Current Approaches and Future Perspectives for Nanobodies in Stroke Diagnostic and Therapy


{kind=link}
{kind=link}
Abstract
1. Stroke and Post-Stroke Inflammation
2. Nanobodies—Single Domain Antibodies
2.1. Structure of Nanobodies and Conventional Antibody Fragments
2.2. Advantages and Limitations of Nanobodies
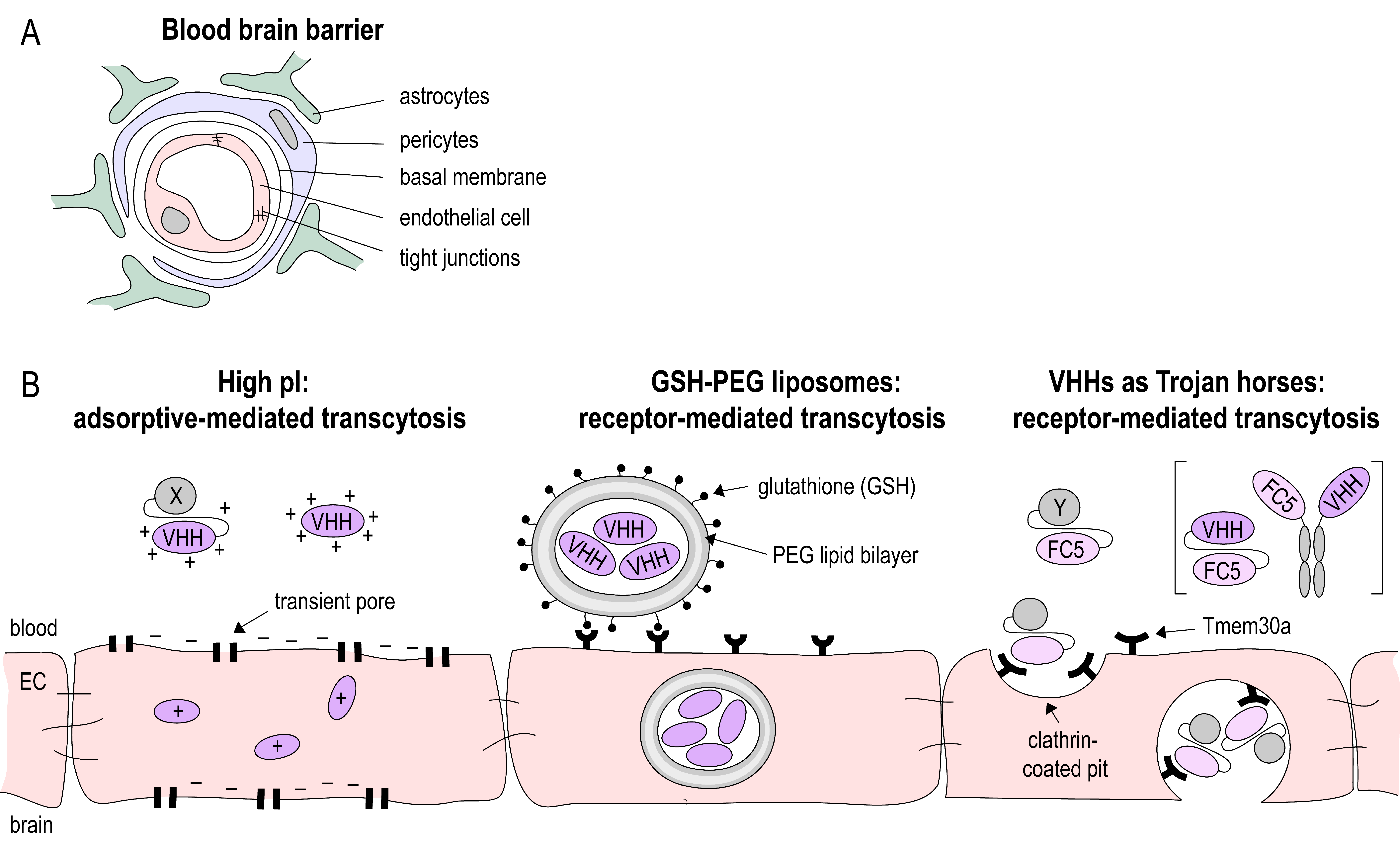
2.3. Nanobodies at the BBB
3. Stroke Imaging—New Job Opportunities for Nanobodies?
3.1. Principles of Stroke Imaging
3.2. Imaging Endothelial Activation
4. Nanobodies as New Thrombolytic Agents
5. Nanobodies to Modulate Post-Stroke Inflammation
5.1. Targeting DAMP Signaling
5.2. Inflammatory Cytokine Neutralization
5.2.1. TNFα
5.2.2. IL-1β
5.3. Cell Migration
5.3.1. CXCR2
5.3.2. CXCR4
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Truelsen, T.; Piechowski-Jozwiak, B.; Bonita, R.; Mathers, C.; Bogousslavsky, J.; Boysen, G. Stroke incidence and prevalence in europe: A review of available data. Eur. J. Neurol. 2006, 13, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Pandian, J.D.; William, A.G.; Kate, M.P.; Norrving, B.; Mensah, G.A.; Davis, S.; Roth, G.A.; Thrift, A.G.; Kengne, A.P.; Kissela, B.M.; et al. Strategies to improve stroke care services in low- and middle-income countries: A systematic review. Neuroepidemiology 2017, 49, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Ay, H.; Furie, K.L.; Singhal, A.; Smith, W.S.; Sorensen, A.G.; Koroshetz, W.J. An evidence-based causative classification system for acute ischemic stroke. Ann. Neurol. 2005, 58, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Bogousslavsky, J.; Caplan, L.R.; Donnan, G.A.; Hennerici, M.G. Classification of stroke subtypes. Cerebrovasc. Dis. 2009, 27, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Grabb, M.C.; Zipfel, G.J.; Choi, D.W. Brain tissue responses to ischemia. J. Clin. Investig. 2000, 106, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus 2007, 22, E2. [Google Scholar] [CrossRef]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.Y.; Akhisaroglu, M.; Fleming, T.; et al. Damp signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Giffard, R.G.; Han, R.Q.; Emery, J.F.; Duan, M.; Pittet, J.F. Regulation of apoptotic and inflammatory cell signaling in cerebral ischemia: The complex roles of heat shock protein 70. Anesthesiology 2008, 109, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.R.; Lu, A.; Tang, Y.; Millhorn, D.E. Multiple molecular penumbras after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 1011–1032. [Google Scholar] [CrossRef] [PubMed]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular histones, cell-free DNA, or nucleosomes: Differences in immunostimulation. Cell. Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 73–90. [Google Scholar]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Chen, X.; Chen, J.; Gautam, S.C.; Xu, Y.; Chopp, M. Mcp-1, mip-1, il-8 and ischemic cerebral tissue enhance human bone marrow stromal cell migration in interface culture. Hematology 2002, 7, 113–117. [Google Scholar] [CrossRef]
- Chamorro, A.; Meisel, A.; Planas, A.M.; Urra, X.; van de Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Haley, M.J.; Lawrence, C.B. The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 2017, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Leukocyte recruitment and ischemic brain injury. Neuromol. Med. 2010, 12, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.K.; Su, C.; Muyldermans, S.; van der Loo, W. Heavy-chain antibodies in camelidae; a case of evolutionary innovation. Immunogenetics 2002, 54, 39–47. [Google Scholar]
- Beghein, E.; Gettemans, J. Nanobody technology: A versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Barthelemy, P.A.; Raab, H.; Appleton, B.A.; Bond, C.J.; Wu, P.; Wiesmann, C.; Sidhu, S.S. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human vh domains. J. Biol. Chem. 2008, 283, 3639–3654. [Google Scholar] [CrossRef]
- Tanha, J.; Nguyen, T.D.; Ng, A.; Ryan, S.; Ni, F.; Mackenzie, R. Improving solubility and refolding efficiency of human v(h)s by a novel mutational approach. Protein Eng. Des. Sel. 2006, 19, 503–509. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Pirez-Schirmer, M.; Rossotti, M.; Badagian, N.; Leizagoyen, C.; Brena, B.M.; Gonzalez-Sapienza, G. Comparison of three antihapten vhh selection strategies for the development of highly sensitive immunoassays for microcystins. Anal. Chem. 2017, 89, 6800–6806. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M. Camelid single-domain antibodies: Historical perspective and future outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed]
- Eden, T.; Menzel, S.; Wesolowski, J.; Bergmann, P.; Nissen, M.; Dubberke, G.; Seyfried, F.; Albrecht, B.; Haag, F.; Koch-Nolte, F. A cdna immunization strategy to generate nanobodies against membrane proteins in native conformation. Front. Immunol. 2017, 8, 1989. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Dekker, S.; Hendriks, R.W.; Panayotou, G.; van Remoortere, A.; San, J.K.; Grosveld, F.; Drabek, D. Generation of heavy-chain-only antibodies in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15130–15135. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.; Zinner, K.; Mucke, N.; Bartoschik, T.; Muyldermans, S.; Hoheisel, J.D. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci. Rep. 2018, 8, 7934. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Hirama, T.; Ding, W.; Mackenzie, R.; Tanha, J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS ONE 2011, 6, e28218. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Single antibody domains as small recognition units: Design and in vitro antigen selection of camelized, human vh domains with improved protein stability. Protein Eng. 1996, 9, 531–537. [Google Scholar] [CrossRef]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen binding and solubility effects upon the veneering of a camel vhh in framework-2 to mimic a vh. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef]
- Ewert, S.; Cambillau, C.; Conrath, K.; Pluckthun, A. Biophysical properties of camelid v(hh) domains compared to those of human v(h)3 domains. Biochemistry 2002, 41, 3628–3636. [Google Scholar] [CrossRef]
- Schumacher, D.; Helma, J.; Schneider, A.F.L.; Leonhardt, H.; Hackenberger, C.P.R. Nanobodies: Chemical functionalization strategies and intracellular applications. Angew. Chem. Int. Ed. Engl. 2018, 57, 2314–2333. [Google Scholar] [CrossRef] [PubMed]
- Caljon, G.; Caveliers, V.; Lahoutte, T.; Stijlemans, B.; Ghassabeh, G.H.; Van Den Abbeele, J.; Smolders, I.; De Baetselier, P.; Michotte, Y.; Muyldermans, S.; et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood-brain barrier. Br. J. Pharmacol. 2012, 165, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha vhh proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthr. Rheumatol. 2006, 54, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; Poelmans, S.; Bontinck, L.; Hemeryck, A.; et al. The preclinical pharmacology of the high affinity anti-il-6r nanobody(r) alx-0061 supports its clinical development in rheumatoid arthritis. Arthr. Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.; Wang, Z.J.; Arbabi-Ghahroudi, M.; Chang, T.A.; Durocher, Y.; Trojahn, U.; Baardsnes, J.; Jaramillo, M.L.; Li, S.; Baral, T.N.; et al. Differential tumor-targeting abilities of three single-domain antibody formats. Cancer Lett. 2010, 289, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014, 28, 4764–4778. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel heavy-chain antibodies: Diverse germline v(h)h and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000, 19, 921–930. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Knobl, P.; Cataland, S.; De Beuf, K.; Callewaert, F.; De Winter, H.; Zeldin, R.K. Caplacizumab reduces the frequency of major thromboembolic events, exacerbations and death in patients with acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2017, 15, 1448–1452. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knobl, P.; Wu, H.; Artoni, A.; Westwood, J.P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase i study of tas266, a novel tetravalent agonistic nanobody(r) targeting the dr5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef]
- Cooper, P.R.; Ciambrone, G.J.; Kliwinski, C.M.; Maze, E.; Johnson, L.; Li, Q.; Feng, Y.; Hornby, P.J. Efflux of monoclonal antibodies from rat brain by neonatal fc receptor, fcrn. Brain Res. 2013, 1534, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Abuqayyas, L.; Balthasar, J.P. Investigation of the role of fcgammar and fcrn in mab distribution to the brain. Mol. Pharm. 2013, 10, 1505–1513. [Google Scholar] [CrossRef]
- Nabuurs, R.J.; Rutgers, K.S.; Welling, M.M.; Metaxas, A.; de Backer, M.E.; Rotman, M.; Bacskai, B.J.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. In vivo detection of amyloid-beta deposits using heavy chain antibody fragments in a transgenic mouse model for alzheimer’s disease. PLoS ONE 2012, 7, e38284. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Bourgeois, J.P.; Celli, S.; Glacial, F.; Le Sourd, A.M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Rougeon, F.; et al. Cell-penetrating anti-gfap vhh and corresponding fluorescent fusion protein vhh-gfp spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012, 26, 3969–3979. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the hiv-1 tat peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Sai, Y.; Kobayashi, H.; Kamata, M.; Wakamiya, T.; Tsuji, A. Structure-internalization relationship for adsorptive-mediated endocytosis of basic peptides at the blood-brain barrier. J. Pharmacol. Exp. Ther. 1997, 280, 410–415. [Google Scholar]
- Li, T.; Vandesquille, M.; Koukouli, F.; Dudeffant, C.; Youssef, I.; Lenormand, P.; Ganneau, C.; Maskos, U.; Czech, C.; Grueninger, F.; et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J. Control. Release 2016, 243, 1–10. [Google Scholar] [CrossRef]
- Iqbal, U.; Trojahn, U.; Albaghdadi, H.; Zhang, J.; O’Connor-McCourt, M.; Stanimirovic, D.; Tomanek, B.; Sutherland, G.; Abulrob, A. Kinetic analysis of novel mono- and multivalent vhh-fragments and their application for molecular imaging of brain tumours. Br. J. Pharmacol. 2010, 160, 1016–1028. [Google Scholar] [CrossRef]
- Rotman, M.; Welling, M.M.; Bunschoten, A.; de Backer, M.E.; Rip, J.; Nabuurs, R.J.; Gaillard, P.J.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. Enhanced glutathione pegylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for alzheimer’s disease. J. Control. Release 2015, 203, 40–50. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Pardridge, W.M. Biotin delivery to brain with a covalent conjugate of avidin and a monoclonal antibody to the transferrin receptor. J. Pharmacol. Exp. Ther. 1992, 263, 897–903. [Google Scholar]
- Thom, G.; Burrell, M.; Haqqani, A.S.; Yogi, A.; Lessard, E.; Brunette, E.; Delaney, C.; Baumann, E.; Callaghan, D.; Rodrigo, N.; et al. Enhanced delivery of galanin conjugates to the brain through bioengineering of the anti-transferrin receptor antibody ox26. Mol. Pharm. 2018, 15, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L.; Yang, J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm Res. 1995, 12, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Pardridge, W.M. Brain and organ uptake in the rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody-iduronidase fusion protein. Mol. Pharm. 2017, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, K.; Hoffmann, M.M.; Dreis, S.; Herbert, E.; Alyautdin, R.N.; Michaelis, M.; Kreuter, J.; Langer, K. Covalent linkage of apolipoprotein e to albumin nanoparticles strongly enhances drug transport into the brain. J. Pharmacol. Exp. Ther. 2006, 317, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pardridge, W.M. Blood-brain barrier targeting of bdnf improves motor function in rats with middle cerebral artery occlusion. Brain Res. 2006, 1111, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Wang, Y.; Pardridge, W.M. Gdnf fusion protein for targeted-drug delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 100, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Abulrob, A.; Sprong, H.; Van Bergen en Henegouwen, P.; Stanimirovic, D. The blood-brain barrier transmigrating single domain antibody: Mechanisms of transport and antigenic epitopes in human brain endothelial cells. J. Neurochem. 2005, 95, 1201–1214. [Google Scholar] [CrossRef]
- Webster, C.I.; Caram-Salas, N.; Haqqani, A.S.; Thom, G.; Brown, L.; Rennie, K.; Yogi, A.; Costain, W.; Brunette, E.; Stanimirovic, D.B. Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J. 2016, 30, 1927–1940. [Google Scholar] [CrossRef]
- Forster, A.; Gass, A.; Kern, R.; Ay, H.; Chatzikonstantinou, A.; Hennerici, M.G.; Szabo, K. Brain imaging in patients with transient ischemic attack: A comparison of computed tomography and magnetic resonance imaging. Eur. Neurol. 2012, 67, 136–141. [Google Scholar] [CrossRef]
- Thomalla, G.; Cheng, B.; Ebinger, M.; Hao, Q.; Tourdias, T.; Wu, O.; Kim, J.S.; Breuer, L.; Singer, O.C.; Warach, S.; et al. Dwi-flair mismatch for the identification of patients with acute ischaemic stroke within 4.5 h of symptom onset (pre-flair): A multicentre observational study. Lancet Neurol. 2011, 10, 978–986. [Google Scholar] [CrossRef]
- Kim, D.E. Chapter 70-principles and methods of molecular imaging in stroke a2-caplan, louis r. In Primer on Cerebrovascular Diseases, 2nd ed.; Biller, J., Leary, M.C., Lo, E.H., Thomas, A.J., Yenari, M., Zhang, J.H., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 332–338. [Google Scholar]
- Quenault, A.; Martinez de Lizarrondo, S.; Etard, O.; Gauberti, M.; Orset, C.; Haelewyn, B.; Segal, H.C.; Rothwell, P.M.; Vivien, D.; Touze, E.; et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain 2017, 140, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Frechou, M.; Beray-Berthat, V.; Raynaud, J.S.; Meriaux, S.; Gombert, F.; Lancelot, E.; Plotkine, M.; Marchand-Leroux, C.; Ballet, S.; Robert, P.; et al. Detection of vascular cell adhesion molecule-1 expression with uspio-enhanced molecular mri in a mouse model of cerebral ischemia. Contrast Media Mol. Imaging 2013, 8, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Gauberti, M.; Montagne, A.; Marcos-Contreras, O.A.; Le Behot, A.; Maubert, E.; Vivien, D. Ultra-sensitive molecular mri of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke 2013, 44, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Deddens, L.H.; van Tilborg, G.A.; van der Toorn, A.; de Vries, H.E.; Dijkhuizen, R.M. Pecam-1-targeted micron-sized particles of iron oxide as mri contrast agent for detection of vascular remodeling after cerebral ischemia. Contrast Media Mol. Imaging 2013, 8, 393–401. [Google Scholar] [CrossRef]
- Deddens, L.H.; van Tilborg, G.A.; van der Toorn, A.; van der Marel, K.; Paulis, L.E.; van Bloois, L.; Storm, G.; Strijkers, G.J.; Mulder, W.J.; de Vries, H.E.; et al. Mri of icam-1 upregulation after stroke: The importance of choosing the appropriate target-specific particulate contrast agent. Mol. Imaging Biol. 2013, 15, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Olafsen, T.; Wu, A.M. Antibody vectors for imaging. Semin. Nucl. Med. 2010, 40, 167–181. [Google Scholar] [CrossRef]
- Bala, G.; Blykers, A.; Xavier, C.; Descamps, B.; Broisat, A.; Ghezzi, C.; Fagret, D.; Van Camp, G.; Caveliers, V.; Vanhove, C.; et al. Targeting of vascular cell adhesion molecule-1 by 18f-labelled nanobodies for pet/ct imaging of inflamed atherosclerotic plaques. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 1001–1008. [Google Scholar] [CrossRef]
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies targeting mouse/human vcam1 for the nuclear imaging of atherosclerotic lesions. Circ. Res. 2012, 110, 927–937. [Google Scholar] [CrossRef]
- Gauberti, M.; Montagne, A.; Quenault, A.; Vivien, D. Molecular magnetic resonance imaging of brain-immune interactions. Front. Cell. Neurosci. 2014, 8, 389. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Caljon, G.; Stijlemans, B.; Saerens, D.; Van Den Abbeele, J.; Muyldermans, S.; Magez, S.; De Baetselier, P. Affinity is an important determinant of the anti-trypanosome activity of nanobodies. PLoS Negl. Trop. Dis. 2012, 6, e1902. [Google Scholar] [CrossRef]
- Vandesquille, M.; Li, T.; Po, C.; Ganneau, C.; Lenormand, P.; Dudeffant, C.; Czech, C.; Grueninger, F.; Duyckaerts, C.; Delatour, B.; et al. Chemically-defined camelid antibody bioconjugate for the magnetic resonance imaging of alzheimer’s disease. MAbs 2017, 9, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, O.; Hornung, R.; Khatri, P.; Kleindorfer, D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the united states: A doubling of treatment rates over the course of 5 years. Stroke 2011, 42, 1952–1955. [Google Scholar] [CrossRef] [PubMed]
- Thomalla, G.; Simonsen, C.Z.; Boutitie, F.; Andersen, G.; Berthezene, Y.; Cheng, B.; Cheripelli, B.; Cho, T.H.; Fazekas, F.; Fiehler, J.; et al. Mri-guided thrombolysis for stroke with unknown time of onset. N. Engl. J. Med. 2018, 379, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, M.L.; Zatloukalova, M.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Gils, A.; Declerck, P.J. In vitro and in vivo characterisation of the profibrinolytic effect of an inhibitory anti-rat tafi nanobody. Thromb. Haemost. 2014, 111, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S. Caplacizumab: First global approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef]
- De Meyer, S.F.; Stoll, G.; Wagner, D.D.; Kleinschnitz, C. Von willebrand factor: An emerging target in stroke therapy. Stroke 2012, 43, 599–606. [Google Scholar] [CrossRef]
- Momi, S.; Tantucci, M.; Van Roy, M.; Ulrichts, H.; Ricci, G.; Gresele, P. Reperfusion of cerebral artery thrombosis by the gpib-vwf blockade with the nanobody alx-0081 reduces brain infarct size in guinea pigs. Blood 2013, 121, 5088–5097. [Google Scholar] [CrossRef]
- Nieswandt, B.; Stoll, G. (Dis)solving the stroke problem by vWF inhibition? Blood 2013, 121, 4972–4974. [Google Scholar] [CrossRef]
- Montaner, J.; Ribo, M.; Monasterio, J.; Molina, C.A.; Alvarez-Sabin, J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke 2003, 34, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.C.; Leenaerts, D.; Brouns, R.; Engelborghs, S.; Ieven, M.; De Deyn, P.P.; Lambeir, A.M.; Hendriks, D. Procarboxypeptidase u (procpu, tafi, procpb2) in cerebrospinal fluid during ischemic stroke is associated with stroke progression, outcome and blood-brain barrier dysfunction. J. Thromb. Haemost. 2018, 16, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Wyseure, T.; Peeters, M.; Vandeputte, N.; Gils, A.; Deckmyn, H.; Vanhoorelbeke, K.; Declerck, P.J.; De Meyer, S.F. Inhibition of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 reduces ischemic brain damage in mice. Stroke 2016, 47, 2419–2422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Weeks, S.D.; Ameloot, P.; Callewaert, N.; Strelkov, S.V.; Declerck, P.J. Elucidation of the molecular mechanisms of two nanobodies that inhibit thrombin-activatable fibrinolysis inhibitor activation and activated thrombin-activatable fibrinolysis inhibitor activity. J. Thromb. Haemost. 2016, 14, 1629–1638. [Google Scholar] [CrossRef]
- Danquah, W.; Meyer-Schwesinger, C.; Rissiek, B.; Pinto, C.; Serracant-Prat, A.; Amadi, M.; Iacenda, D.; Knop, J.H.; Hammel, A.; Bergmann, P.; et al. Nanobodies that block gating of the p2x7 ion channel ameliorate inflammation. Sci. Transl. Med. 2016, 8, 366ra162. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, G.; Sarchielli, P.; Caso, V.; Paciaroni, M.; Floridi, A.; Floridi, A.; Gallai, V. Different cytokine levels in thrombolysis patients as predictors for clinical outcome. Eur. J. Neurol. 2004, 11, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Amadio, S.; Gianfriddo, M.; Vannucchi, M.G.; Volonte, C.; Bernardi, G.; Pedata, F.; Sancesario, G. P2x7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 2006, 26, 974–982. [Google Scholar] [CrossRef]
- Arbeloa, J.; Perez-Samartin, A.; Gottlieb, M.; Matute, C. P2x7 receptor blockade prevents atp excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol. Dis. 2012, 45, 954–961. [Google Scholar] [CrossRef]
- Chu, K.; Yin, B.; Wang, J.; Peng, G.; Liang, H.; Xu, Z.; Du, Y.; Fang, M.; Xia, Q.; Luo, B. Inhibition of p2x7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 2012, 9, 69. [Google Scholar] [CrossRef]
- Kaiser, M.; Penk, A.; Franke, H.; Krugel, U.; Norenberg, W.; Huster, D.; Schaefer, M. Lack of functional p2x7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal. 2016, 12, 453–463. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Kitamura, Y.; Takata, K.; Hide, I.; Nakata, Y.; Taniguchi, T. Possible involvement of p2x7 receptor activation in microglial neuroprotection against focal cerebral ischemia in rats. Biol. Pharm. Bull. 2008, 31, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Le Feuvre, R.A.; Brough, D.; Touzani, O.; Rothwell, N.J. Role of p2x7 receptors in ischemic and excitotoxic brain injury in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; Stokes, L.; Sluyter, R. The p2x7 receptor channel: Recent developments and the use of p2x7 antagonists in models of disease. Pharmacol. Rev. 2014, 66, 638–675. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Biber, K.; Finsen, B. Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sawamoto, K.; Suzuki, S.; Suzuki, N.; Adachi, K.; Kawase, T.; Mihara, M.; Ohsugi, Y.; Abe, K.; Okano, H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: Possible involvement of stat3 activation in the protection of neurons. J. Neurochem. 2005, 94, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, O.; Tarabin, V.; Suzuki, S.; Attigah, N.; Coserea, I.; Schneider, A.; Vogel, J.; Prinz, S.; Schwab, S.; Monyer, H.; et al. Regulation of body temperature and neuroprotection by endogenous interleukin-6 in cerebral ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Intiso, D.; Zarrelli, M.M.; Lagioia, G.; Di Rienzo, F.; Checchia De Ambrosio, C.; Simone, P.; Tonali, P.; Cioffi Dagger, R.P. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patients. Neurol. Sci. 2004, 24, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Losy, J. Early tnf-alpha levels correlate with ischaemic stroke severity. Acta Neurol. Scand. 2001, 104, 288–295. [Google Scholar] [CrossRef]
- Dziewulska, D.; Mossakowski, M.J. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin. Neuropathol. 2003, 22, 35–40. [Google Scholar] [PubMed]
- Zelova, H.; Hosek, J. Tnf-alpha signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Martin, D.; Hallenbeck, J.M. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J. Cereb. Blood Flow Metab. 1997, 17, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Y.; Gong, C.; Qin, Z.; Liu, X.H.; Lorris Betz, A. Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res. Mol. Brain Res. 1999, 69, 135–143. [Google Scholar] [CrossRef]
- Meistrell, M.E., 3rd; Botchkina, G.I.; Wang, H.; Di Santo, E.; Cockroft, K.M.; Bloom, O.; Vishnubhakat, J.M.; Ghezzi, P.; Tracey, K.J. Tumor necrosis factor is a brain damaging cytokine in cerebral ischemia. Shock 1997, 8, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Tasaki, K.; Ruetzler, C.A.; Hallenbeck, J.M. Tnf-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997, 17, 483–490. [Google Scholar] [CrossRef]
- Lis, K.; Kuzawinska, O.; Balkowiec-Iskra, E. Tumor necrosis factor inhibitors—State of knowledge. Arch. Med. Sci. 2014, 10, 1175–1185. [Google Scholar] [CrossRef]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent llama single-domain antibody fragments against tumor necrosis factor have picomolar potencies due to intramolecular interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef]
- Kalden, J.R.; Schulze-Koops, H. Immunogenicity and loss of response to tnf inhibitors: Implications for rheumatoid arthritis treatment. Nat. Rev. Rheumatol. 2017, 13, 707–718. [Google Scholar] [CrossRef]
- Efimov, G.A.; Kruglov, A.A.; Khlopchatnikova, Z.V.; Rozov, F.N.; Mokhonov, V.V.; Rose-John, S.; Scheller, J.; Gordon, S.; Stacey, M.; Drutskaya, M.S.; et al. Cell-type-restricted anti-cytokine therapy: Tnf inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011. [Google Scholar] [CrossRef]
- Steeland, S.; Puimege, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; De Rycke, R.; Toussaint, W.; Vanhoutte, L.; Vanhove, C.; De Vos, F.; Vandenbroucke, R.E.; Libert, C. Tnfr1 inhibition with a nanobody protects against EAE development in mice. Sci. Rep. 2017, 7, 13646. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef]
- Liu, T.; McDonnell, P.C.; Young, P.R.; White, R.F.; Siren, A.L.; Hallenbeck, J.M.; Barone, F.C.; Feurestein, G.Z. Interleukin-1 beta mRNA expression in ischemic rat cortex. Stroke 1993, 24, 1746–1750, discussion 1750–1741. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Barone, F.C.; Aiyar, N.V.; Feuerstein, G.Z. Interleukin-1 receptor and receptor antagonist gene expression after focal stroke in rats. Stroke 1997, 28, 155–161, discussion 161–152. [Google Scholar] [CrossRef] [PubMed]
- Pradillo, J.M.; Murray, K.N.; Coutts, G.A.; Moraga, A.; Oroz-Gonjar, F.; Boutin, H.; Moro, M.A.; Lizasoain, I.; Rothwell, N.J.; Allan, S.M. Reparative effects of interleukin-1 receptor antagonist in young and aged/co-morbid rodents after cerebral ischemia. Brain Behav. Immun. 2017, 61, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, N.J.; Ross, J.; Rothwell, N.J.; Loddick, S.A. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br. J. Pharmacol. 2003, 140, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009, CD005121. [Google Scholar] [CrossRef] [PubMed]
- Emsley, H.C.; Smith, C.J.; Georgiou, R.F.; Vail, A.; Hopkins, S.J.; Rothwell, N.J.; Tyrrell, P.J.; Acute Stroke, I. A randomised phase ii study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef]
- Boutin, H.; LeFeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of il-1alpha and il-1beta in ischemic brain damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [CrossRef]
- Stroemer, R.P.; Rothwell, N.J. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J. Cereb. Blood Flow Metab. 1998, 18, 833–839. [Google Scholar] [CrossRef]
- Liberale, L.; Diaz-Canestro, C.; Bonetti, N.R.; Paneni, F.; Akhmedov, A.; Beer, J.H.; Montecucco, F.; Luscher, T.F.; Camici, G.G. Post-ischaemic administration of the murine canakinumab-surrogate antibody improves outcome in experimental stroke. Eur. Heart J. 2018, 39, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Shi, X.; Zhou, B.; Teng, J.; Zhang, C.; Liu, S.; Lian, J.; Luo, B.; Zhao, G.; Lu, H.; et al. Interleukin 8 (cxcl8)-cxc chemokine receptor 2 (cxcr2) axis contributes to mir-4437-associated recruitment of granulocytes and natural killer cells in ischemic stroke. Mol. Immunol. 2018, 101, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Garau, A.; Bertini, R.; Colotta, F.; Casilli, F.; Bigini, P.; Cagnotto, A.; Mennini, T.; Ghezzi, P.; Villa, P. Neuroprotection with the cxcl8 inhibitor repertaxin in transient brain ischemia. Cytokine 2005, 30, 125–131. [Google Scholar] [CrossRef]
- Connell, B.J.; Gordon, J.R.; Saleh, T.M. Elr-cxc chemokine antagonism is neuroprotective in a rat model of ischemic stroke. Neurosci. Lett. 2015, 606, 117–122. [Google Scholar] [CrossRef]
- Brait, V.H.; Rivera, J.; Broughton, B.R.; Lee, S.; Drummond, G.R.; Sobey, C.G. Chemokine-related gene expression in the brain following ischemic stroke: No role for cxcr2 in outcome. Brain Res. 2011, 1372, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.E.; Dombrecht, B.; Manini, J.; Willis, J.; Vlerick, D.; De Taeye, S.; Van den Heede, K.; Roobrouck, A.; Grot, E.; Kent, T.C.; et al. Potent and efficacious inhibition of cxcr2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol. Pharmacol. 2015, 87, 251–262. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Li, Y.; Yang, G.Y. Roles of chemokine cxcl12 and its receptors in ischemic stroke. Curr. Drug Targets 2012, 13, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Yu, S.J.; Shia, K.S.; Wu, C.H.; Song, J.S.; Kuan, H.H.; Yeh, K.C.; Chen, C.T.; Bae, E.; Wang, Y. A novel cxcr4 antagonist cx549 induces neuroprotection in stroke brain. Cell. Transpl. 2017, 26, 571–583. [Google Scholar] [CrossRef]
- Walter, H.L.; van der Maten, G.; Antunes, A.R.; Wieloch, T.; Ruscher, K. Treatment with amd3100 attenuates the microglial response and improves outcome after experimental stroke. J. Neuroinflamm. 2015, 12, 24. [Google Scholar] [CrossRef]
- Ruscher, K.; Kuric, E.; Liu, Y.; Walter, H.L.; Issazadeh-Navikas, S.; Englund, E.; Wieloch, T. Inhibition of cxcl12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.H.; Heukers, R.; Brink, H.J.; Arsova, A.; Maussang, D.; Cutolo, P.; Strubbe, B.; Vischer, H.F.; Bachelerie, F.; Smit, M.J. Cxcr4-specific nanobodies as potential therapeutics for whim syndrome. J. Pharmacol. Exp. Ther. 2017, 363, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Jahnichen, S.; Blanchetot, C.; Maussang, D.; Gonzalez-Pajuelo, M.; Chow, K.Y.; Bosch, L.; De Vrieze, S.; Serruys, B.; Ulrichts, H.; Vandevelde, W.; et al. Cxcr4 nanobodies (vhh-based single variable domains) potently inhibit chemotaxis and hiv-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20565–20570. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jank, L.; Pinto-Espinoza, C.; Duan, Y.; Koch-Nolte, F.; Magnus, T.; Rissiek, B. Current Approaches and Future Perspectives for Nanobodies in Stroke Diagnostic and Therapy. Antibodies 2019, 8, 5. https://doi.org/10.3390/antib8010005
Jank L, Pinto-Espinoza C, Duan Y, Koch-Nolte F, Magnus T, Rissiek B. Current Approaches and Future Perspectives for Nanobodies in Stroke Diagnostic and Therapy. Antibodies. 2019; 8(1):5. https://doi.org/10.3390/antib8010005
Chicago/Turabian StyleJank, Larissa, Carolina Pinto-Espinoza, Yinghui Duan, Friedrich Koch-Nolte, Tim Magnus, and Björn Rissiek. 2019. "Current Approaches and Future Perspectives for Nanobodies in Stroke Diagnostic and Therapy" Antibodies 8, no. 1: 5. https://doi.org/10.3390/antib8010005
APA StyleJank, L., Pinto-Espinoza, C., Duan, Y., Koch-Nolte, F., Magnus, T., & Rissiek, B. (2019). Current Approaches and Future Perspectives for Nanobodies in Stroke Diagnostic and Therapy. Antibodies, 8(1), 5. https://doi.org/10.3390/antib8010005