Single-Domain Antibodies and Their Formatting to Combat Viral Infections


Abstract
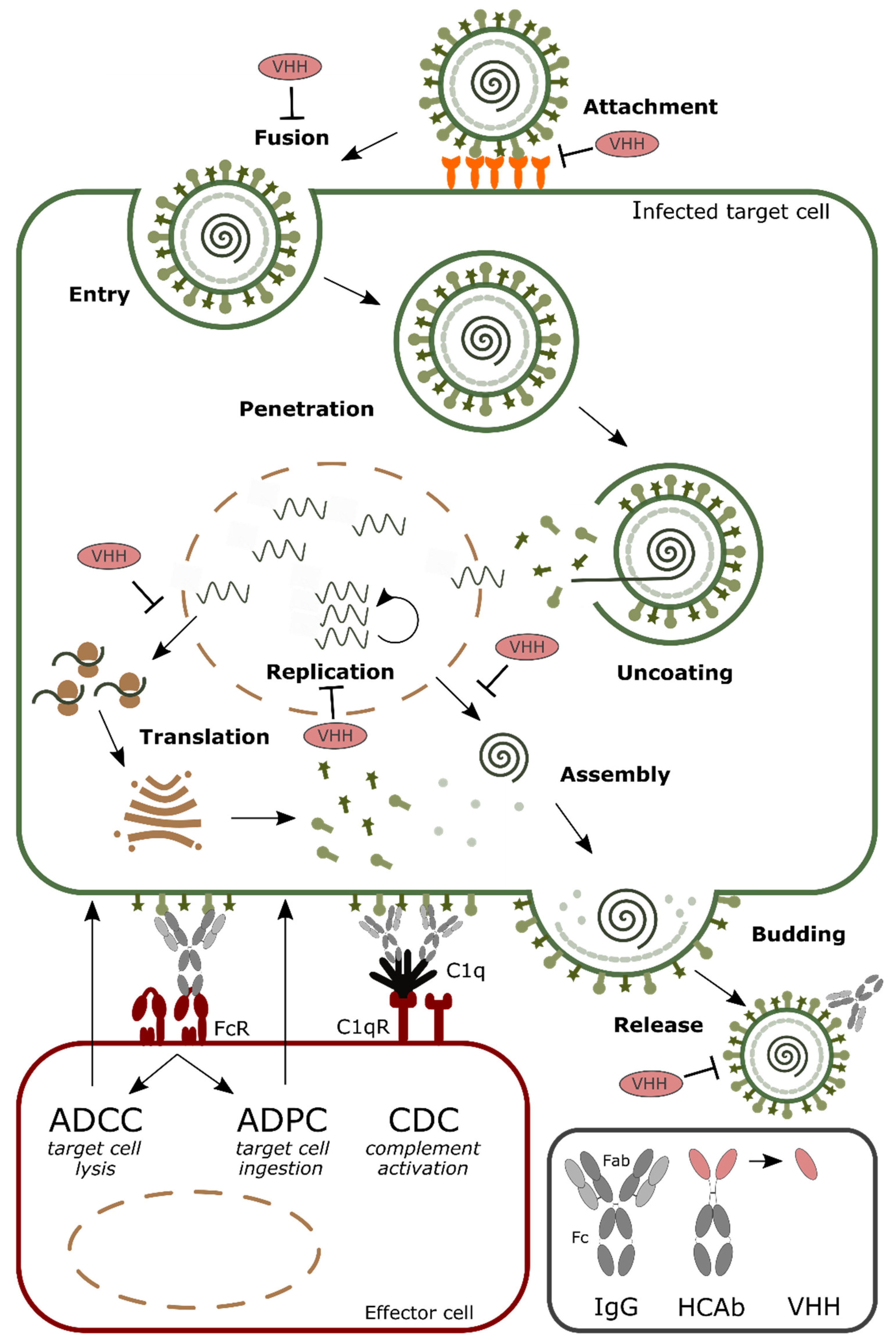
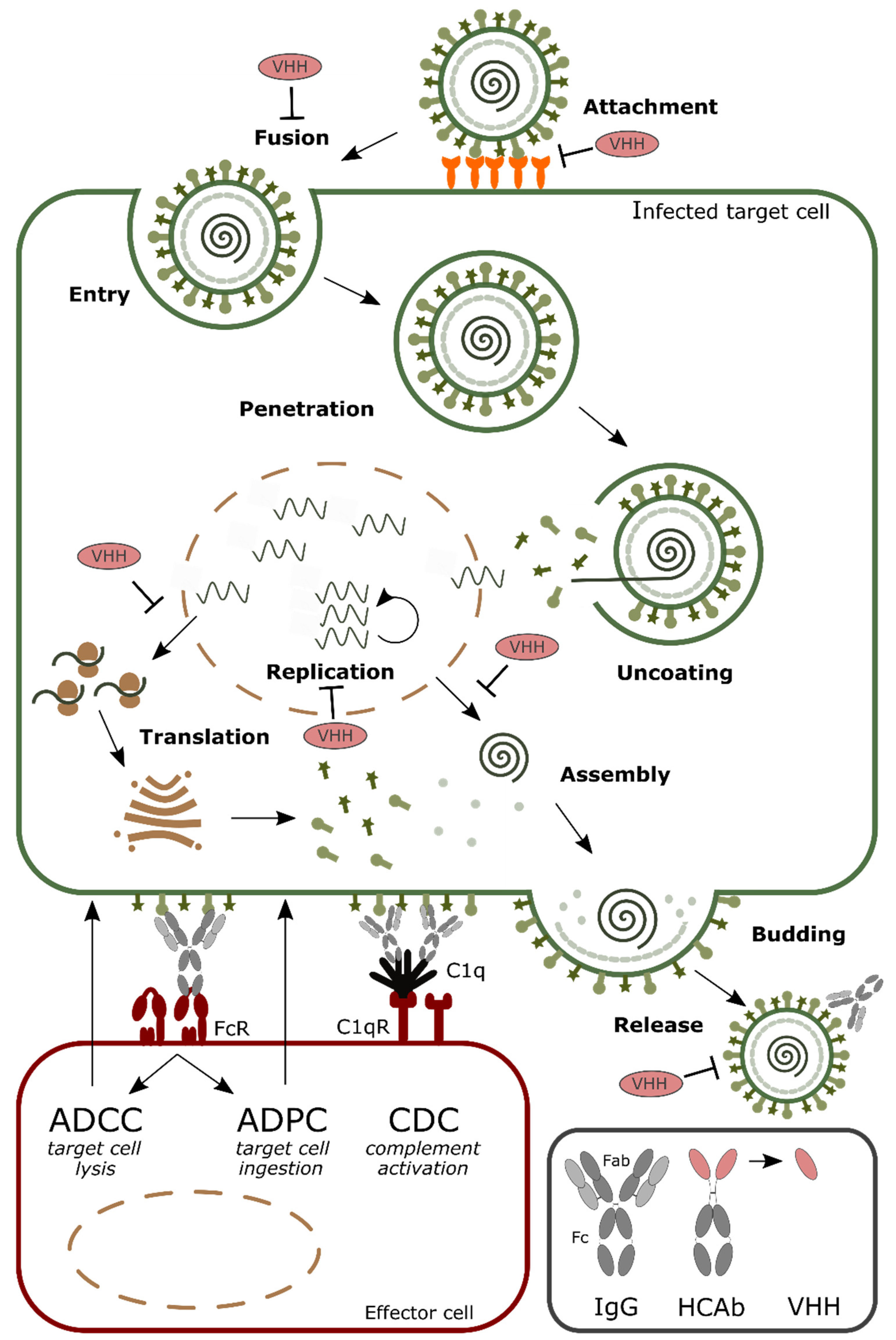
:1. Introduction
2. Formatting of VHHs to Increase the Half-Life in Circulation
3. Increasing Valency to Improve Potency
4. Arming VHHs with Effector Functions
5. Targeting and Delivery of VHHs
6. Antiviral Single Domain Antibodies as Tools for Diagnostic and Antigen Display
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sapienza, G.; Rossotti, M.A.; Tabares-da Rosa, S. Single-Domain Antibodies as Versatile Affinity Reagents for Analytical and Diagnostic Applications. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018, 31, 267–275. [Google Scholar] [CrossRef]
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins Struct. Funct. Bioinform. 2018, 86, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; MacKenzie, C.R. Antigen recognition by single-domain antibodies: Structural latitudes and constraints. MAbs 2018, 10, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Sequence and Structure of VH Domain from Naturally Occurring Camel Heavy Chain Immunoglobulins Lacking Light Chains. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7831284 (accessed on 2 November 2018).
- Vu, K.B.; Ghahroudi, M.A.; Wyns, L.; Muyldermans, S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 1997, 34, 1121–1131. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Ruuls, R.C.; Nijman, I.J.; Niewold, T.A.; Frenken, L.G.; de Geus, B. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 2000, 37, 579–590. [Google Scholar] [CrossRef]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Sabir, J.S.M.; Atef, A.; El-Domyati, F.M.; Edris, S.; Hajrah, N.; Alzohairy, A.M.; Bahieldin, A. Construction of naïve camelids VHH repertoire in phage display-based library. C. R. Biol. 2014, 337, 244–249. [Google Scholar] [CrossRef]
- Goldman, E.R.; Anderson, G.P.; Liu, J.L.; Delehanty, J.B.; Sherwood, L.J.; Osborn, L.E.; Cummins, L.B.; Hayhurst, A. Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal. Chem. 2006, 78, 8245–8255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, Z.; Shao, L.; Kong, X.; Hou, X.; Tian, D.; Sun, Y.; Xiao, Y.; Yu, L. Nanobody-derived nanobiotechnology tool kits for diverse biomedical and biotechnology applications. Int. J. Nanomed. 2016, 11, 3287–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Bencurova, E.; Pulzova, L.; Flachbartova, Z.; Bhide, M. A rapid and simple pipeline for synthesis of mRNA-ribosome-V(H)H complexes used in single-domain antibody ribosome display. Mol. Biosyst. 2015, 11, 1515–1524. [Google Scholar] [CrossRef]
- Hufton, S.E.; Risley, P.; Ball, C.R.; Major, D.; Engelhardt, O.G.; Poole, S. The breadth of cross sub-type neutralisation activity of a single domain antibody to influenza hemagglutinin can be increased by antibody valency. PLoS ONE 2014, 9, e103294. [Google Scholar] [CrossRef]
- Forsman, A.; Beirnaert, E.; Aasa-Chapman, M.M.I.; Hoorelbeke, B.; Hijazi, K.; Koh, W.; Tack, V.; Szynol, A.; Kelly, C.; McKnight, A.; et al. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol. 2008, 82, 12069–12081. [Google Scholar] [CrossRef]
- Zhao, G.; He, L.; Sun, S.; Qiu, H.; Tai, W.; Chen, J.; Li, J.; Chen, Y.; Guo, Y.; Wang, Y.; et al. A Novel Nanobody Targeting Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Receptor-Binding Domain Has Potent Cross-Neutralizing Activity and Protective Efficacy against MERS-CoV. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Hultberg, A.; Temperton, N.J.; Rosseels, V.; Koenders, M.; Gonzalez-Pajuelo, M.; Schepens, B.; Ibañez, L.I.; Vanlandschoot, P.; Schillemans, J.; Saunders, M.; et al. Llama-derived single domain antibodies to build multivalent, superpotent and broadened neutralizing anti-viral molecules. PLoS ONE 2011, 6, e17665. [Google Scholar] [CrossRef]
- Rossey, I.; Gilman, M.S.A.; Kabeche, S.C.; Sedeyn, K.; Wrapp, D.; Kanekiyo, M.; Chen, M.; Mas, V.; Spitaels, J.; Melero, J.A.; et al. Potent single-domain antibodies that arrest respiratory syncytial virus fusion protein in its prefusion state. Nat. Commun. 2017, 8, 14158. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Schmidt, F.I.; Hanke, L.; Cragnolini, J.; Cavallari, M.; Altenburg, A.; Brewer, R.; Ingram, J.; Shoemaker, C.; Ploegh, H.L. Intracellular Expression of Camelid Single-Domain Antibodies Specific for Influenza Virus Nucleoprotein Uncovers Distinct Features of Its Nuclear Localization. J. Virol. 2014, 89, 2792–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercruysse, T.; Pardon, E.; Vanstreels, E.; Steyaert, J.; Daelemans, D. An Intrabody Based on a Llama Single-Domain Antibody Targeting the N-terminal α-Helical Multimerization Domain of HIV-1 Rev Prevents Viral Production. J. Biol. Chem. 2010, 285, 21768–21780. [Google Scholar] [CrossRef] [PubMed]
- Boons, E.; Li, G.; Vanstreels, E.; Vercruysse, T.; Pannecouque, C.; Vandamme, A.-M.; Daelemans, D. A stably expressed llama single-domain intrabody targeting Rev displays broad-spectrum anti-HIV activity. Antivir. Res. 2014, 112, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh Gh., G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 2002. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; George, A.J.; Adams, G.P.; Stafford, W.F.; Jamar, F.; Tai, M.S.; McCartney, J.E.; Oppermann, H.; Heelan, B.T.; Peters, A.M.; et al. Single-chain Fv radioimmunotargeting. Q. J. Nucl. Med. 1996, 40, 320–333. [Google Scholar] [CrossRef]
- Batra, S.K.; Jain, M.; Wittel, U.A.; Chauhan, S.C.; Colcher, D. Pharmacokinetics and biodistribution of genetically engineered antibodies. Curr. Opin. Biotechnol. 2002, 13, 603–608. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs 2009, 23, 93–109. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Solt, C.B.; Fijten, H.; Setten, M.C. Prolonged in vivo residence times of llama single-domain antibody fragments in pigs by binding to porcine immunoglobulins. Vaccine 2005, 23, 4926–4934. [Google Scholar] [CrossRef]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc receptor and IgG-based therapeutics. MAbs 2011, 3, 422–430. [Google Scholar] [CrossRef]
- Fan, K.; Jiang, B.; Guan, Z.; He, J.; Yang, D.; Xie, N.; Nie, G.; Xie, C.; Yan, X. Fenobody: A {Ferritin-Displayed} Nanobody with High Apparent Affinity and {Half-Life} Extension. Anal. Chem. 2018, 90, 5671–5677. [Google Scholar] [CrossRef] [PubMed]
- Mu, B.; Huang, X.; Bu, P.; Zhuang, J.; Cheng, Z.; Feng, J.; Yang, D.; Dong, C.; Zhang, J.; Yan, X. Influenza virus detection with pentabody-activated nanoparticles. J. Virol. Methods 2010, 169, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; van Solt, C.B.; Fijten, H.P.D.; van Keulen, L.; Rosalia, R.A.; Weerdmeester, K.; Cornelissen, A.H.M.; Bruin, M.G.M.; Eblé, P.L.; Dekker, A. Passive immunization of guinea pigs with llama single-domain antibody fragments against foot-and-mouth disease. Vet. Microbiol. 2007, 120, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Tabata, Y.; Ikada, Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J. Pharm. Sci. 1994, 83, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Gaberc-Porekar, V.; Zore, I.; Podobnik, B.; Menart, V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr. Opin. Drug Discov. Dev. 2008, 11, 242–250. [Google Scholar]
- Kubetzko, S. Protein PEGylation Decreases Observed Target Association Rates via a Dual Blocking Mechanism. Mol. Pharmacol. 2005, 68, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Bendele, A.; Seely, J.; Richey, C.; Sennello, G.; Shopp, G. Short communication: Renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol. Sci. 1998, 42, 152–157. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion Proteins for {Half-Life} Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef]
- Tang, L.; Persky, A.M.; Hochhaus, G.; Meibohm, B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004, 93, 2184–2204. [Google Scholar] [CrossRef]
- Constantinou, A.; Epenetos, A.A.; Hreczuk-Hirst, D.; Jain, S.; Wright, M.; Chester, K.A.; Deonarain, M.P. Site-specific polysialylation of an antitumor single-chain Fv fragment. Bioconjug. Chem. 2009, 20, 924–931. [Google Scholar] [CrossRef]
- Berson, S.A.; Yalow, R.S.; Schreiber, S.S.; Post, J. Tracer experiments with I131 labeled human serum albumin: Distribution and degradation studies. J. Clin. Investig. 1953, 32, 746–768. [Google Scholar] [CrossRef] [PubMed]
- Peters, T. Serum Albumin. Adv. Protein Chem. 1985, 37, 161–245. [Google Scholar] [CrossRef] [PubMed]
- Hoefman, S.; Ottevaere, I.; Baumeister, J.; Sargentini-Maier, M. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies 2015, 4, 141–156. [Google Scholar] [CrossRef]
- Terryn, S.; Francart, A.; Lamoral, S.; Hultberg, A.; Rommelaere, H.; Wittelsberger, A.; Callewaert, F.; Stohr, T.; Meerschaert, K.; Ottevaere, I.; et al. Protective Effect of Different {Anti-Rabies} Virus {VHH} Constructs against Rabies Disease in Mice. PLoS ONE 2014, 9, e109367. [Google Scholar] [CrossRef]
- Kim, J. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. AJP Gastrointest. Liver Physiol. 2006, 290, G352–G360. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Okba, N.M.A.; Gutierrez-Alvarez, J.; Drabek, D.; van Dieren, B.; Widagdo, W.; Lamers, M.M.; Widjaja, I.; Fernandez-Delgado, R.; Sola, I.; et al. Chimeric camel/human heavy-chain antibodies protect against MERS-CoV infection. Sci. Adv. 2018, 4, eaas9667. [Google Scholar] [CrossRef]
- Yau, K.Y.F.; Dubuc, G.; Li, S.; Hirama, T.; Mackenzie, C.R.; Jermutus, L.; Hall, J.C.; Tanha, J. Affinity maturation of a V(H)H by mutational hotspot randomization. J. Immunol. Methods 2005, 297, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Saerens, D.; Ghassabeh, G.H.; Muyldermans, S. Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 2008, 8, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Lutje Hulsik, D.; Liu, Y.; Strokappe, N.M.; Battella, S.; El Khattabi, M.; McCoy, L.E.; Sabin, C.; Hinz, A.; Hock, M.; Macheboeuf, P.; et al. A gp41 MPER-specific Llama VHH Requires a Hydrophobic CDR3 for Neutralization but not for Antigen Recognition. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.M.; Ibañez, L.I.; Van den Hoecke, S.; De Baets, S.; Smet, A.; Roose, K.; Schepens, B.; Descamps, F.J.; Fiers, W.; Muyldermans, S.; et al. Single-domain antibodies targeting neuraminidase protect against an H5N1 influenza virus challenge. J. Virol. 2014, 88, 8278–8296. [Google Scholar] [CrossRef] [PubMed]
- Matz, J.; Kessler, P.; Bouchet, J.; Combes, O.; Ramos, O.H.P.; Barin, F.; Baty, D.; Martin, L.; Benichou, S.; Chames, P. Straightforward selection of broadly neutralizing single-domain antibodies targeting the conserved CD4 and coreceptor binding sites of HIV-1 gp120. J. Virol. 2013, 87, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, L.I.; De Filette, M.; Hultberg, A.; Verrips, T.; Temperton, N.; Weiss, R.A.; Vandevelde, W.; Schepens, B.; Vanlandschoot, P.; Saelens, X. Nanobodies with in vitro neutralizing activity protect mice against H5N1 influenza virus infection. J. Infect. Dis. 2011, 203, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Schepens, B.; Ibañez, L.I.; De Baets, S.; Hultberg, A.; Bogaert, P.; De Bleser, P.; Vervalle, F.; Verrips, T.; Melero, J.; Vandevelde, W.; et al. Nanobodies® specific for respiratory syncytial virus fusion protein protect against infection by inhibition of fusion. J. Infect. Dis. 2011, 204, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and Characterization of ALX-0171, a Potent Novel Therapeutic Nanobody for the Treatment of Respiratory Syncytial Virus Infection. Antimicrob. Agents Chemother. 2015, 60, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, C.; Mas, V.; Detalle, L.; Depla, E.; Cano, O.; Vázquez, M.; Stortelers, C.; Melero, J.A. Trivalency of a Nanobody Specific for the Human Respiratory Syncytial Virus Fusion Glycoprotein Drastically Enhances Virus Neutralization and Impacts Escape Mutant Selection. Antimicrob. Agents Chemother. 2016, 60, 6498–6509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillib, S.V.; Ivanova, T.I.; Vasilev, L.A.; Rutovskaya, M.V.; Saakyan, S.A.; Gribova, I.Y.; Tutykhina, I.L.; Sedova, E.S.; Lysenko, A.A.; Shmarov, M.M.; et al. Formatted single-domain antibodies can protect mice against infection with influenza virus (H5N2). Antivir. Res. 2013, 97, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Boruah, B.M.; Liu, D.; Ye, D.; Gu, T.; Jiang, C.; Qu, M.; Wright, E.; Wang, W.; He, W.; Liu, C.; et al. Single Domain Antibody Multimers Confer Protection against Rabies Infection. PLoS ONE 2013, 8, e71383. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Van den Hoecke, S.; Ehrhardt, K.; Kolpe, A.; El Bakkouri, K.; Deng, L.; Grootaert, H.; Schoonooghe, S.; Smet, A.; Bentahir, M.; Roose, K.; et al. Hierarchical and Redundant Roles of Activating FcγRs in Protection against Influenza Disease by M2e-Specific IgG1 and IgG2a Antibodies. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- El Bakkouri, K.; Descamps, F.; De Filette, M.; Smet, A.; Festjens, E.; Birkett, A.; Van Rooijen, N.; Verbeek, S.; Fiers, W.; Saelens, X. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 2011, 186, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Jans, J.; Vissers, M.; Heldens, J.G.M.; de Jonge, M.I.; Levy, O.; Ferwerda, G. Fc gamma receptors in respiratory syncytial virus infections: implications for innate immunity. Rev. Med. Virol. 2014, 24, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.R.; Dowd, K.A.; Engle, M.; Tesh, R.B.; Johnson, S.; Pierson, T.C.; Diamond, M.S. Poorly neutralizing cross-reactive antibodies against the fusion loop of West Nile virus envelope protein protect in vivo via Fcgamma receptor and complement-dependent effector mechanisms. J. Virol. 2011, 85, 11567–11580. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Nybakken, G.E.; Thompson, B.S.; Engle, M.J.; Marri, A.; Fremont, D.H.; Diamond, M.S. Antibodies against West Nile Virus Nonstructural Protein NS1 Prevent Lethal Infection through Fc γ Receptor-Dependent and -Independent Mechanisms. J. Virol. 2006, 80, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Cocklin, S.L.; Schmitz, J.E. The role of Fc receptors in HIV infection and vaccine efficacy. Curr. Opin. HIV AIDS 2014, 9, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Boesch, A.W.; Brown, E.; Ackerman, M.E. The role of Fc Receptors in HIV Prevention and Therapy. Immunol. Rev. 2015, 268, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Bobkov, V.; Zarca, A.M.; Van Hout, A.; Arimont, M.; Doijen, J.; Bialkowska, M.; Toffoli, E.; Klarenbeek, A.; van der Woning, B.; van der Vliet, H.J.; et al. Nanobody-Fc constructs targeting chemokine receptor CXCR4 potently inhibit signaling and CXCR4-mediated HIV-entry and induce antibody effector functions. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.W.; Siadat-Pajouh, M.; Krishnaney, A.A.; Greenberg, H.B. Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science 1996, 272, 104–107. [Google Scholar] [CrossRef]
- Feng, N.; Lawton, J.A.; Gilbert, J.; Kuklin, N.; Vo, P.; Prasad, B.V.V.; Greenberg, H.B. Inhibition of rotavirus replication by a non-neutralizing, rotavirus VP6-specific IgA mAb. J. Clin. Investig. 2002, 109, 1203–1213. [Google Scholar] [CrossRef]
- Aladin, F.; Einerhand, A.W.C.; Bouma, J.; Bezemer, S.; Hermans, P.; Wolvers, D.; Bellamy, K.; Frenken, L.G.J.; Gray, J.; Iturriza-Gómara, M. In vitro neutralisation of rotavirus infection by two broadly specific recombinant monovalent llama-derived antibody fragments. PLoS ONE 2012, 7, e32949. [Google Scholar] [CrossRef]
- Garaicoechea, L.; Olichon, A.; Marcoppido, G.; Wigdorovitz, A.; Mozgovoj, M.; Saif, L.; Surrey, T.; Parreño, V. Llama-derived single-chain antibody fragments directed to rotavirus VP6 protein possess broad neutralizing activity in vitro and confer protection against diarrhea in mice. J. Virol. 2008, 82, 9753–9764. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; Jäkel, M.; Sultana, S.; Alam, N.H.; Bardhan, P.K.; Chisti, M.J.; Salam, M.A.; Theis, W.; Hammarström, L.; Frenken, L.G.J. Anti-Rotavirus Protein Reduces Stool Output in Infants With Diarrhea: A Randomized Placebo-Controlled Trial. Gastroenterology 2013, 145, 740–748.e8. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; Tam, J.C.H.; Watkinson, R.E.; Bidgood, S.R.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, N.S.; Friesen, R.H.E.; Zhu, X.; Jongeneelen, M.; Blokland, S.; Vermond, J.; van Eijgen, A.; Tang, C.; van Diepen, H.; Obmolova, G.; et al. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 2018, 362, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Günaydın, G.; Alvarez, B.; Lin, Y.; Hammarström, L.; Marcotte, H. Co-expression of anti-rotavirus proteins (llama VHH antibody fragments) in Lactobacillus: Development and functionality of vectors containing two expression cassettes in tandem. PLoS ONE 2014, 9, e96409. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Pace, C.S.; Yao, X.; Yu, F.; Padte, N.N.; Huang, Y.; Seaman, M.S.; Li, Q.; Ho, D.D. Rational design and characterization of the novel, broad and potent bispecific HIV-1 neutralizing antibody iMabm36. J. Acquir. Immune Defic. Syndr. 2014, 66, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, E.M.; Zhang, H.; Desai, P.J.; Biragyn, A.; Markham, R.B. Antiviral activity of a single-domain antibody immunotoxin binding to glycoprotein D of herpes simplex virus 2. Antimicrob. Agents Chemother. 2015, 59, 527–535. [Google Scholar] [CrossRef]
- Wang, S.X.; Michiels, J.; Ariën, K.K.; New, R.; Vanham, G.; Roitt, I. Inhibition of HIV Virus by Neutralizing Vhh Attached to Dual Functional Liposomes Encapsulating Dapivirine. Nanoscale Res. Lett. 2016, 11, 350. [Google Scholar] [CrossRef]
- Pant, N.; Marcotte, H.; Hermans, P.; Bezemer, S.; Frenken, L.; Johansen, K.; Hammarström, L. Lactobacilli producing bispecific llama-derived anti-rotavirus proteins in vivo for rotavirus-induced diarrhea. Future Microbiol. 2011, 6, 583–593. [Google Scholar] [CrossRef]
- Álvarez, B.; Krogh-Andersen, K.; Tellgren-Roth, C.; Martínez, N.; Günaydın, G.; Lin, Y.; Martín, M.C.; Álvarez, M.A.; Hammarström, L.; Marcotte, H. An Exopolysaccharide-Deficient Mutant of Lactobacillus rhamnosus GG Efficiently Displays a Protective Llama Antibody Fragment against Rotavirus on Its Surface. Appl. Environ. Microbiol. 2015, 81, 5784–5793. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, W.; Matz, J.; Ye, C.; Bracq, L.; Delon, J.; Kimata, J.T.; Chen, Z.; Benichou, S.; Zhou, P. The Glycosylphosphatidylinositol-Anchored Variable Region of Llama Heavy Chain-Only Antibody JM4 Efficiently Blocks both Cell-Free and T Cell-T Cell Transmission of Human Immunodeficiency Virus Type 1. J. Virol. 2016, 90, 10642–10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, P.M.; Vanover, D.; Lindsay, K.E.; Bawage, S.S.; Kirschman, J.L.; Bhosle, S.; Lifland, A.W.; Zurla, C.; Santangelo, P.J. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 2018, 9, 3999. [Google Scholar] [CrossRef] [PubMed]
- Thueng-in, K.; Thanongsaksrikul, J.; Srimanote, P.; Bangphoomi, K.; Poungpair, O.; Maneewatch, S.; Choowongkomon, K.; Chaicumpa, W. Cell penetrable humanized-VH/V(H)H that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLoS ONE 2012, 7, e49254. [Google Scholar] [CrossRef] [PubMed]
- Phalaphol, A.; Thueng-In, K.; Thanongsaksrikul, J.; Poungpair, O.; Bangphoomi, K.; Sookrung, N.; Srimanote, P.; Chaicumpa, W. Humanized-VH/VHH that inhibit HCV replication by interfering with the virus helicase activity. J. Virol. Methods 2013, 194, 289–299. [Google Scholar] [CrossRef]
- Glab-Ampai, K.; Malik, A.A.; Chulanetra, M.; Thanongsaksrikul, J.; Thueng-In, K.; Srimanote, P.; Tongtawe, P.; Chaicumpa, W. Inhibition of HCV replication by humanized-single domain transbodies to NS4B. Biochem. Biophys. Res. Commun. 2016, 476, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Tarr, A.W.; Lafaye, P.; Meredith, L.; Damier-Piolle, L.; Urbanowicz, R.A.; Meola, A.; Jestin, J.-L.; Brown, R.J.P.; McKeating, J.A.; Rey, F.A.; et al. An alpaca nanobody inhibits hepatitis C virus entry and cell-to-cell transmission. Hepatology 2013, 58, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.A.; Tao, R.N.; DePorter, S.M.; Spiegel, D.A.; McNaughton, B.R. A Nanobody Activation Immunotherapeutic That Selectively Destroys HER2-Positive Breast Cancer Cells. ChemBioChem 2016, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Rozan, C.; Cornillon, A.; Pétiard, C.; Chartier, M.; Behar, G.; Boix, C.; Kerfelec, B.; Robert, B.; Pèlegrin, A.; Chames, P.; et al. Single-domain antibody-based and linker-free bispecific antibodies targeting FcγRIII induce potent antitumor activity without recruiting regulatory T cells. Mol. Cancer Ther. 2013, 12, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, J.; Zhu, X.; Tang, X.; Bao, Y.; Sun, X.; Huang, Y.; Tian, F.; Liu, X.; Yang, L. Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential. Int. J. Nanomed. 2017, 12, 1969–1983. [Google Scholar] [CrossRef]
- Li, T.; Qi, S.; Unger, M.; Hou, Y.N.; Deng, Q.W.; Liu, J.; Lam, C.M.C.; Wang, X.W.; Xin, D.; Zhang, P.; et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci. Rep. 2016, 6, 27055. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, J.M.; Pant, N.; Wolvers, D.; Bezemer, S.; Hermans, P.W.; Bellamy, K.; Sarker, S.A.; van der Logt, C.P.E.; Svensson, L.; Verrips, C.T.; et al. Reduction in morbidity of rotavirus induced diarrhoea in mice by yeast produced monovalent llama-derived antibody fragments. Vaccine 2006, 24, 4130–4137. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, S.Y.; Tabatabai, J.; Schnitzler, P.; Farah, C.; Rameil, S.; Sander, P.; Koromyslova, A.; Hansman, G.S. Development of a Nanobody-Based Lateral Flow Immunoassay for Detection of Human Norovirus. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Hu, Y.; Li, G.; Ou, W.; Mao, P.; Xin, S.; Wan, Y. Combining magnetic nanoparticle with biotinylated nanobodies for rapid and sensitive detection of influenza H3N2. Nanoscale Res. Lett. 2014, 9, 528. [Google Scholar] [CrossRef] [PubMed]
- Peyret, H.; Gehin, A.; Thuenemann, E.C.; Blond, D.; El Turabi, A.; Beales, L.; Clarke, D.; Gilbert, R.J.C.; Fry, E.E.; Stuart, D.I.; et al. Tandem fusion of hepatitis B core antigen allows assembly of virus-like particles in bacteria and plants with enhanced capacity to accommodate foreign proteins. PLoS ONE 2015, 10, e0120751. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]

{kind=link}
| VHH Format | Functionality | Virus | Reference |
Homobivalent VHHs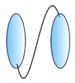 | - Enhance and broaden antiviral activity | RSV Influenza Rabies HIV | Hultberg et al. [21] Schepens et al. [55] Detalle et al. [56] Palomo et al. [57] Hultberg et al. [21] Cardoso et al. [52] Hufton et al. [18] Ibanez et al. [54] Hultberg et al. [21] Terryn et al. [45] Hulsik et al. [51] Matz et al. [53] |
| Bispecific VHHs - VHH linked to anti albumin VHH 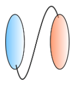 - VHH linked to anti IgG VHH 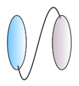 - VHH linked to VHH which binds different epitopes on same target 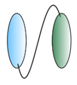 | - Half-life extension - Half-life extension - Enhance and broaden antiviral activity | Rabies FMDV RSV Rabies HIV | Terryn et al. [45] Harmsen et al. [30] Hultberg et al. [21] Hultberg et al. [21] Matz et al. [53] |
| VHH Format | Functionality | Virus | Reference |
PEGylation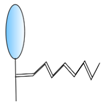 | - Half-life extension | FMDV | Harmsen et al. [34] |
VHH linked to IgG Fc region | - Half-life extension - Enhance and broaden antiviral activity - Effector function | MERS Influenza HIV Rotavirus | Raj et al. [48] Zhao et al. [20] Cardoso et al. [52] Laursen et al. [75] Bobkov et al. [68] Günaydın et al. [76] |
VHH linked to ferritin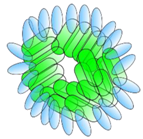 | - Half-life extension | Influenza | Fan et al. [32] |
VHH linked to GCN4 | - Enhance and broaden antiviral activity | Influenza | Tillib et al. [58] |
VHH linked to COMP48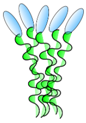 | - Enhance and broaden antiviral activity | Rabies | Boruah et al. [59] |
VHH linked to IgG | - Effector function - Targeting | HIV | Sun et al. [77] |
| VHH Format | Functionality | Virus | Reference |
VHH linked to cytotoxic domain | - Effector function | HSV-2 | Geoghegan et al. [78] |
VHH linked to liposome | - Effector function - Targeting | HIV | Wang et al. [79] |
VHH linked to bacteria | - Targeting | Rotavirus | Pant et al. [80] Günaydın et al. [76] Alvarez et al. [81] |
VHH linked to GPI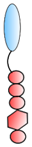 | - Targeting | HIV RSV | Liu et al. [82] Tiwari et al. [83] |
VHH linked to cell-penetrating peptide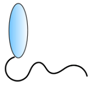 | - Targeting | HCV | Thueng-in et al. [84] Phalaphol et al. [85] Glab-ampai et al. [86] Tarr et al. [87] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vlieger, D.; Ballegeer, M.; Rossey, I.; Schepens, B.; Saelens, X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies 2019, 8, 1. https://doi.org/10.3390/antib8010001
De Vlieger D, Ballegeer M, Rossey I, Schepens B, Saelens X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies. 2019; 8(1):1. https://doi.org/10.3390/antib8010001
Chicago/Turabian StyleDe Vlieger, Dorien, Marlies Ballegeer, Iebe Rossey, Bert Schepens, and Xavier Saelens. 2019. "Single-Domain Antibodies and Their Formatting to Combat Viral Infections" Antibodies 8, no. 1: 1. https://doi.org/10.3390/antib8010001
APA StyleDe Vlieger, D., Ballegeer, M., Rossey, I., Schepens, B., & Saelens, X. (2019). Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies, 8(1), 1. https://doi.org/10.3390/antib8010001