Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients

Abstract
1. Introduction
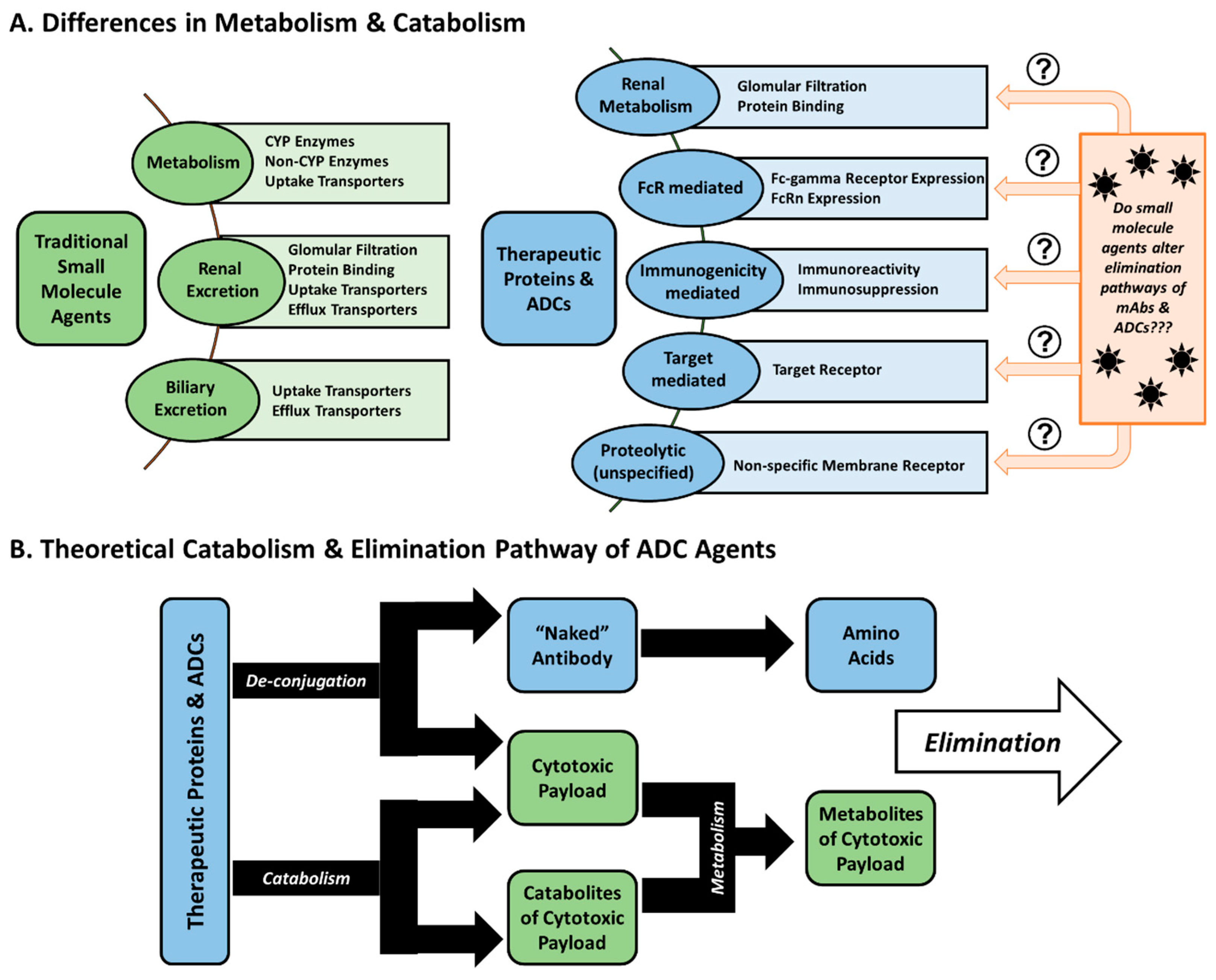
2. Pharmacokinetic Considerations
2.1. Pharmacokinetic Disposition
2.2. Innate Immune System: Mononuclear Phagocyte System
3. Physical Characteristics of mAbs & ADCS
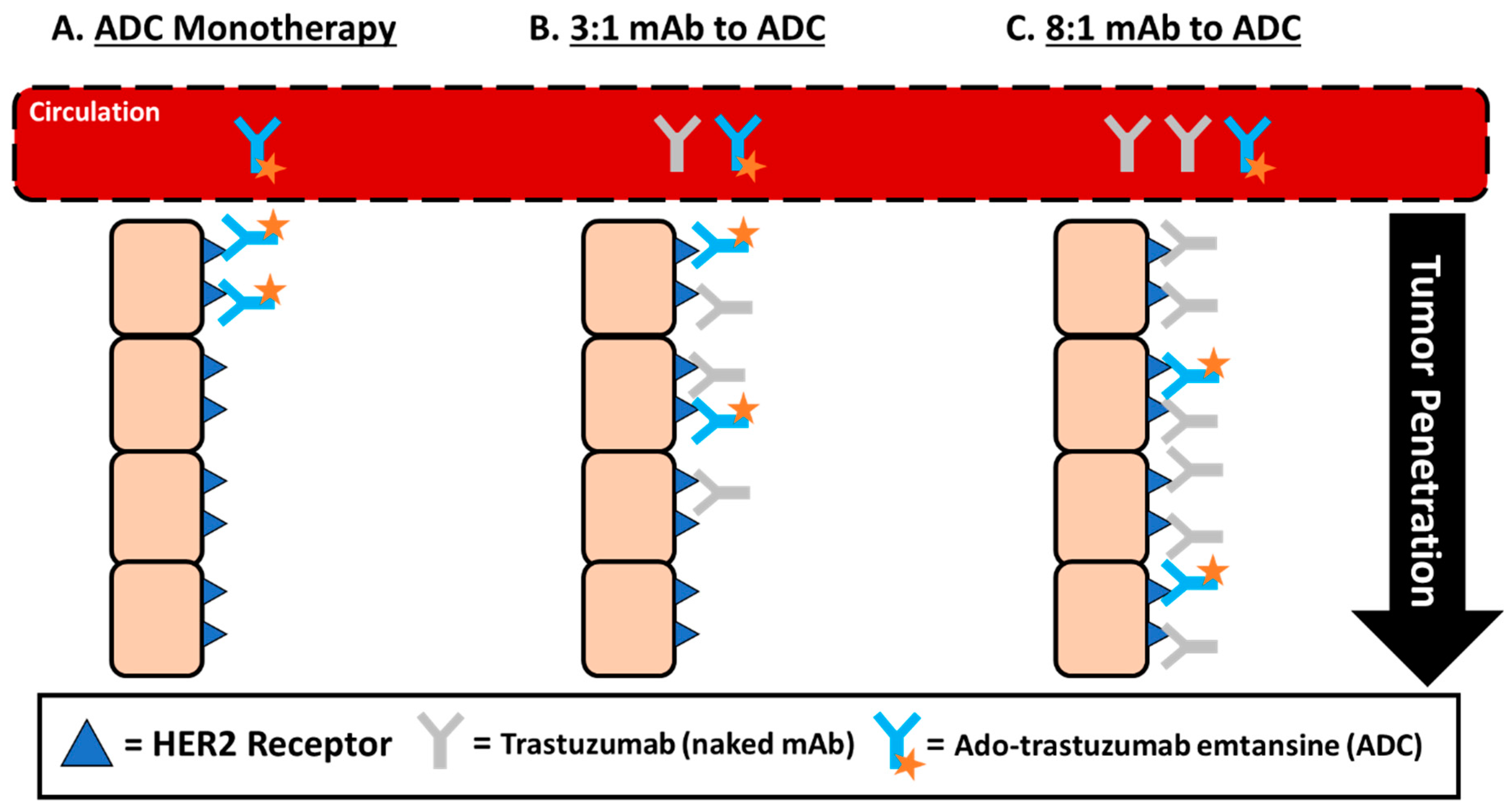
3.1. Size
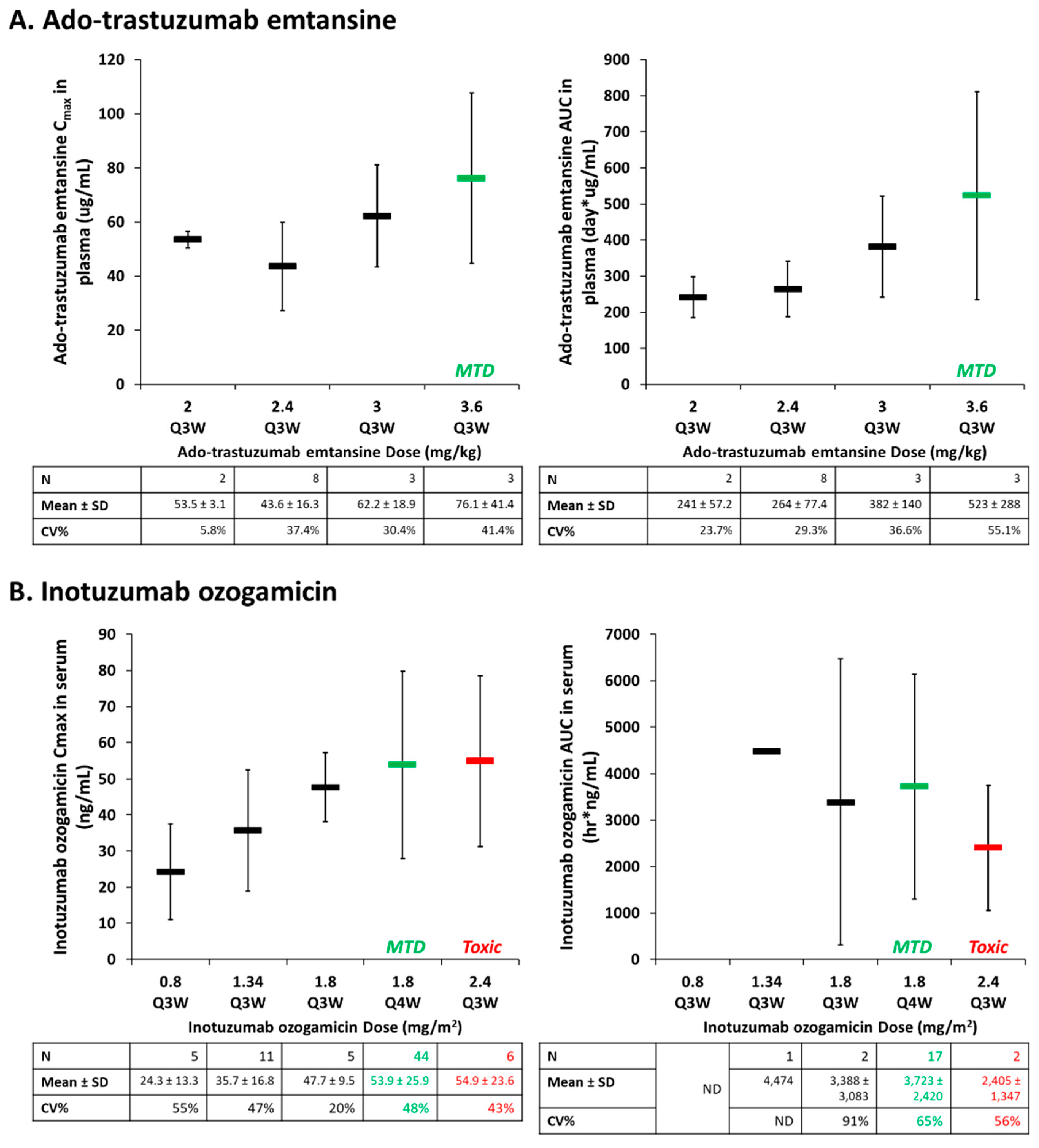
3.2. Drug-Antibody Ratio (Dar)
3.3. Surface Engineering & Chemical Alterations
3.4. Charge and pH Engineering
4. Host-Associated Factors and Disease Status
4.1. Presence of Liver Metastases
4.2. Sex and Body Habitus
4.3. Biochemical Mediators of Immunity in Blood
4.4. Renal or Hepatic Impairment
4.5. Neonatal Fc Receptor (FcRn)
4.6. Fc-Gamma Receptors (FcγR)
5. Pharmacologic-Associated Factors
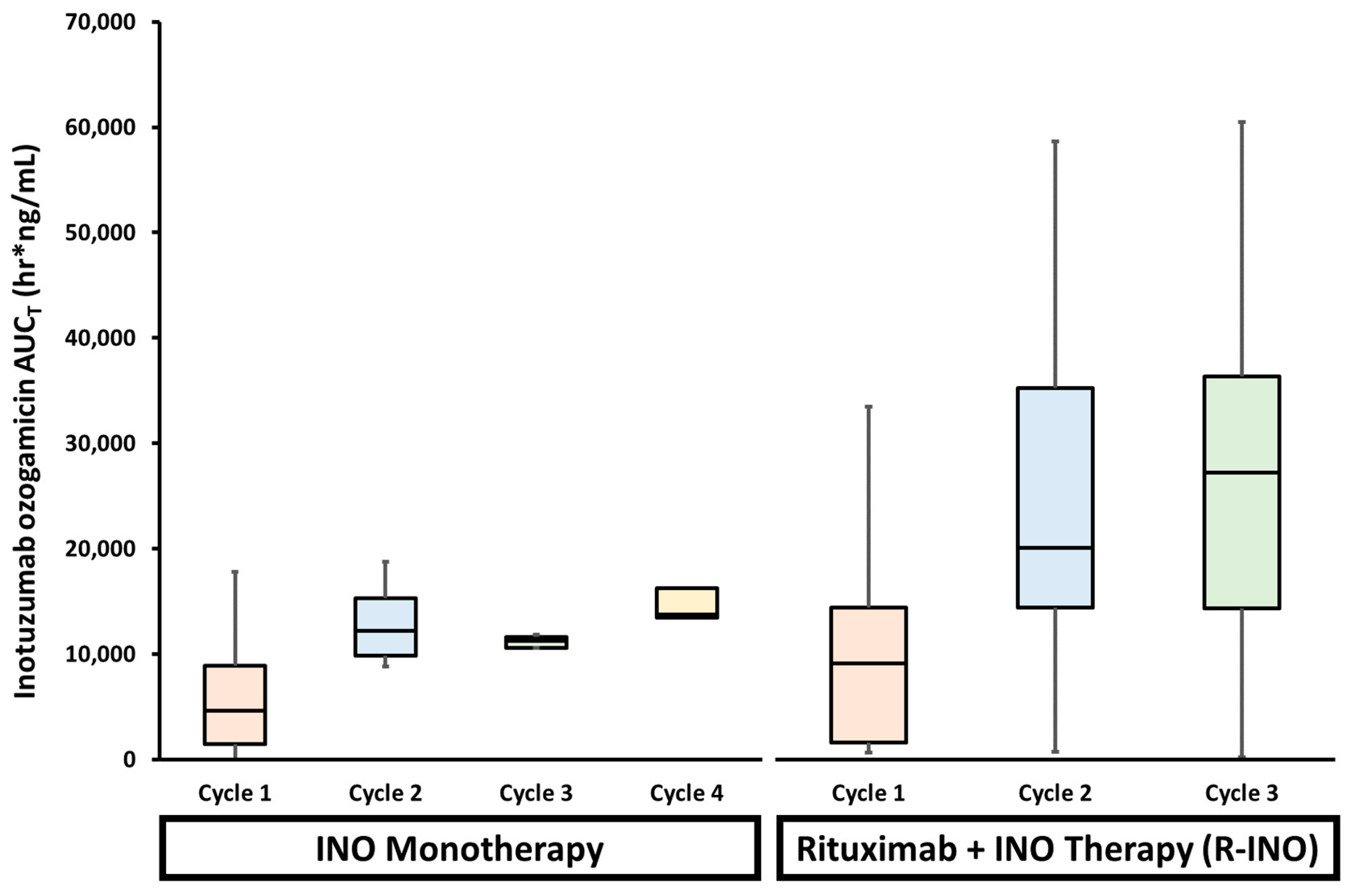
5.1. Drug–Drug Interactions (DDIs)
5.2. Ocular Toxicity
6. Preclinical Model Considerations
6.1. General Limitations in Preclinical Tumor Implantation
6.2. Differences in Clinically-Relevant Covariates
6.3. Differences in Immune Cells & Phagocytes In Pre-Clinical Models
6.4. Prediction of Human ADC PK Using Allometry
6.5. Alternative Strategies: Humans as a Model?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADA | anti-drug antibodies |
| ADC | antibody-drug conjugate |
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| AUC | area under the curve |
| BBB | blood-brain barrier |
| BSA | body surface area |
| CDC | complement-dependent cytotoxicity |
| CMA | carrier-mediated agent |
| CL | clearance |
| DAR | drug–antibody ratio |
| DDI | drug–drug interaction |
| DM1 | mertansine |
| DM4 | ravtansine |
| ESRD | end stage renal disease |
| Fabs | Fab fragments |
| FcγR | Fc-gamma receptors |
| FcRn | neonatal Fc receptor |
| HGF | hepatocyte growth factor |
| iv | intravenously |
| KO | knockout |
| mAb | monoclonal antibody |
| MMAE | monomethyl auristatin E |
| MMAF | monomethyl auristatin F |
| MPS | mononuclear phagocyte system |
| NHP | non-human primate |
| NSCLC | non-small cell lung cancer |
| NP | nanoparticle |
| PD | pharmacodynamic |
| PDX | patient-derived xenografts |
| PEG | polyethylene glycol |
| PFS | progression-free survival |
| pI | isoelectric point |
| PK | pharmacokinetic |
| SABV | “sex as a biologic variable” |
| sc | subcutaneously |
| scFv | single chain variable fragment |
| TAM | tumor-associated macrophage |
| WT | wild type |
References
- Khera, E.; Thurber, G.M. Pharmacokinetic and Immunological Considerations for Expanding the Therapeutic Window of Next-Generation Antibody-Drug Conjugates. Biodrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Ordas, I.; Mould, D.R.; Feagan, B.G.; Sandborn, W.J. Anti-TNF monoclonal antibodies in inflammatory bowel disease: Pharmacokinetics-based dosing paradigms. Clin. Pharmacol. Ther. 2012, 91, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ji, P.; Li, Z.; Roy, P.; Sahajwalla, C.G. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J. Clin. Pharmacol. 2013, 53, 314–325. [Google Scholar] [CrossRef]
- Tabrizi, M.; Bornstein, G.G.; Suria, H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 2010, 12, 33–43. [Google Scholar] [CrossRef]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling Components of the Tumor Microenvironment to Enhance Cancer Therapy. Front. Oncol. 2015, 5, 214. [Google Scholar] [CrossRef]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef]
- Van Rijt, S.H.; Bein, T.; Meiners, S. Medical nanoparticles for next generation drug delivery to the lungs. Eur. Respir. J. 2014, 44, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Tannock, I.F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Pea, F.; Lipman, J. The clinical relevance of plasma protein binding changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Scheife, R. Protein binding: What does it mean? DICP Ann. Pharm. 1989, 23, S27–S31. [Google Scholar] [CrossRef]
- Breij, E.C.; de Goeij, B.E.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Bellosta, S.; Baldessin, L.; Boccia, D.; Racagni, G.; Corsini, A. Pharmacokinetics interactions of monoclonal antibodies. Pharmacol. Res. 2016, 111, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J. Immunol. 2013, 190, 4315–4323. [Google Scholar] [CrossRef]
- Kasturirangan, S.; Rainey, G.J.; Xu, L.; Wang, X.; Portnoff, A.; Chen, T.; Fazenbaker, C.; Zhong, H.; Bee, J.; Zeng, Z.; et al. Targeted Fcgamma Receptor (FcgammaR)-mediated Clearance by a Biparatopic Bispecific Antibody. J. Biol. Chem. 2017, 292, 4361–4370. [Google Scholar] [CrossRef]
- Lucas, A.T.; Madden, A.J.; Zamboni, W.C. Formulation and physiologic factors affecting the pharmacology of carrier-mediated anticancer agents. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1419–1433. [Google Scholar] [CrossRef]
- Lucas, A.T.; Price, L.S.L.; Schorzman, A.N.; Storrie, M.; Piscitelli, J.A.; Razo, J.; Zamboni, W.C. Factors Affecting the Pharmacology of Antibody–Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef]
- Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Dubrot, J.; Milheiro, F.; Alfaro, C.; Palazon, A.; Martinez-Forero, I.; Perez-Gracia, J.L.; Morales-Kastresana, A.; Romero-Trevejo, J.L.; Ochoa, M.C.; Hervas-Stubbs, S.; et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol. Immunother. 2010, 59, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Sadauskas, E.; Wallin, H.; Stoltenberg, M.; Vogel, U.; Doering, P.; Larsen, A.; Danscher, G. Kupffer cells are central in the removal of nanoparticles from the organism. Part. Fibre Toxicol. 2007, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Suzman, D.L.; Pelosof, L.; Rosenberg, A.; Avigan, M.I. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, K.M.; MacParland, S.A.; Ma, X.Z.; Spetzler, V.N.; Echeverri, J.; Ouyang, B.; Fadel, S.M.; Sykes, E.A.; Goldaracena, N.; Kaths, J.M.; et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 2016, 15, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Vexler, V.; Yu, L.; Pamulapati, C.; Garrido, R.; Grimm, H.P.; Sriraman, P.; Bohini, S.; Schraeml, M.; Singh, U.; Brandt, M.; et al. Target-mediated drug disposition and prolonged liver accumulation of a novel humanized anti-CD81 monoclonal antibody in cynomolgus monkeys. mAbs 2013, 5, 776–786. [Google Scholar] [CrossRef]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20–mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef]
- Eigenmann, M.J.; Fronton, L.; Grimm, H.P.; Otteneder, M.B.; Krippendorff, B.F. Quantification of IgG monoclonal antibody clearance in tissues. mAbs 2017, 9, 1007–1015. [Google Scholar] [CrossRef]
- Milenic, D.E.; Wong, K.J.; Baidoo, K.E.; Nayak, T.K.; Regino, C.A.; Garmestani, K.; Brechbiel, M.W. Targeting HER2: A report on the in vitro and in vivo pre-clinical data supporting trastuzumab as a radioimmunoconjugate for clinical trials. mAbs 2010, 2, 550–564. [Google Scholar] [CrossRef]
- Yang, Z.X.; Cao, H.; Xing, C.G.; Wei, S.H.; Jiang, G.Q.; Liu, Z.L. Visualization and body distribution of [(1)(3)(1)I]-herceptin in nude mice with BT-474 breast carcinoma. Genet. Mol. Res. 2014, 13, 6804–6812. [Google Scholar] [CrossRef]
- Glassman, P.M.; Balthasar, J.P. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol. Med. 2014, 11, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.P.M.; Magerm, D.E.; Khan, M.K. Synthesis and Biodisposition of Dendrimer Composite Nanoparticles; Pan Stanford Publishing: Singapore, Singapore, 2010. [Google Scholar]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [PubMed]
- Schell, R.F.; Sidone, B.J.; Caron, W.P.; Walsh, M.D.; White, T.F.; Zamboni, B.A.; Ramanathan, R.K.; Zamboni, W.C. Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Washington, C.B.; Lu, J.F.; Lieberman, G.; Banken, L.; Klein, P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother. Pharmacol. 2005, 56, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Modi, S.; LoRusso, P.M.; Pegram, M.; Guardino, E.; Althaus, B.; Lu, D.; Strasak, A.; Elias, A. Phase 1b/2a study of trastuzumab emtansine (T-DM1), paclitaxel, and pertuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res. 2016, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: Results of a phase I study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef]
- Baselga, J. Phase I and II clinical trials of trastuzumab. Ann. Oncol. 2001, 12, S49–S55. [Google Scholar] [CrossRef]
- Green, M.D. Clinical Pharmacology Review of herceptin (98-0369); Drugs@FDA; Food and Drug Administration: Silver Spring, MD, USA, 1998.
- Deslandes, A. Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies. mAbs 2014, 6, 859–870. [Google Scholar] [CrossRef]
- Li, H.; Han, T.H.; Hunder, N.N.; Jang, G.; Zhao, B. Population Pharmacokinetics of Brentuximab Vedotin in Patients With CD30-Expressing Hematologic Malignancies. J. Clin. Pharmacol. 2017, 57, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Quartino, A.L.; Hillenbach, C.; Li, J.; Li, H.; Wada, R.D.; Visich, J.; Li, C.; Heinzmann, D.; Jin, J.Y.; Lum, B.L. Population pharmacokinetic and exposure-response analysis for trastuzumab administered using a subcutaneous “manual syringe” injection or intravenously in women with HER2-positive early breast cancer. Cancer Chemother. Pharmacol. 2016, 77, 77–88. [Google Scholar] [CrossRef]
- Kai, M.P.; Brighton, H.E.; Fromen, C.A.; Shen, T.W.; Luft, J.C.; Luft, Y.E.; Keeler, A.W.; Robbins, G.R.; Ting, J.P.; Zamboni, W.C.; et al. Tumor Presence Induces Global Immune Changes and Enhances Nanoparticle Clearance. ACS Nano 2016, 10, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ramanathan, R.K.; Zamboni, B.A.; Strychor, S.; Ramalingam, S.; Edwards, R.P.; Friedland, D.M.; Stoller, R.G.; Belani, C.P.; Maruca, L.J.; et al. Population pharmacokinetics of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. J. Clin. Pharmacol. 2012, 52, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, G.; Wu, X.; Livingston, P.; Scholz, W.; Kearns, C.; Maffuid, P. Abstract A73: Antitumor activity of MVT-5873, a monoclonal antibody targeting sialyl Lewisa, alone and in combination with gemcitabine/nab-paclitaxel in a BxPC3 human pancreatic cancer xenograft model. Cancer Res. 2016, 76, A73. [Google Scholar] [CrossRef]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef]
- Kamath, A.V.; Iyer, S. Challenges and advances in the assessment of the disposition of antibody-drug conjugates. Biopharm. Drug Dispos. 2016, 37, 66–74. [Google Scholar] [CrossRef]
- Alley, S.C.; Zhang, X.; Okeley, N.M.; Anderson, M.; Law, C.L.; Senter, P.D.; Benjamin, D.R. The pharmacologic basis for antibody-auristatin conjugate activity. J. Pharmacol. Exp. Ther. 2009, 330, 932–938. [Google Scholar] [CrossRef]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Bumbaca, D.; Valle, N.R.; Kozak, K.R.; Fourie, A.; Chuh, J.; Koppada, N.; Saad, O.; et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjugate Chem. 2011, 22, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Frazer-Abel, A.; Reynolds, P.R.; Giclas, P.C.; Chappell, A.; Pangburn, M.K.; Younis, H.; Henry, S.P. Mechanistic understanding for the greater sensitivity of monkeys to antisense oligonucleotide-mediated complement activation compared with humans. J. Pharmacol. Exp. Ther. 2014, 351, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Forde, P.M.; Hammers, H.J.; Emens, L.A.; Taube, J.M.; Topalian, S.L. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015, 42, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Sznol, M.; Ferrucci, P.F.; Hogg, D.; Atkins, M.B.; Wolter, P.; Guidoboni, M.; Lebbe, C.; Kirkwood, J.M.; Schachter, J.; Daniels, G.A.; et al. Pooled Analysis Safety Profile of Nivolumab and Ipilimumab Combination Therapy in Patients With Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3815–3822. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals 2018, 11. [Google Scholar] [CrossRef]
- Leelawattanachai, J.; Kwon, K.W.; Michael, P.; Ting, R.; Kim, J.Y.; Jin, M.M. Side-by-Side Comparison of Commonly Used Biomolecules That Differ in Size and Affinity on Tumor Uptake and Internalization. PLoS ONE 2015, 10, e0124440. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-Drug Conjugates and Small Molecule-Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef]
- Deng, S.; Lin, Z.; Li, W. Recent Advances in Antibody-Drug Conjugates for Breast Cancer Treatment. Curr. Med. Chem. 2017, 24, 2505–2527. [Google Scholar] [CrossRef] [PubMed]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.M.; Diez-Posada, S.; Stewart, A.C. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef]
- Govindan, S.V.; Sharkey, R.M.; Goldenberg, D.M. Prospects and progress of antibody-drug conjugates in solid tumor therapies. Expert Opin. Biol. Ther. 2016, 16, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Catcott, K.C.; McShea, M.A.; Bialucha, C.U.; Miller, K.L.; Hicks, S.W.; Saxena, P.; Gesner, T.G.; Woldegiorgis, M.; Lewis, M.E.; Bai, C.; et al. Microscale screening of antibody libraries as maytansinoid antibody-drug conjugates. mAbs 2016, 8, 513–523. [Google Scholar] [CrossRef]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef]
- Kamath, A.V.; Iyer, S. Preclinical Pharmacokinetic Considerations for the Development of Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3470–3479. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tang, F.; Yang, Y.; Zhao, L.; Zhou, H.; Dong, J.; Huang, W. Real-Time Analysis on Drug-Antibody Ratio of Antibody-Drug Conjugates for Synthesis, Process Optimization, and Quality Control. Sci. Rep. 2017, 7, 7763. [Google Scholar] [CrossRef] [PubMed]
- Cilliers, C.; Menezes, B.; Nessler, I.; Linderman, J.; Thurber, G.M. Improved Tumor Penetration and Single-Cell Targeting of Antibody-Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018, 78, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H.; Lowman, H.B. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef]
- Kim, K.M.; McDonagh, C.F.; Westendorf, L.; Brown, L.L.; Sussman, D.; Feist, T.; Lyon, R.; Alley, S.C.; Okeley, N.M.; Zhang, X.; et al. Anti-CD30 diabody-drug conjugates with potent antitumor activity. Mol. Cancer Ther. 2008, 7, 2486–2497. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient Preparation of Site-Specific Antibody-Drug Conjugates Using Cysteine Insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef]
- Sochaj, A.M.; Swiderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015, 33, 775–784. [Google Scholar] [CrossRef]
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kim, J. Advances in the Development of Site-Specific Antibody-Drug Conjugation. Anti-Cancer Agents Med. Chem. 2015, 15, 828–836. [Google Scholar] [CrossRef]
- Hotzel, I.; Theil, F.P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Bumbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Tibbitts, J.; Canter, D.; Graff, R.; Smith, A.; Khawli, L.A. Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. mAbs 2016, 8, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A. Biological importance of glycosylation. Dev. Biol. Stand. 1998, 96, 43–47. [Google Scholar]
- Shibata-Koyama, M.; Iida, S.; Misaka, H.; Mori, K.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated rituximab potentiates human neutrophil phagocytosis through its high binding for FcgammaRIIIb and MHC class II expression on the phagocytotic neutrophils. Exp. Hematol. 2009, 37, 309–321. [Google Scholar] [CrossRef]
- Stork, R.; Zettlitz, K.A.; Muller, D.; Rether, M.; Hanisch, F.G.; Kontermann, R.E. N-glycosylation as novel strategy to improve pharmacokinetic properties of bispecific single-chain diabodies. J. Biol. Chem. 2008, 283, 7804–7812. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef]
- Wright, A.; Morrison, S.L. Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J. Exp. Med. 1994, 180, 1087–1096. [Google Scholar] [CrossRef]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjugate Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef]
- Le, N.P.; Bowden, T.A.; Struwe, W.B.; Crispin, M. Immune recruitment or suppression by glycan engineering of endogenous and therapeutic antibodies. Biochim. Biophys. Acta 2016, 1860, 1655–1668. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chen, W.; Feng, Y.; Dimitrov, D.S. Crystallizable Fragment Glycoengineering for Therapeutic Antibodies Development. Front. Immunol. 2017, 8, 1554. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Katoh, T.; Saldova, R.; O’Flaherty, R.; Izumi, T.; Mimura-Kimura, Y.; Utsunomiya, T.; Mizukami, Y.; Yamamoto, K.; Matsumoto, T.; et al. Glycosylation engineering of therapeutic IgG antibodies: Challenges for the safety, functionality and efficacy. Protein Cell 2018, 9, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Gefen, T.; Vaya, J.; Khatib, S.; Harkevich, N.; Artoul, F.; Heller, E.D.; Pitcovski, J.; Aizenshtein, E. The impact of PEGylation on protein immunogenicity. Int. Immunopharmacol. 2013, 15, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Mero, A.; Clementi, C.; Veronese, F.M.; Pasut, G. Covalent conjugation of poly(ethylene glycol) to proteins and peptides: Strategies and methods. Methods Mol. Biol. 2011, 751, 95–129. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. Biodrugs Clin. Immunother. Biopharm. Gene Ther. 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Caliceti, P.; Veronese, F.M. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv. Drug Deliv. Rev. 2003, 55, 1261–1277. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef]
- Zacchigna, M.; Di Luca, G.; Cateni, F.; Maurich, V.; Ballico, M.; Bonora, G.M.; Drioli, S. New MultiPEG-conjugated theophylline derivatives: Synthesis and pharmacological evaluations. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2007, 30, 343–350. [Google Scholar] [CrossRef]
- Drioli, S.; Bonora, G.M.; Ballico, M. New syntheses of branched, multifunctional high-molecular weight poly(ethylene glycol)s or (MultiPEG)s. Open Org. Chem. J. 2008, 2, 17–25. [Google Scholar] [CrossRef]
- Sreedhara, A.; Lau, K.; Li, C.; Hosken, B.; Macchi, F.; Zhan, D.; Shen, A.; Steinmann, D.; Schoneich, C.; Lentz, Y. Role of surface exposed tryptophan as substrate generators for the antibody catalyzed water oxidation pathway. Mol. Pharm. 2013, 10, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Yin, J.; Joyce, M.; Lau, K.; Wecksler, A.T.; Deperalta, G.; Yi, L.; John Wang, Y.; Kabakoff, B.; Kishore, R.S. Effect of ambient light on IgG1 monoclonal antibodies during drug product processing and development. Eur. J. Pharm. Biopharm. 2016, 100, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Torosantucci, R.; Schoneich, C.; Jiskoot, W. Oxidation of therapeutic proteins and peptides: Structural and biological consequences. Pharm. Res. 2014, 31, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vlasak, J.; Li, Y.; Pristatsky, P.; Fang, Y.; Pittman, T.; Roman, J.; Wang, Y.; Prueksaritanont, T.; Ionescu, R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol. Immunol. 2011, 48, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Feng, J.; Lin, H.Y.; Mullapudi, S.; Bishop, E.; Tous, G.I.; Casas-Finet, J.; Hakki, F.; Strouse, R.; Schenerman, M.A. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal. Chem. 2007, 79, 2797–2805. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.P.; Fielder, P.J.; Khawli, L.A. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjugate Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef]
- Gangopadhyay, A.; Petrick, A.T.; Thomas, P. Modification of antibody isoelectric point affects biodistribution of 111-indium-labeled antibody. Nucl. Med. Biol. 1996, 23, 257–261. [Google Scholar] [CrossRef]
- Ten Kate, C.I.; Fischman, A.J.; Rubin, R.H.; Fucello, A.J.; Riexinger, D.; Wilkinson, R.A.; Du, L.; Khaw, B.A.; Strauss, H.W. Effect of isoelectric point on biodistribution and inflammation: Imaging with indium-111-labelled IgG. Eur. J. Nucl. Med. 1990, 17, 305–309. [Google Scholar] [CrossRef]
- Bickel, U.; Lee, V.M.Y.; Pardridge, W.M. Pharmacokinetic differences between 111in-and 125i-labeled cationized monoclonal antibody against β-amyloid in mouse and dog. Drug Deliv. 1995, 2, 128–135. [Google Scholar] [CrossRef]
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D.; et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs 2010, 2, 613–624. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Bickel, U.; Buciak, J.; Yang, J.; Diagne, A.; Aepinus, C. Cationization of a monoclonal antibody to the human immunodeficiency virus REV protein enhances cellular uptake but does not impair antigen binding of the antibody. Immunol. Lett. 1994, 42, 191–195. [Google Scholar] [CrossRef]
- Wall, D.A.; Maack, T. Endocytic uptake, transport, and catabolism of proteins by epithelial cells. Am. J. Physiol. 1985, 248, C12–C20. [Google Scholar] [CrossRef] [PubMed]
- Robieux, I.; Sorio, R.; Borsatti, E.; Cannizzaro, R.; Vitali, V.; Aita, P.; Freschi, A.; Galligioni, E.; Monfardini, S. Pharmacokinetics of vinorelbine in patients with liver metastases. Clin. Pharmacol. Ther. 1996, 59, 32–40. [Google Scholar] [CrossRef]
- Twelves, C.J.; O’Reilly, S.M.; Coleman, R.E.; Richards, M.A.; Rubens, R.D. Weekly epirubicin for breast cancer with liver metastases and abnormal liver biochemistry. Br. J. Cancer 1989, 60, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Hellmann, M.D.; Hamid, O.; Tsai, K.K.; Loo, K.L.; Gubens, M.A.; Rosenblum, M.; Harview, C.L.; Taube, J.M.; Handley, N. Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol Res. 2017, 5, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Franconi, F.; Rosano, G.; Campesi, I. Need for gender-specific pre-analytical testing: The dark side of the moon in laboratory testing. Int. J. Cardiol. 2015, 179, 514–535. [Google Scholar] [CrossRef]
- Clayton, J.A. Applying the new SABV (sex as a biological variable) policy to research and clinical care. Physiol Behav. 2018, 187, 2–5. [Google Scholar] [CrossRef]
- NOT-OD-15-102: Consideration of Sex as a Biological Variable in NIH-funded Research. Available online: https://grants.nih.gov/grants/guide/notice-files/NOT-OD-15-102.html (accessed on 18 October 2018).
- Korth-Bradley, J.M.; Dowell, J.A.; King, S.P.; Liu, H.; Berger, M.S. Impact of age and gender on the pharmacokinetics of gemtuzumab ozogamicin. Pharmacotherapy 2001, 21, 1175–1180. [Google Scholar] [CrossRef]
- Gibiansky, L.; Passey, C.; Roy, A.; Bello, A.; Gupta, M. Model-based pharmacokinetic analysis of elotuzumab in patients with relapsed/refractory multiple myeloma. J. Pharmacokinet. Pharmacodyn. 2016, 43, 243–257. [Google Scholar] [CrossRef]
- Lu, D.; Girish, S.; Gao, Y.; Wang, B.; Yi, J.H.; Guardino, E.; Samant, M.; Cobleigh, M.; Rimawi, M.; Conte, P.; et al. Population pharmacokinetics of trastuzumab emtansine (T-DM1), a HER2-targeted antibody-drug conjugate, in patients with HER2-positive metastatic breast cancer: Clinical implications of the effect of covariates. Cancer Chemother. Pharmacol. 2014, 74, 399–410. [Google Scholar] [CrossRef]
- Gupta, M.; Lorusso, P.M.; Wang, B.; Yi, J.H.; Burris, H.A., 3rd; Beeram, M.; Modi, S.; Chu, Y.W.; Agresta, S.; Klencke, B.; et al. Clinical implications of pathophysiological and demographic covariates on the population pharmacokinetics of trastuzumab emtansine, a HER2-targeted antibody-drug conjugate, in patients with HER2-positive metastatic breast cancer. J. Clin. Pharmacol. 2012, 52, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Caron, W.P.; Lay, J.C.; Fong, A.M.; La-Beck, N.M.; Kumar, P.; Newman, S.E.; Zhou, H.; Monaco, J.H.; Clarke-Pearson, D.L.; Brewster, W.R.; et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J. Pharmacol. Exp. Ther. 2013, 347, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.C.; Phuangsab, A.; Van Alten, P.J.; Walter, R.J. Steroid sex hormones and macrophage function: Regulation of chemiluminescence and phagocytosis. Am. J. Reprod. Immunol. 1996, 35, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Darr, D.B.; Santos, C.M.; Ross, M.; Valdivia, A.; Jordan, J.L.; Midkiff, B.R.; Cohen, S.; Nikolaishvili-Feinberg, N.; Miller, C.R.; et al. Effects of tumor microenvironment heterogeneity on nanoparticle disposition and efficacy in breast cancer tumor models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6083–6095. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Tarrant, T.K.; White, T.F.; Barrow, D.A.; Santos, C.M.; Timoshchenko, R.G.; Hanna, S.K.; Ramanathan, R.K.; Lee, C.R.; Bae-Jump, V.L.; et al. Roles of chemokines CCL2 and CCL5 in the pharmacokinetics of PEGylated liposomal doxorubicin in vivo and in patients with recurrent epithelial ovarian cancer. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Vernon-Roberts, B. The effects of steroid hormones on macrophage activity. Int. Rev. Cytol. 1969, 25, 131–159. [Google Scholar] [PubMed]
- Atkinson, J.P.; Schreiber, A.D.; Frank, M.M. Effects of corticosteroids and splenectomy on the immune clearance and destruction of erythrocytes. J. Clin. Investig. 1973, 52, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Netti, F.; Schreiber, A.D. Effect of estradiol and steroid analogues on the clearance of immunoglobulin G-coated erythrocytes. J. Clin. Investig. 1985, 75, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Ruiz, P.; Bernal, J.A.; Escobar, M.; Garcia-Egido, A.; Lopez-Saez, J.J. Enhancement of splenic-macrophage Fcgamma receptor expression by treatment with estrogens. Clin. Diagn. Lab. Immunol. 2001, 8, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Ruiz, P.; Briceno, F.; Lopez, R.; Michan, A. Treatment with progesterone analogues decreases macrophage Fcgamma receptors expression. Clin. Immunol. Immunopathol. 1998, 89, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Holdstock, G.; Chastenay, B.F.; Krawitt, E.L. Effects of testosterone, oestradiol and progesterone on immune regulation. Clin. Exp. Immunol. 1982, 47, 449–456. [Google Scholar] [PubMed]
- Schreiber, A.D.; Nettl, F.M.; Sanders, M.C.; King, M.; Szabolcs, P.; Friedman, D.; Gomez, F. Effect of endogenous and synthetic sex steroids on the clearance of antibody-coated cells. J. Immunol. 1988, 141, 2959–2966. [Google Scholar] [PubMed]
- Werb, Z.; Foley, R.; Munck, A. Interaction of glucocorticoids with macrophages. Identification of glucocorticoid receptors in monocytes and macrophages. J. Exp. Med. 1978, 147, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.K.; Mitcho, Y.L.; Rath, N.C. Effect of estradiol on interleukin 1 synthesis by macrophages. Int. J. Immunopharmacol. 1988, 10, 247–252. [Google Scholar] [PubMed]
- Bhalla, A.K.; Amento, E.P.; Krane, S.M. Differential effects of 1,25-dihydroxyvitamin D3 on human lymphocytes and monocyte/macrophages: Inhibition of interleukin-2 and augmentation of interleukin-1 production. Cell. Immunol. 1986, 98, 311–322. [Google Scholar] [CrossRef]
- Blifeld, C.; Prehn, J.L.; Jordan, S.C. Stimulus-specific 1,25(OH)2D3 modulation of TNF and IL-1-beta gene expression in human peripheral blood mononuclear cells and monocytoid cell lines. Transplantation 1991, 51, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Boltz-Nitulescu, G.; Willheim, M.; Spittler, A.; Leutmezer, F.; Tempfer, C.; Winkler, S. Modulation of IgA, IgE, and IgG Fc receptor expression on human mononuclear phagocytes by 1 alpha,25-dihydroxyvitamin D3 and cytokines. J. Leukoc. Biol. 1995, 58, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, C.; Jin, C.H.; Yamaguchi, Y.; Tomida, M.; Hozumi, M.; Matsuda, T.; Hirano, T.; Kishimoto, T.; Suda, T. Production of interleukin 6 and its relation to the macrophage differentiation of mouse myeloid leukemia cells (M1) treated with differentiation-inducing factor and 1 alpha,25-dihydroxyvitamin D3. Biochem. Biophys. Res. Commun. 1989, 158, 660–666. [Google Scholar] [CrossRef]
- Saggese, G.; Federico, G.; Balestri, M.; Toniolo, A. Calcitriol inhibits the PHA-induced production of IL-2 and IFN-gamma and the proliferation of human peripheral blood leukocytes while enhancing the surface expression of HLA class II molecules. J. Endocrinol. Investig. 1989, 12, 329–335. [Google Scholar] [CrossRef]
- Taimi, M.; Defacque, H.; Commes, T.; Favero, J.; Caron, E.; Marti, J.; Dornand, J. Effect of retinoic acid and vitamin D on the expression of interleukin-1 beta, tumour necrosis factor-alpha and interleukin-6 in the human monocytic cell line U937. Immunology 1993, 79, 229–235. [Google Scholar] [PubMed]
- Keophiphath, M.; Rouault, C.; Divoux, A.; Clement, K.; Lacasa, D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Luther, S.A.; Cyster, J.G. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2001, 2, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry: Pharmacokinetics in Patients with Impaired Renal Function—Study Design Data Analysis, and Impact on Dosing and Labeling. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf (accessed on 16 September 2018).
- Guidance for Industry: Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design Data Analysis, and Impact on Dosing and Labeling. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf (accessed on 16 September 2018).
- Aldoss, I.T.; Plumb, T.; Zhen, W.K.; Lydiatt, D.D.; Ganti, A.K. Cetuximab in hemodialysis: A case report. Head Neck 2009, 31, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Micallef, R.A.; Barrett-Lee, P.J.; Donovan, K.; Ashraf, M.; Williams, L. Trastuzumab in patients on haemodialysis for renal failure. Clin. Oncol. 2007, 19, 559. [Google Scholar] [CrossRef]
- Sato, Y.; Doden, K.; Nishida, Y.; Shimizu, S.; Tanaka, N.; Yagi, D.; Asaumi, Y.; Hirano, Y.; Maeda, K.; Miyanaga, T.; et al. Bevacizumab therapy for a colorectal cancer patient or hemodialysis with hepatic metastasis. Gan Kagaku Ryoho. Cancer Chemother. 2013, 40, 647–650. [Google Scholar]
- Shi, S. Biologics: An update and challenge of their pharmacokinetics. Curr. Drug Metab. 2014, 15, 271–290. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Seidling, H.M. Pharmacokinetic predictions for patients with renal impairment: Focus on peptides and protein drugs. Br. J. Clin. Pharmacol. 2012, 74, 66–74. [Google Scholar] [CrossRef]
- Covell, D.G.; Barbet, J.; Holton, O.D.; Black, C.D.; Parker, R.J.; Weinstein, J.N. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Cancer Res. 1986, 46, 3969–3978. [Google Scholar]
- Gu, J.; Congdon, E.E.; Sigurdsson, E.M. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J. Biol. Chem. 2013, 288, 33081–33095. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Sato, Y.; Okada, T.; Chang, K.; Endo, T.; Morrison, S. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology 2000, 10, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; Kennedy, P.J.; Santos, H.A.; Barrias, C.; Sarmento, B. A comprehensive review of the neonatal Fc receptor and its application in drug delivery. Pharmacol. Ther. 2016, 161, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. mAbs 2014, 6, 502–508. [Google Scholar] [CrossRef]
- Reardon, D.A.; Lassman, A.B.; van den Bent, M.; Kumthekar, P.; Merrell, R.; Scott, A.M.; Fichtel, L.; Sulman, E.P.; Gomez, E.; Fischer, J.; et al. Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro-Oncology 2017, 19, 965–975. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef]
- Zhang, Y.; Pardridge, W.M. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J. Neuroimmunol. 2001, 114, 168–172. [Google Scholar] [CrossRef]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Balthasar, J.P. Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 2016, 43, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Le, T.; Rock, B.M.; Rock, D.A.; Siu, S.; Huard, J.N.; Conner, K.P.; Milburn, R.R.; O’Neill, J.W.; Tometsko, M.E.; et al. Altering Antibody-Drug Conjugate Binding to the Neonatal Fc Receptor Impacts Efficacy and Tolerability. Mol. Pharm. 2016, 13, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 2012, 39, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Wroblewski, V.J. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J. Biol. Chem. 2007, 282, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Hinton, P.R.; Xiong, J.M.; Johlfs, M.G.; Tang, M.T.; Keller, S.; Tsurushita, N. An engineered human IgG1 antibody with longer serum half-life. J. Immunol. 2006, 176, 346–356. [Google Scholar] [CrossRef]
- Vugmeyster, Y.; Xu, X.; Theil, F.P.; Khawli, L.A.; Leach, M.W. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J. Biol. Chem. 2012, 3, 73–92. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Jiang, W.; Wroblewski, V.J. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: Relationship to pharmacokinetics in mice and primates. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Brachet, G.; Respaud, R.; Arnoult, C.; Henriquet, C.; Dhommee, C.; Viaud-Massuard, M.C.; Heuze-Vourc’h, N.; Joubert, N.; Pugniere, M.; Gouilleux-Gruart, V. Increment in Drug Loading on an Antibody-Drug Conjugate Increases Its Binding to the Human Neonatal Fc Receptor in Vitro. Mol. Pharm. 2016, 13, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcgamma receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M. Incorporation of FcRn-mediated disposition model to describe the population pharmacokinetics of therapeutic monoclonal IgG antibody in clinical patients. Biopharm. Drug Dispos. 2016, 37, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Ternant, D.; Arnoult, C.; Pugniere, M.; Dhommee, C.; Drocourt, D.; Perouzel, E.; Passot, C.; Baroukh, N.; Mulleman, D.; Tiraby, G.; et al. IgG1 Allotypes Influence the Pharmacokinetics of Therapeutic Monoclonal Antibodies through FcRn Binding. J. Immunol. 2016, 196, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Mancardi, D.A.; Albanesi, M.; Jonsson, F.; Iannascoli, B.; Van Rooijen, N.; Kang, X.; England, P.; Daeron, M.; Bruhns, P. The high-affinity human IgG receptor FcgammaRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood 2013, 121, 1563–1573. [Google Scholar] [CrossRef]
- Van der Poel, C.E.; Spaapen, R.M.; van de Winkel, J.G.; Leusen, J.H. Functional characteristics of the high affinity IgG receptor, FcgammaRI. J. Immunol. 2011, 186, 2699–2704. [Google Scholar] [CrossRef]
- Abuqayyas, L.; Zhang, X.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcgammaR on the pharmacokinetics and anti-platelet effects of MWReg30, a monoclonal anti-GPIIb antibody. Int. J. Pharm. 2013, 444, 185–192. [Google Scholar] [CrossRef]
- Abuqayyas, L.; Balthasar, J.P. Application of knockout mouse models to investigate the influence of FcγR on the tissue distribution and elimination of 8C2, a murine IgG1 monoclonal antibody. Int. J. Pharm. 2012, 439, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Pegram, M.; Ngo, D. Application and potential limitations of animal models utilized in the development of trastuzumab (Herceptin): A case study. Adv. Drug Deliv. Rev. 2006, 58, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.H.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. Immunother. Cancer 2014, 2, 29. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Li, F.; Ulrich, M.; Jonas, M.; Stone, I.J.; Linares, G.; Zhang, X.; Westendorf, L.; Benjamin, D.R.; Law, C.L. Tumor-Associated Macrophages Can Contribute to Antitumor Activity through FcgammaR-Mediated Processing of Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Kenny, J.R.; Liu, M.M.; Chow, A.T.; Earp, J.C.; Evers, R.; Slatter, J.G.; Wang, D.D.; Zhang, L.; Zhou, H. Therapeutic protein drug-drug interactions: Navigating the knowledge gaps-highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA workshop. AAPS J. 2013, 15, 933–940. [Google Scholar] [CrossRef]
- Agus, D.B.; Gordon, M.S.; Taylor, C.; Natale, R.B.; Karlan, B.; Mendelson, D.S.; Press, M.F.; Allison, D.E.; Sliwkowski, M.X.; Lieberman, G.; et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2534–2543. [Google Scholar] [CrossRef]
- Cortes, J.; Swain, S.M.; Kudaba, I.; Hauschild, M.; Patel, T.; Grincuka, E.; Masuda, N.; McNally, V.; Ross, G.; Brewster, M.; et al. Absence of pharmacokinetic drug-drug interaction of pertuzumab with trastuzumab and docetaxel. Anti-Cancer Drugs 2013, 24, 1084–1092. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Yoshino, T.; Fuse, N.; Ura, T.; Takahari, D.; Feng, H.P.; Shimamoto, T.; Noguchi, K.; Ohtsu, A. Phase 1 pharmacokinetic study of MK-0646 (dalotuzumab), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in combination with cetuximab and irinotecan in Japanese patients with advanced colorectal cancer. Cancer Chemother. Pharmacol. 2013, 72, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, L.; Gonzalez, A.; Leger, O.; Beck, A.; Pauwels, P.J.; Haeuw, J.F.; Corvaia, N. A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances the antitumor activity of vinorelbine and anti-epidermal growth factor receptor therapy against human cancer xenografts. Int. J. Cancer 2005, 113, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; McKern, N.M.; Ward, C.W. Signalling by the type 1 insulin-like growth factor receptor: Interplay with the epidermal growth factor receptor. Growth Factors 2004, 22, 89–95. [Google Scholar] [CrossRef]
- Atzori, F.; Tabernero, J.; Cervantes, A.; Prudkin, L.; Andreu, J.; Rodriguez-Braun, E.; Domingo, A.; Guijarro, J.; Gamez, C.; Rodon, J.; et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6304–6312. [Google Scholar] [CrossRef]
- Sclafani, F.; Kim, T.Y.; Cunningham, D.; Kim, T.W.; Tabernero, J.; Schmoll, H.J.; Roh, J.K.; Kim, S.Y.; Park, Y.S.; Guren, T.K.; et al. A Randomized Phase II/III Study of Dalotuzumab in Combination With Cetuximab and Irinotecan in Chemorefractory, KRAS Wild-Type, Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2015, 107, djv258. [Google Scholar] [CrossRef] [PubMed]
- Fayad, L.; Offner, F.; Smith, M.R.; Verhoef, G.; Johnson, P.; Kaufman, J.L.; Rohatiner, A.; Advani, A.; Foran, J.; Hess, G.; et al. Safety and clinical activity of a combination therapy comprising two antibody-based targeting agents for the treatment of non-Hodgkin lymphoma: Results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Kheir, W.J.; Sniegowski, M.C.; El-Sawy, T.; Li, A.; Esmaeli, B. Ophthalmic complications of targeted cancer therapy and recently recognized ophthalmic complications of traditional chemotherapy. Surv. Ophthalmol. 2014, 59, 493–502. [Google Scholar] [CrossRef]
- Renouf, D.J.; Velazquez-Martin, J.P.; Simpson, R.; Siu, L.L.; Bedard, P.L. Ocular toxicity of targeted therapies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3277–3286. [Google Scholar] [CrossRef]
- Beeram, M.; Krop, I.E.; Burris, H.A.; Girish, S.R.; Yu, W.; Lu, M.W.; Holden, S.N.; Modi, S. A phase 1 study of weekly dosing of trastuzumab emtansine (T-DM1) in patients with advanced human epidermal growth factor 2-positive breast cancer. Cancer 2012, 118, 5733–5740. [Google Scholar] [CrossRef]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and Activity of Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, in Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer: A Phase I Expansion Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Dupuis, J.; Tilly, H.; Morschhauser, F.; Laine, F.; Houot, R.; Haioun, C.; Copie, C.; Varga, A.; Lambert, J.; et al. A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Forero-Torres, A.; Ramchandren, R.; Pal, S.K.; Ansell, S.M.; Infante, J.R.; de Vos, S.; Hamlin, P.A.; Kim, S.K.; Whiting, N.C.; et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Motzer, R.J.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; Kim, S.K.; et al. Phase I Trials of Anti-ENPP3 Antibody-Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4399–4406. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Takano, Y.; Shigeyasu, C.; Imoto, S.; Yamada, M. Abnormal Corneal Lesions Induced by Trastuzumab Emtansine: An Antibody-Drug Conjugate for Breast Cancer. Cornea 2016, 35, 1378–1380. [Google Scholar] [CrossRef]
- Eaton, J.S.; Miller, P.E.; Mannis, M.J.; Murphy, C.J. Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2015, 31, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Translational research: 4 ways to fix the clinical trial. Nature 2011, 477, 526–528. [Google Scholar] [CrossRef]
- Matthews, R.A. Medical progress depends on animal models—Doesn’t it? J. R. Soc. Med. 2008, 101, 95–98. [Google Scholar] [CrossRef]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; Macleod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of treatment effects between animal experiments and clinical trials: Systematic review. BMJ (Clin. Res. Ed.) 2007, 334, 197. [Google Scholar] [CrossRef]
- Food and Drug Administration, U.S. Department of Health and Human Services. Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products; Food and Drug Administration, U.S. Department of Health and Human Services: Washington, DC, USA, 2004.
- Thomas, D. Oncology Clinical Trials—Secrets of Success. In BIOtechNOW; Biotechnology Industry Organization: Washington, DC, USA, 2012. [Google Scholar]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef]
- Plowman, J.; Dykes, D.J.; Hollingshead, M.; Simpson-Herren, L.; Alley, M.C. Human Tumor Xenograft Models; Humana Press: Totowa, NJ, USA, 1997. [Google Scholar]
- Bertotti, A.; Trusolino, L. From bench to bedside: Does preclinical practice in translational oncology need some rebuilding? J. Natl. Cancer Inst. 2013, 105, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; de Sauvage, F.; Egeblad, M.; Olive, K.P.; Tuveson, D.; Weiss, W. Recapitulating human cancer in a mouse. Nat. Biotechnol. 2013, 31, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: A bridge to the clinic. Investig. New Drugs 1999, 17, 343–359. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Building a better Trap. Proc. Natl. Acad. Sci. USA 2003, 100, 8624–8625. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lin, D.; Gout, P.W.; Collins, C.C.; Xu, Y.; Wang, Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv. Drug Deliv. Rev. 2014, 79–80, 222–237. [Google Scholar] [CrossRef]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 106. [Google Scholar] [CrossRef]
- Steel, G.G.; Courtenay, V.D.; Peckham, M.J. The response to chemotherapy of a variety of human tumour xenografts. Br. J. Cancer 1983, 47, 1–13. [Google Scholar] [CrossRef]
- Lucas, A.T.; White, T.F.; Deal, A.M.; Herity, L.B.; Song, G.; Santos, C.M.; Zamboni, W.C. Profiling the relationship between tumor-associated macrophages and pharmacokinetics of liposomal agents in preclinical murine models. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 471–482. [Google Scholar] [CrossRef]
- Sassoon, I.; Blanc, V. Antibody-drug conjugate (ADC) clinical pipeline: A review. Methods Mol. Biol. 2013, 1045, 1–27. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, G.; Gupta, M.; Feng, Y.; Statkevich, P.; Roy, A. Exposure-Response Analysis of Nivolumab in Patients With Previously Treated or Untreated Advanced Melanoma. J. Clin. Pharmacol. 2017, 57, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Bouchlaka, M.N.; Sckisel, G.D.; Chen, M.; Mirsoian, A.; Zamora, A.E.; Maverakis, E.; Wilkins, D.E.; Alderson, K.L.; Hsiao, H.H.; Weiss, J.M.; et al. Aging predisposes to acute inflammatory induced pathology after tumor immunotherapy. J. Exp. Med. 2013, 210, 2223–2237. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef]
- Hurez, V.; Daniel, B.J.; Sun, L.; Liu, A.J.; Ludwig, S.M.; Kious, M.J.; Thibodeaux, S.R.; Pandeswara, S.; Murthy, K.; Livi, C.B.; et al. Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res. 2012, 72, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Pfister, G.; Weiskopf, D.; Lazuardi, L.; Kovaiou, R.D.; Cioca, D.P.; Keller, M.; Lorbeg, B.; Parson, W.; Grubeck-Loebenstein, B. Naive T cells in the elderly: Are they still there? Ann. N. Y. Acad. Sci. 2006, 1067, 152–157. [Google Scholar] [CrossRef]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed]
- De Heredia, F.P.; Gomez-Martinez, S.; Marcos, A. Obesity, inflammation and the immune system. Proc. Nutr. Soc. 2012, 71, 332–338. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Tomanek-Chalkley, A.; Askeland, E.J.; Kucaba, T.; Griffith, T.S.; Norian, L.A. Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J. Immunol. 2012, 189, 1311–1321. [Google Scholar] [CrossRef]
- Macia, L.; Delacre, M.; Abboud, G.; Ouk, T.S.; Delanoye, A.; Verwaerde, C.; Saule, P.; Wolowczuk, I. Impairment of dendritic cell functionality and steady-state number in obese mice. J. Immunol. 2006, 177, 5997–6006. [Google Scholar] [CrossRef]
- Milner, J.J.; Sheridan, P.A.; Karlsson, E.A.; Schultz-Cherry, S.; Shi, Q.; Beck, M.A. Diet-induced obese mice exhibit altered heterologous immunity during a secondary 2009 pandemic H1N1 infection. J. Immunol. 2013, 191, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Schneider, H.J.; Friedrich, N.; Klotsche, J.; Pieper, L.; Nauck, M.; John, U.; Dorr, M.; Felix, S.; Lehnert, H.; Pittrow, D.; et al. The predictive value of different measures of obesity for incident cardiovascular events and mortality. J. Clin. Endocrinol. Metab. 2010, 95, 1777–1785. [Google Scholar] [CrossRef]
- Henry, S.P.; Monteith, D.; Levin, A.A. Antisense oligonucleotide inhibitors for the treatment of cancer: 2. Toxicological properties of phosphorothioate oligodeoxynucleotides. Anti-Cancer Drug Des. 1997, 12, 395–408. [Google Scholar]
- Haley, P.J. Species differences in the structure and function of the immune system. Toxicology 2003, 188, 49–71. [Google Scholar] [CrossRef]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, M.; Schoeden, G. Macrophage biology and immunology: Man is not a mouse. J. Leukoc. Biol. 2007, 81, 579. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B. Nitric oxide production and nitric oxide synthase type 2 expression by human mononuclear phagocytes: A review. Mol. Med. 1998, 4, 557–591. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, M.; Schoedon, G. Species differences in macrophage NO production are important. Nat. Immunol. 2002, 3, 102. [Google Scholar] [CrossRef]
- Bornstein, G.G.; Klakamp, S.L.; Andrews, L.; Boyle, W.J.; Tabrizi, M. Surrogate approaches in development of monoclonal antibodies. Drug Discov. Today 2009, 14, 1159–1165. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef]
- Mechetina, L.V.; Najakshin, A.M.; Alabyev, B.Y.; Chikaev, N.A.; Taranin, A.V. Identification of CD16-2, a novel mouse receptor homologous to CD16/Fc gamma RIII. Immunogenetics 2002, 54, 463–468. [Google Scholar] [CrossRef]
- Rogers, K.A.; Scinicariello, F.; Attanasio, R. IgG Fc receptor III homologues in nonhuman primate species: Genetic characterization and ligand interactions. J. Immunol. 2006, 177, 3848–3856. [Google Scholar] [CrossRef] [PubMed]
- Dartois, C.; Freyer, G.; Michallet, M.; Henin, E.; You, B.; Darlavoix, I.; Vermot-Desroches, C.; Tranchand, B.; Girard, P. Exposure-effect population model of inolimomab, a monoclonal antibody administered in first-line treatment for acute graft-versus-host disease. Clin. Pharmacokinet. 2007, 46, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Hinton, P.R.; Johlfs, M.G.; Xiong, J.M.; Hanestad, K.; Ong, K.C.; Bullock, C.; Keller, S.; Tang, M.T.; Tso, J.Y.; Vasquez, M.; et al. Engineered human IgG antibodies with longer serum half-lives in primates. J. Biol. Chem. 2004, 279, 6213–6216. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Tsen, M.F.; Ghetie, V.; Ward, E.S. Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur. J. Immunol. 1994, 24, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, Y.; Hong, H.J. Antibody engineering for the development of therapeutic antibodies. Mol. Cells 2005, 20, 17–29. [Google Scholar] [PubMed]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc receptor and IgG-based therapeutics. mAbs 2011, 3, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Devanaboyina, S.C.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [CrossRef]
- Gurbaxani, B.; Dela Cruz, L.L.; Chintalacharuvu, K.; Morrison, S.L. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol. Immunol. 2006, 43, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.A.; Benson, K.; Davis-Bruno, K.; Dempster, M.; Finch, G.L.; Harlow, P.; Haggerty, H.G.; Hart, T.; Kinter, L.; Leighton, J.K.; et al. Nonclinical aspects of biopharmaceutical development: Discussion of case studies at a PhRMA-FDA workshop. Int. J. Toxicol. 2008, 27, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.A.; Tseng, C.M.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Zhou, J.; Mateos, F.; Ober, R.J.; Ward, E.S. Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J. Mol. Biol. 2005, 345, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Petkova, S.B.; Akilesh, S.; Sproule, T.J.; Christianson, G.J.; Al Khabbaz, H.; Brown, A.C.; Presta, L.G.; Meng, Y.G.; Roopenian, D.C. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: Potential application in humorally mediated autoimmune disease. Int. Immunol. 2006, 18, 1759–1769. [Google Scholar] [CrossRef]
- Tam, S.H.; McCarthy, S.G.; Brosnan, K.; Goldberg, K.M.; Scallon, B.J. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. mAbs 2013, 5, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H.; DiLea, C. First-time-in-human dose selection: Allometric thoughts and perspectives. J. Clin. Pharmacol. 1995, 35, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Adult Healthy Volunteer; USFDA: Silver Spring, MD, USA, 2005.
- Felici, A.; Verweij, J.; Sparreboom, A. Dosing strategies for anticancer drugs: The good, the bad and body-surface area. Eur. J. Cancer 2002, 38, 1677–1684. [Google Scholar] [CrossRef]
- Mahmood, I. Interspecies scaling: Predicting clearance of anticancer drugs in humans. A comparative study of three different approaches using body weight or body surface area. Eur. J. Drug Metab. Pharmacokinet. 1996, 21, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. mAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Caron, W.P.; Clewell, H.; Dedrick, R.; Ramanathan, R.K.; Davis, W.L.; Yu, N.; Tonda, M.; Schellens, J.H.; Beijnen, J.H.; Zamboni, W.C. Allometric scaling of pegylated liposomal anticancer drugs. J. Pharmacokinet. Pharmacodyn. 2011, 38, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, D.; Wong, A.; Drake, E.; Reyes, A.E., 2nd; Lin, B.C.; Stephan, J.P.; Desnoyers, L.; Shen, B.Q.; Dennis, M.S. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. mAbs 2011, 3, 376–386. [Google Scholar] [CrossRef]
- Vugmeyster, Y.; Szklut, P.; Wensel, D.; Ross, J.; Xu, X.; Awwad, M.; Gill, D.; Tchistiakov, L.; Warner, G. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm. Res. 2011, 28, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Iyer, S.; Theil, F.P.; Mortensen, D.L.; Fielder, P.J.; Prabhu, S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: What have we learned? mAbs 2011, 3, 61–66. [Google Scholar] [CrossRef]
- Ling, J.; Zhou, H.; Jiao, Q.; Davis, H.M. Interspecies scaling of therapeutic monoclonal antibodies: Initial look. J. Clin. Pharmacol. 2009, 49, 1382–1402. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, S.; Schellens, J.H. The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. Br. J. Cancer 2007, 97, 577–581. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Brand Name | Manufacturer | Highest Phase of Studies Open | Number of Open Studies | Target Antigen | Linker | Payload | Indications |
| Moxetumomab pasudotox-tdfk | Lumoxiti | AstraZeneca | Approved–2018 | 15 | CD22 | Cleavable (disulfide) | Pseudomonas exotoxin A | Hematological |
| Gemtuzumab ozogamicin | Mylotarg | Pfizer | Approved–2017 | 83 | CD33 | Cleavable (acid labile) | Calicheamicin | Hematological |
| Inotuzumab ozogamicin | Besponsa | Pfizer | Approved–2017 | 32 | CD22 | Cleavable (acid labile) | Calicheamicin | Hematological |
| Trastuzumab emtansine | Kadcyla | Genentech | Approved–2013 | 85 | HER2 | Non-cleavable | DM1 | Solid |
| Brentuximab vedotin | Adcetris | Seattle Genetics | Approved–2011 | 143 | CD30 | Cleavable (protease) | MMAE | Hematological |
| Generic Name | Investigational Name | Manufacturer | Highest Phase of Studies Open | Number of Open Studies | Target Antigen | Linker | Payload | Indications |
| Depatuxizumab mafodotin | ABT-414 | Abbvie | III | 4 | EGFR | Non-cleavable | MMAF | Solid |
| Enfortumab vedotin | ASG-22CE | Astellas Pharma | III | 5 | Nectin 4 | Cleavable (protease) | MMAE | Solid |
| Margetuximab | MGAH22 | MacroGenics | III | 3 | HER2 | Cleavable (thioether) | DM1 | Solid |
| Mirvetuximab soravtansine | IMGN-853 | ImmunoGen | III | 6 | FOLRI 1 | Cleavable (disulfide) | DM4 | Solid |
| Polatuzumab vedotin | DCDS-4501A | Genentech | III | 9 | CD79b | Cleavable (protease) | MMAE | Hematological |
| Rovalpituzumab tesirine | SC0001-SCX | Stemcentrx | III | 9 | DLL3 | Cleavable (protease) | SCX | Solid |
| Sacituzumab govitecan | IMMU-132 | Immunomedics | III | 4 | TROP2 EGP1 | Cleavable (acid labile) | SN-38 | Solid |
| Trastuzumab deruxtecan | DS-8201 | Daiichi Sankyo Inc. | III | 11 | ERBB2 | Cleavable (Protease) | Topoisomerase I inhibitor | Solid |
| Trastuzumab duocarmazine | SYD985 | Synthon Biopharmaceuticals | III | 2 | ERBB2 | Cleavable (Protease) | Duocarmycin | Solid |
| PSMA-PyL | 18F-DCFPyL | Progenics | II/III | 27 | Fluorinated PSMA | Cleavable (Protease) | MMAE | Solid |
| - | AGS-16C3F | Agensys | II | 1 | AGS-16/ENPP3 | Non-cleavable | MMAF | Solid |
| Anetumab Ravtansine | BAY 94-9343 | Bayer Healthcare | II | 11 | Mesothelin | Cleavable (disulfide) | DM4 | Solid |
| Labetuzumab govitecan | IMMU-130 | Immunomedics | II | 2 | CEACAM5 | Cleavable (acid labile) | SN-38 | Solid |
| Tisotumab Vedotin | HuMax-TF | Genmab Seattle Genetics | II | 3 | Tissue Factor | Cleavable (disulfide) | MMAE | Solid |
| - | CX-2009 | Cytomx | I/II | 1 | CD166 | Cleavable (protease) | DM4 | Solid |
| Enapotamab vedotin | HuMax-AXL | Genmab | I/II | 1 | AXL | Cleavable (protease) | MMAE | Solid |
| Indatuximab ravtansine | BT-062 | Biotest | I/II | 1 | CD138 | Cleavable (disulfide) | DM4 | Hematological |
| Pinatuzumab vedotin | DCDT-2980S | Genentech | I/II | 1 | CD22 | Cleavable (protease) | MMAE | Hematological |
| Generic Name | Brand Name | Type of Antibody | Antibody Isotype | Target Antigen |
|---|---|---|---|---|
| Oncology | ||||
| Nivolumab | Opdivo | Human | IgG4 | PD-1R |
| Daratumumab | Darzalex | Human | IgG1 | CD38 |
| Ofatumumab | Arzerra | Human | IgG1 | CD20 |
| Durvalumab | Imfinzi | Human | IgG1 | PD-L1 |
| Ipilimumab | Yervoy | Human | IgG1 | CTLA-4 |
| Necitumumab | Portrazza | Human | IgG1 | EGFR |
| Ramucirumab | Cyramza | Human | IgG1 | VEGFR2 |
| Olaratumab | Lartruvo | Human | IgG1 | PDGFRa |
| Panitumumab | Vectibix | Human | IgG2 | EGFR |
| Avelumab | Bavencio | Human | IgG1 | PD-L1 |
| Cemiplimab-rwlc | Libtayo | Human | IgG4 | PD-1 |
| Atezolizumab | Tecentriq | Humanized | IgG1 | PD-L1 |
| Elotuzumab | Empliciti | Humanized | IgG1 | SLAMF7 |
| Obinutuzumab | Gazyva | Humanized | IgG1 | CD20 |
| Pembrolizumab | Keytruda | Humanized | IgG4 | PD-1R |
| Bevacizumab | Avastin | Humanized | IgG1 | VEGF |
| Bevacizumab-awwb | Mvasi | Humanized | IgG1 | VEGF |
| Pertuzumab | Perjeta | Humanized | IgG1 | HER2 |
| Alemtuzumab | Campath Lemtrada | Humanized | IgG1 | CD52 |
| Trastuzumab | Herceptin | Humanized | IgG1 | HER2 |
| Trastuzumab-dkst | Ogivri | Humanized | IgG1 | HER2 |
| Trastuzumab-pkrb | Herzuma | Humanized | IgG1 | HER2 |
| Blinatumomab | Blincyto | Humanized | IgG1 | CD19 |
| Rituximab/Hyaluronidase | Rituxan Hycela | Chimeric | IgG1 | CD20 |
| Rituximab | Rituxan | Chimeric | IgG1 | CD20 |
| Rituximab-abbs | Truxima | Chimeric | IgG1 | CD20 |
| Dinutuximab | Unituxin | Chimeric | IgG1 | disialoganglioside GD2 |
| Cetuximab | Erbitux | Chimeric | IgG1 | EGFR |
| Inflammatory Diseases | ||||
| Ustekinumab | Stelara | Human | IgG1 | IL-12/IL-23 |
| Secukinumab | Cosentyx | Human | IgG1 | IL6 |
| Belimumab | Benlysta | Human | IgG1 | BLyS |
| Guselkumab | Tremfya | Human | IgG1 | IL23 |
| Adalimumab | Humira | Human | IgG1 | TNFa |
| Adalimumab-atto | Amjevita | Human | IgG1 | TNFa |
| Adalimumab-adbm | Cyltezo | Human | IgG1 | TNFa |
| Adalimumab-adaz | Hyrimoz | Human | IgG1 | TNFa |
| Golimumab | Simponi | Human | IgG1 | TNFa |
| Sarilumab | Kevzara | Human | IgG1 | IL6R |
| Dupilumab | Dupixent | Human | IgG4 | IL4Ra |
| Brodalumab | Siliq | Human | IgG2 | IL-17a |
| Vedolizumab | Entyvio | Humanized | IgG1 | a4b7 integrin |
| Certolizumab pegol | Cimzia | Humanized | Fab | TNFa |
| Ixekizumab | Taltz | Humanized | IgG4 | IL-17a |
| Tocilizumab | Actemra | Humanized | IgG1 | IL-6 receptor |
| Natalizumab | Tysarbi | Humanized | IgG4 | a4-integrin |
| Efalizumab | Raptiva | Humanized | IgG1 | CD11a |
| Tildrakizumab-asmn | Ilumya | Humanized | IgG1 | IL-23 |
| Infliximab | Remicade | Chimeric | IgG1 | TNFa |
| Infliximab-abda | Renflexis | Chimeric | IgG1 | TNFa |
| Infliximab-dyyb | Inflectra | Chimeric | IgG1 | TNFa |
| Infliximab-qbtx | Ixifi | Chimeric | IgG1 | TNFa |
| Organ Transplant | ||||
| Daclizumab | Zinbryta | Humanized | IgG1 | CD25 |
| Basiliximab | Simulect | Chimeric | IgG1 | CD25 |
| Muromonab-CD3 | Orthoclone-OKT3 | Murine | IgG2a | CD3 |
| Miscellaneous Conditions | ||||
| Canakinumab | Ilaris | Human | IgG1 | IL1B |
| Denosumab | Prolia Xgeva | Human | IgG2 | RANKL |
| Bezlotoxumab | Zinplava | Human | IgG1 | C. difficile toxin B |
| Alirocumab | Praluent | Human | IgG1 | PCSK9 |
| Evolocumab | Repatha | Human | IgG2 | PCSK9 |
| Erenumab-aooe | Aimovig | Human | IgG2 | CGRP |
| Burosumab-twza | Crysvita | Human | IgG1 | FGF23 |
| Emapalumab-lzsg | Gamifant | Human | IgG1 | IFNg |
| Raxibacumab | Raxibacumab | Human | IgG1 | B. anthracis toxin |
| Lanadelumab-flyo | Takhzyro | Human | IgG1 | Kallikrein |
| Ocrelizumab | Ocrevus | Humanized | gG1 | CD20 |
| Omalizumab | Xolair | Humanized | IgG1 | IgE |
| Reslizumab | Cinqair | Humanized | IgG4 | IL5 |
| Daclizumab | Zinbryta | Humanized | IgG1 | IL2R |
| Mepolizumab | Nucala | Humanized | IgG1 | IL5 |
| Ranibizumab | Lucentis | Humanized | IgG1 | VEGFR1, VEGFR2 |
| Idarucizumab | Praxabind | Humanized | IgG1 | Dabigatran |
| Fremanezumab-vfrm | Ajovy | Humanized | IgG2 | CGRP |
| Galcanezumab-gnim | Emgality | Humanized | IgG4 | CGRP |
| Benralizumab | Fasenra | Humanized | IgG1 | IL-5Ra |
| Emicizumab-kxwh | Hemlibra | Humanized | IgG4 | Factor IXa & Factor X |
| Mogamulizumab-kpkc | Poteligeo | Humanized | IgG1 | CCR4 |
| Mepolizumab | Nucala | Humanized | IgG1 | IL-5 |
| Ibalizumab-uiyk | Trogarzo | Humanized | IgG4 | HIV-1 |
| Obiltoxaximab | Anthem | Chimeric | IgG1 | Anthrax toxin |
| Siltuximab | Sylvant | Chimeric | IgG1 | IL-6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, A.T.; Robinson, R.; Schorzman, A.N.; Piscitelli, J.A.; Razo, J.F.; Zamboni, W.C. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies 2019, 8, 3. https://doi.org/10.3390/antib8010003
Lucas AT, Robinson R, Schorzman AN, Piscitelli JA, Razo JF, Zamboni WC. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies. 2019; 8(1):3. https://doi.org/10.3390/antib8010003
Chicago/Turabian StyleLucas, Andrew T., Ryan Robinson, Allison N. Schorzman, Joseph A. Piscitelli, Juan F. Razo, and William C. Zamboni. 2019. "Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients" Antibodies 8, no. 1: 3. https://doi.org/10.3390/antib8010003
APA StyleLucas, A. T., Robinson, R., Schorzman, A. N., Piscitelli, J. A., Razo, J. F., & Zamboni, W. C. (2019). Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies, 8(1), 3. https://doi.org/10.3390/antib8010003