Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
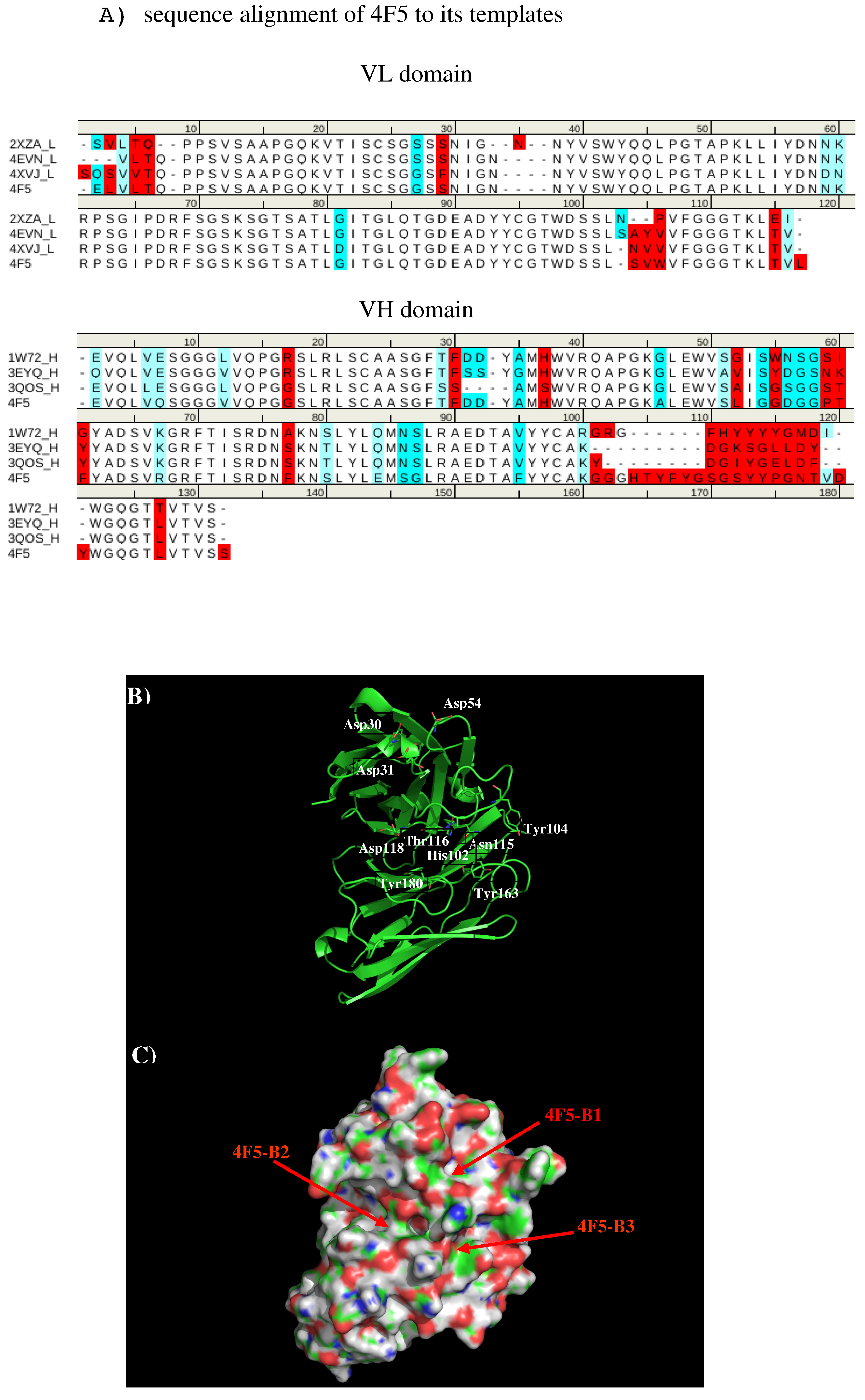
2.1. Homology Modeling of the Antibody 4F5
2.2. MCSS of Functional Groups
2.3. Identification of Sequence Pattern
2.4. Search for Epitopes Based on the Sequence Pattern
3. Results
3.1. Recognition of HA1 by Antibody HC19.
3.2. Recognition of HA1 by Antibodies CR9114 and FI6V3.
3.3. Recognition of HA1 to Antibody BH151
3.4. Prediction of Epitopes of HA1 to Antibody 4F5
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schafer, J.R.; Kawaoka, Y.; Bean, W.J.; Suss, J.; Senne, D.; Webster, R.G. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology 1993, 194, 781. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Fanning, T.G.; Hultin, J.V.; Taubenberger, J.K. Origin and evolution of the 1918 "Spanish" influenza virus hemagglutinin gene. Proc. Natl. Acad. Sci. USA. 1999, 96, 1651. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, Y.; Krauss, S.; Webster, R.G. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J. Virol. 1989, 63, 4603. [Google Scholar] [PubMed]
- Bean, W.J.; Schell, M.; Katz, J.; Kawaoka, Y.; Naeve, C.; Gorman, O.; Webster, R.G. Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J. Virol. 1992, 66, 1129. [Google Scholar] [PubMed]
- Doerr, H.W.; Varwig, D.; Allwinn, R.; Cinatl, J. Will the next human influenza pandemic be caused by the virus of the avian flu A/H5N1? Arguments pro and counter. Med. Microbiol. Immunol. 2006, 195, 45. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, G.J. Avian flu and influenza pandemics in human populations. J. Trop. Pediatr. 2004, 50, 192. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Asha, K.; Khanna, M.; Ronsard, L.; Meseko, C.A.; Sanicas, M. The emerging influenza virus threat: Status and new prospects for its therapy and control. Arch. Virol. 2018, 163, 831. [Google Scholar] [CrossRef]
- Beigel, J.; Bray, M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 2008, 78, 91. [Google Scholar] [CrossRef]
- Lowen, A.C.; Palese, P. Influenza virus transmission: Basic science and implications for the use of antiviral drugs during a pandemic. Infect. Disord. Drug Targets 2007, 7, 318. [Google Scholar] [CrossRef]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA 2006, 295, 891. [Google Scholar] [CrossRef]
- Kiso, M.; Mitamura, K.; Sakai-Tagawa, Y.; Shiraishi, K.; Kawakami, C.; Kimura, K.; Hayden, F.G.; Sugaya, N.; Kawaoka, Y. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 2004, 364, 759. [Google Scholar] [CrossRef]
- Hanson, B.J.; Boon, A.C.; Lim, A.P.; Webb, A.; Ooi, E.E.; Webby, R.J. Passive immunoprophylaxis and therapy with humanized monoclonal antibody specific for influenza A H5 hemagglutinin in mice. Respir. Res. 2006, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.; Ballesteros, A.; Qiu, Q.; Sang, L.P.; Shashikumar, S.; Casares, S.; Brumeanu, T.D. Generation and testing anti-influenza human monoclonal antibodies in a new humanized mouse model (DRAGA: HLA-A2. HLA-DR4. Rag1 KO. IL-2Rγc KO. NOD). Hum. Vaccin. Immunother. 2018, 14, 14–345. [Google Scholar] [CrossRef]
- Chiu, F.F.; Venkatesan, N.; Wu, C.R.; Chou, A.H.; Chen, H.W.; Lian, S.P.; Liu, S.J.; Huang, C.C.; Lian, W.C.; Chong, P.; et al. Immunological study of HA1 domain of hemagglutinin of influenza H5N1 virus. Biochem. Biophys. Res. Commun. 2009, 383, 27. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.A.; Friesen, R.H.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246. [Google Scholar] [CrossRef] [PubMed]
- Fleury, D.; Daniels, R.S.; Skehel, J.J.; Knossow, M.; Bizebard, T. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins 2000, 40, 572. [Google Scholar]
- Fleury, D.; Barrere, B.; Bizebard, T.; Daniels, R.S.; Skehel, J.J.; Knossow, M. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat. Struct. Biol. 1999, 6, 530. [Google Scholar] [CrossRef]
- Fleury, D.; Wharton, S.A.; Skehel, J.J.; Knossow, M.; Bizebard, T. Antigen distortion allows influenza virus to escape neutralization. Nat. Struct. Biol. 1998, 5, 119. [Google Scholar] [CrossRef]
- Gerhard, W.; Yewdell, J.; Frankel, M.E.; Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 1981, 290, 713. [Google Scholar] [CrossRef]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343. [Google Scholar] [CrossRef] [PubMed]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Bosecke, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, X.; Zhang, Q.; Zeng, X.; Shi, Z.; Jin, Q.; Zhan, F.; Xu, Y.; Liu, Z.; Feng, Z.; et al. Human 4F5 single-chain Fv antibody recognizing a conserved HA1 epitope has broad neutralizing potency against H5N1 influenza A viruses of different clades. Antiviral Res. 2013, 99, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zeng, X.; Zhang, L.; Peng, H.; Jiao, Y.; Zeng, J.; Treutlein, H.R. Computational identification of epitopes in the glycoproteins of novel bunyavirus (SFTS virus) recognized by a human monoclonal antibody (MAb 4-5). J. Comput. Aided Mol. Des. 2013, 27, 539. [Google Scholar] [CrossRef] [PubMed]
- Caflisch, A.; Karplus, M. Acid and thermal denaturation of barnase investigated by molecular dynamics simulations. J. Mol. Biol. 1995, 252, 672. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Legge, F.S.; Lebani, K.; Mahler, S.M.; Young, P.R.; Watterson, D.; Treutlein, H.R.; Zeng, J. Computational Identification of Antibody Epitopes on the Dengue Virus NS1 Protein. Molecules 2017, 22, 607. [Google Scholar] [CrossRef]
- Tan, X.; Liu, N.; Legge, F.S.; Yang, M.H.; Zeng, J. Computational identification of antibody epitopes of human papillomavirus 16 (HPV16) L1 proteins. J. Theore. Comput. Chem. 2018, 17, 1850017. [Google Scholar] [CrossRef]
- Caflisch, A. Computational combinatorial ligand design: Application to human alpha-thrombin. J. Comput. Aided Mol. Des. 1996, 10, 372. [Google Scholar] [CrossRef]
- Zeng, J.; Treutlein, H.R.; Rudy, G.B. Predicting sequences and structures of MHC-binding peptides: A computational combinatorial approach. J. Comput. Aided Mol. Des. 2001, 15, 573. [Google Scholar] [CrossRef]
- Zeng, J. Mini-review: Computational structure-based design of inhibitors that target protein surfaces. Comb. Chem. High Throughput Screen 2000, 3, 355. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Nheu, T.; Zorzet, A.; Catimel, B.; Nice, E.; Maruta, H.; Burgess, A.W.; Treutlein, H.R. Design of inhibitors of Ras—Raf interaction using a computational combinatorial algorithm. Protein Eng. 2001, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Treutlein, H.R. A method for computational combinatorial peptide design of inhibitors of Ras protein. Protein Eng. 1999, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, I.; Carletti, E.; Kurkova, I.; Nachon, F.; Nicolet, Y.; Mitkevich, V.A.; Debat, H.; Avalle, B.; Belogurov, A.A. Jr.; Kuznetsov, N.; et al. Reactibodies generated by kinetic selection couple chemical reactivity with favorable protein dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 15954. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; McTamney, P.M.; Yassine, H.M.; Whittle, J.R.; Guo, X.; Boyington, J.C.; Wei, C.J.; Nabel, G.J. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 2012, 489, 566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pierce, B.G.; Wang, Q.; Keck, Z.Y.; Fuerst, T.R.; Foung, S.K.; Mariuzza, R.A. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 2015, 290, 10117. [Google Scholar] [CrossRef] [PubMed]
- Hulsmeyer, M.; Chames, P.; Hillig, R.C.; Stanfield, R.L.; Held, G.; Coulie, P.G.; Alings, C.; Wille, G.; Saenger, W.; Uchanska-Ziegler, B.; et al. A major histocompatibility complex-peptide-restricted antibody and T cell receptor molecules recognize their target by distinct binding modes: Crystal structure of human leukocyte antigen (HLA)-A1-MAGE-A1 in complex with FAB-HYB3. J. Biol. Chem. 2005, 280, 2972. [Google Scholar] [CrossRef]
- Thomson, C.A.; Bryson, S.; McLean, G.R.; Creagh, A.L.; Pai, E.F.; Schrader, J.W. Germline V-genes sculpt the binding site of a family of antibodies neutralizing human cytomegalovirus. Embo. J. 2008, 27, 2592. [Google Scholar] [CrossRef]
- Malia, T.J.; Obmolova, G.; Almagro, J.C.; Gilliland, G.L.; Teplyakov, A. Crystal structure of human germline antibody 3-23/B3. Mol. Immunol. 2011, 48, 1586. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All atom empirical potential for molecular modelling and dynamic studies of proteins. J. Phys. Chem. B. 1998, 102, 3586. [Google Scholar] [CrossRef] [PubMed]
- Simonson, T.; Brunger, A.T. Solvation free energies estimated from macroscopic continuum theory: An accuracy assessment. J. Phys. Chem. 1994, 98, 4683. [Google Scholar] [CrossRef]
- Jiao, J.; Legge, F.S.; Zeng, X.; Treutlein, H.R.; Zeng, J. Antibody Recognition of Shiga Toxins (Stxs): Computational Identification of the Epitopes of Stx2 Subunit A to the Antibodies 11E10 and S2C4. PLoS ONE 2014, 9, e88191. [Google Scholar] [CrossRef] [PubMed]
- PyMol: The PyMol Molecular Graphics System; Version 1.5.0.4; Schrodinger LLC: Cambridge, MA, USA, 2012.
- Tan, Y.; Ng, Q.; Jia, Q.; Kwang, J.; He, F. A Novel Humanized Antibody Neutralizes H5N1 Influenza Virus via Two Different Mechanisms. J. Virol. 2015, 89, 3712. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional group | Abbreviation | Amino Acids | |
|---|---|---|---|
| Charged (-) | Acetate ion | ACET | ASP, GLU |
| Charged (+) | Methylguanidinium | MGUA | ARG |
| Charged (+) | Methylammonium | MAMM | LYS |
| Polar | Acetamide | ACEM | ASN,GLN |
| Polar | Methanol | MEOH | SER,THR |
| Hydrophobic | Methanethiol | MESH | CYS,MET |
| Aromatic Polar | Phenol | PHEN | TYR |
| Aromatic Polar | Indole | INDO | TRP |
| Aromatic Polar | Imidazole | IMIA | HIS |
| Aromatic Hydrophobic | Benzene | BENZ | PHE |
| Hydrophobic | Ibutane | IBUT | VAL, ILE, LEU, ALA |
| Antibodies | L Chain | H Chain |
|---|---|---|
| AbHC19 | Asn33, Tyr34, Asn36, Asn55,Pro58, Trp93 | Asn32, Trp52,Ala53, Arg97, Asp98, Trp100, Tyr107 |
| AbCR9114 | Arg31,Tyr36,Tyr49,Pro55,Trp91 | Asn25,Phe48,Ser50,Arg91, Asn94,Tyr96,Ser97 |
| AbBH151 | Ser30,Tyr49,Ala55,Arg91,Ser92,Tyr94 | Tyr32,Phe33,Asn57,Arg98, Gly101,Arg102 |
| 4F5 | Tyr32,Phe59,Gly101,His102,Tyr104,Phe105,Tyr106,ser108,Tyr112 | Tyr33,Asp51,Pro56 |
| Binding Surface | S2 | 9.00 Å | S1 |
|---|---|---|---|
| MCSS Minima Pattern | BENZ | PHEN | |
| PHEN | ACET | ||
| INDO | |||
| MAMM | |||
| MGUA | |||
| Sequence Pattern | F | Gap of one amino acid | Y |
| Y | D/E | ||
| W | |||
| W | |||
| R/K |
| Binding Surface | B1 | 11.50 Å | B2 |
|---|---|---|---|
| MCSS Minima Pattern | BENZ | BENZ | |
| PHEN | PHEN | ||
| INDO | INDO | ||
| MGUA | ACET | ||
| ACET | |||
| Sequence Pattern | F | Gap of 2 amino acid | F |
| Y | Y | ||
| W | W | ||
| R | D/E | ||
| D/E |
| Binding Surface | B1 | 9.50 Å | B2 |
|---|---|---|---|
| MCSS Minima Pattern | ACET | BENZ | |
| PHEN | |||
| ACET | |||
| Sequence Pattern | D/E | Gap of one amino acid | F |
| Y | |||
| D/E | |||
| Binding Surface | B1 | 13.00 Å | B2 |
|---|---|---|---|
| MCSS Minima Pattern | BENZ | BENZ | |
| IMIA | IMIA | ||
| PHEN | PHEN | ||
| INDO | INDO | ||
| ACEM | |||
| MAMM | |||
| MGUA | |||
| Sequence Pattern | F | Gap of two amino acid | F |
| H | H | ||
| Y | Y | ||
| W | W | ||
| Q/N | |||
| R/K |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Legge, F.S.; Huang, C.; Zhang, X.; Jiao, Y.; Treutlein, H.R.; Zeng, J. Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies. Antibodies 2019, 8, 2. https://doi.org/10.3390/antib8010002
Zeng X, Legge FS, Huang C, Zhang X, Jiao Y, Treutlein HR, Zeng J. Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies. Antibodies. 2019; 8(1):2. https://doi.org/10.3390/antib8010002
Chicago/Turabian StyleZeng, Xiaoyan, Fiona S. Legge, Chao Huang, Xiao Zhang, Yongjun Jiao, Herbert R. Treutlein, and Jun Zeng. 2019. "Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies" Antibodies 8, no. 1: 2. https://doi.org/10.3390/antib8010002
APA StyleZeng, X., Legge, F. S., Huang, C., Zhang, X., Jiao, Y., Treutlein, H. R., & Zeng, J. (2019). Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies. Antibodies, 8(1), 2. https://doi.org/10.3390/antib8010002