | 0.1083 | 0.1130 | ||||||||
| Gal2 | 0.0000 | 1.0320 | 0.3722 | 0.9600 | 0.4808 | 0.3009 | 0.5121 | 0.8288 | 0.2810 | 0.6227 | 0.3219 | |||||||||
| Goa1 | 0.0000 | X | 0.0873 | 0.0911 | 0.1052 | 0.1098 | 0.1332 | 0.0480 | 0.0488 | 0.0509 | ||||||||||
| Goa2 | 0.0000 | 0.0641 | 0.1086 | 0.2352 | 0.1399 | 0.3309 | 0.0912 | 0.4349 | 0.0503 | |||||||||||
| Gor1 | 0.0000 | X | 0.0274 | 0.0286 | 0.0278 | 0.0498 | 0.0732 | 0.0764 | ||||||||||||
| Gor2 | 0.0000 | 0.2123 | 0.0313 | 0.4702 | 0.1998 | 0.5354 | 0.1589 | |||||||||||||
| Lem1 | 0.0000 | X | 0.0696 | 0.0727 | 0.0913 | 0.0953 | ||||||||||||||
| Lem2 | 0.0000 | 0.3877 | 0.2311 | 0.2671 | 0.1902 | |||||||||||||||
| Mou1 | 0.0000 | X | 0.1342 | 0.0461 | ||||||||||||||||
| Mou2 | 0.0000 | 0.1572 | 0.0096 | |||||||||||||||||
| Rab1 | 0.0000 | X | ||||||||||||||||||
| Rab2 | 0 |
| Frequent PSPs | Hum3 | Opo3 | Rat3 | Chi3 | Gal3 | Goa3 | Gor3 | Lem3 | Mou3 | Rab3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Hum3 | 0.0000 | 0.4038 | 0.3671 | 0.0862 | 0.2843 | 0.2511 | 0.0321 | 0.0902 | 0.0886 | 0.2035 |
| Opo3 | 0.0000 | 0.0985 | 0.3341 | 0.3403 | 0.2307 | 0.3878 | 0.3613 | 0.3587 | 0.2832 | |
| Rat3 | 0.0000 | 0.3544 | 0.3240 | 0.2989 | 0.3602 | 0.3191 | 0.3950 | 0.2700 | ||
| Chi3 | 0.0000 | 0.3165 | 0.2248 | 0.0803 | 0.1286 | 0.1156 | 0.2220 | |||
| Gal3 | 0.0000 | 0.4034 | 0.2841 | 0.2408 | 0.3766 | 0.3145 | ||||
| Goa3 | 0.0000 | 0.2537 | 0.3055 | 0.1336 | 0.1417 | |||||
| Gor3 | 0.0000 | 0.0795 | 0.0787 | 0.2127 | ||||||
| Lem3 | 0.0000 | 0.2023 | 0.2653 | |||||||
| Mou3 | 0.0000 | 0.1283 | ||||||||
| Rab3 | 0.0000 |
| Opo- Ssum | Rat | Chimp- Anzee | Gallus | Goat | Gorilla | Lemur | Mouse | Rabbit | CORREL | |
|---|---|---|---|---|---|---|---|---|---|---|
| MEGA | 0.4823 (0.6600) | 0.2985 (0.0000) | 0.0000 (0.0000) | 0.7308 (1.0000) | 0.1599 (0.2188) | 0.0000 (0.0000) | 0.0600 (0.0821) | 0.2336 (0.3197) | 0.2659 (0.3639) | X |
| Z.Y.Mo. [35] | 0.2696 (0.9026) | 0.1198 (0.3939) | 0.0309 (0.0920) | 0.2983 (1.0000) | 0.2114 (0.7049) | 0.0038 (0.0000) | 0.1604 (0.5318) | 0.1470 (0.4863) | 0.1670 (0.5542) | 0.8327 |
| H.J.Yu. [36] | 25.9952 (0.9531) | 27.0102 (1.0000) | 5.3704 (0.0000) | 23.5869 (0.8418) | 26.8209 (0.9913) | 5.3704 (0.0000) | 25.2515 (0.9187) | 25.8007 (0.9441) | 20.5706 (0.7024) | 0.5294 |
| Blastn | 0.9567 (0.9567) | 0.7315 (0.7315) | 0.0000 (0.0000) | 1.0000 (1.0000) | 0.5486 (0.5486) | 0.0000 (0.0000) | 0.8874 (0.8874) | 0.6008 (0.6008) | 0.4235 (0.4235) | 0.7323 |
| FPE | 0.1237 (0.4876) | 0.0664 (0.2617) | 0.0000 (0.0000) | 0.2537 (1.0000) | 0.0820 (0.3232) | 0.0000 (0.0000) | 0.0478 (0.1884) | 0.0663 (0.2613) | 0.0664 (0.2617) | 0.9505 |
| Our method | 0.2739 (0.5697) | 0.1876 (0.3902) | 0.0000 (0.0000) | 0.4808 (1.0000) | 0.1086 (0.2259) | 0.0000 (0.0000) | 0.0313 (0.0651) | 0.1998 (0.4156) | 0.1589 (0.3305) | 0.9887 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Zhao, L.; Li, Z.; Dong, X. Genetic Similarity Analysis Based on Positive and Negative Sequence Patterns of DNA. Symmetry 2020, 12, 2090. https://doi.org/10.3390/sym12122090
Lu Y, Zhao L, Li Z, Dong X. Genetic Similarity Analysis Based on Positive and Negative Sequence Patterns of DNA. Symmetry. 2020; 12(12):2090. https://doi.org/10.3390/sym12122090
Chicago/Turabian StyleLu, Yue, Long Zhao, Zhao Li, and Xiangjun Dong. 2020. "Genetic Similarity Analysis Based on Positive and Negative Sequence Patterns of DNA" Symmetry 12, no. 12: 2090. https://doi.org/10.3390/sym12122090
APA StyleLu, Y., Zhao, L., Li, Z., & Dong, X. (2020). Genetic Similarity Analysis Based on Positive and Negative Sequence Patterns of DNA. Symmetry, 12(12), 2090. https://doi.org/10.3390/sym12122090