Enhancers Improve the AID-Induced Hypermutation in Episomal Vector for Antibody Affinity Maturation in Mammalian Cell Display

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
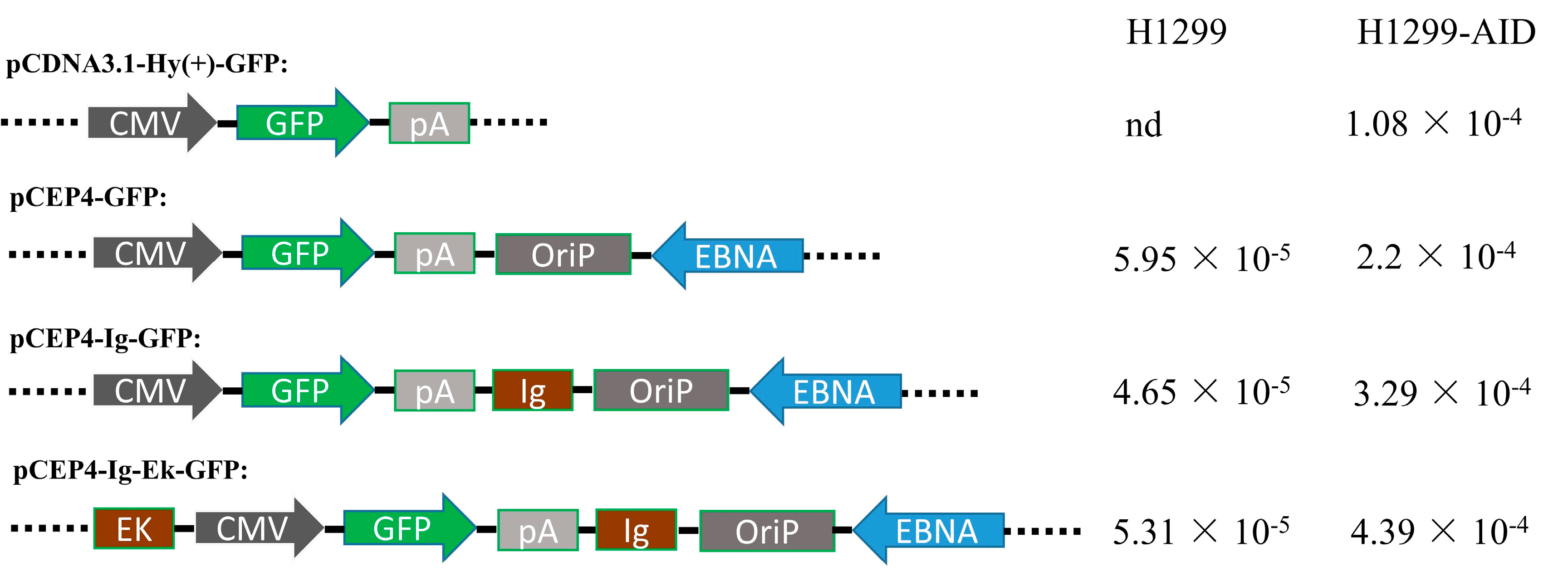
2.1. Plasmids Construction
2.2. Cell Culture
2.3. Stable Cell Lines Selection and Mutation Rate Detection
2.4. Flow Cytometric Analysis and cell Sorting
2.5. Antibody Mutants Sequencing and Expression
2.6. SDS-PAGE and Western Blot
2.7. Affinity Quantification of Antibody Mutants
3. Results
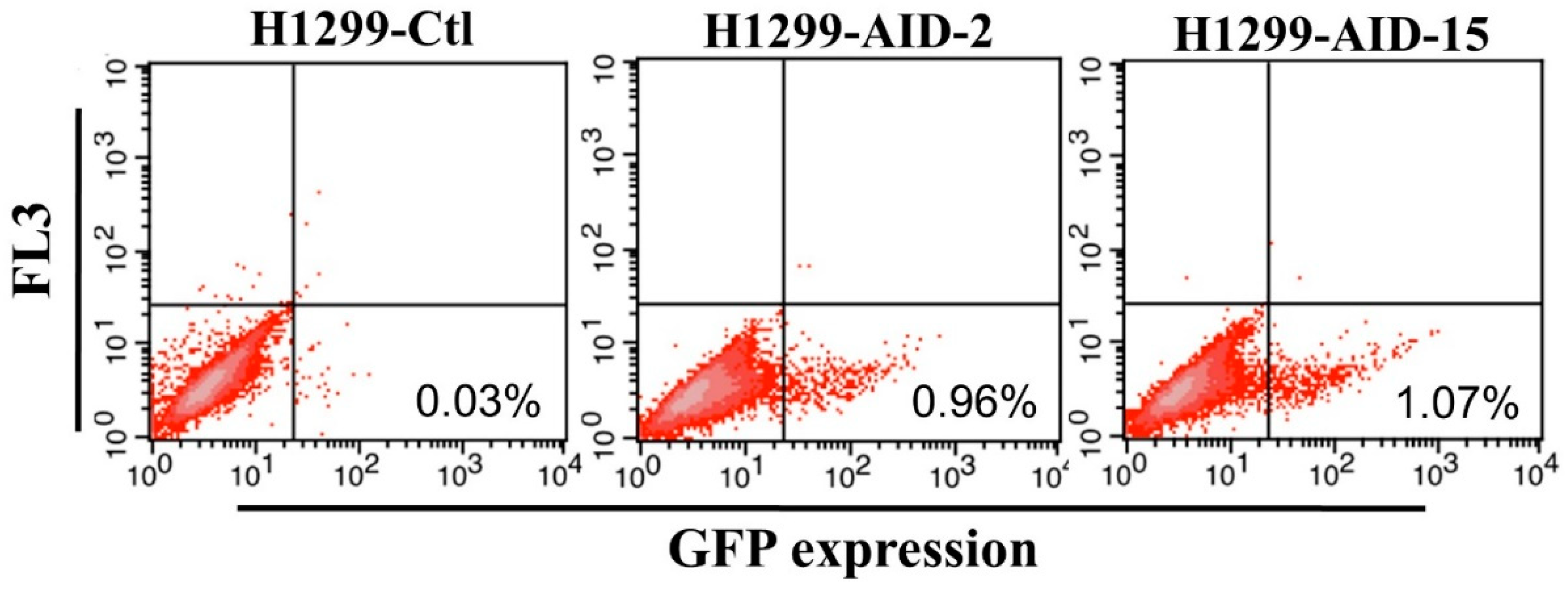
3.1. Generation of High AID Expression H1299 Cell Clones
3.2. Enhancers Increased AID-Induced Hypermutation on Episomal Vectors
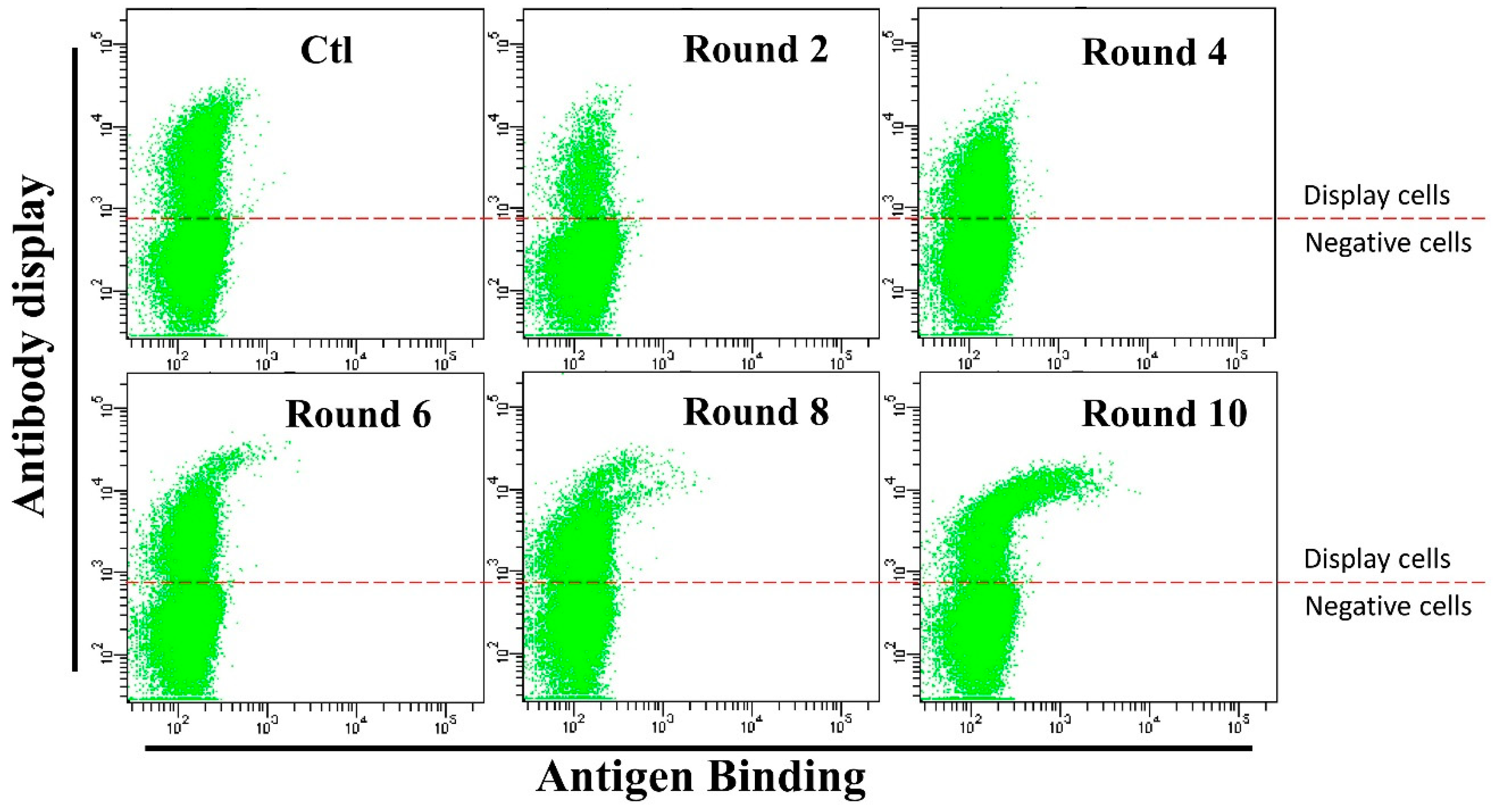
3.3. Anti-HMGB1 Full-Length Antibody Display and Affinity Maturation
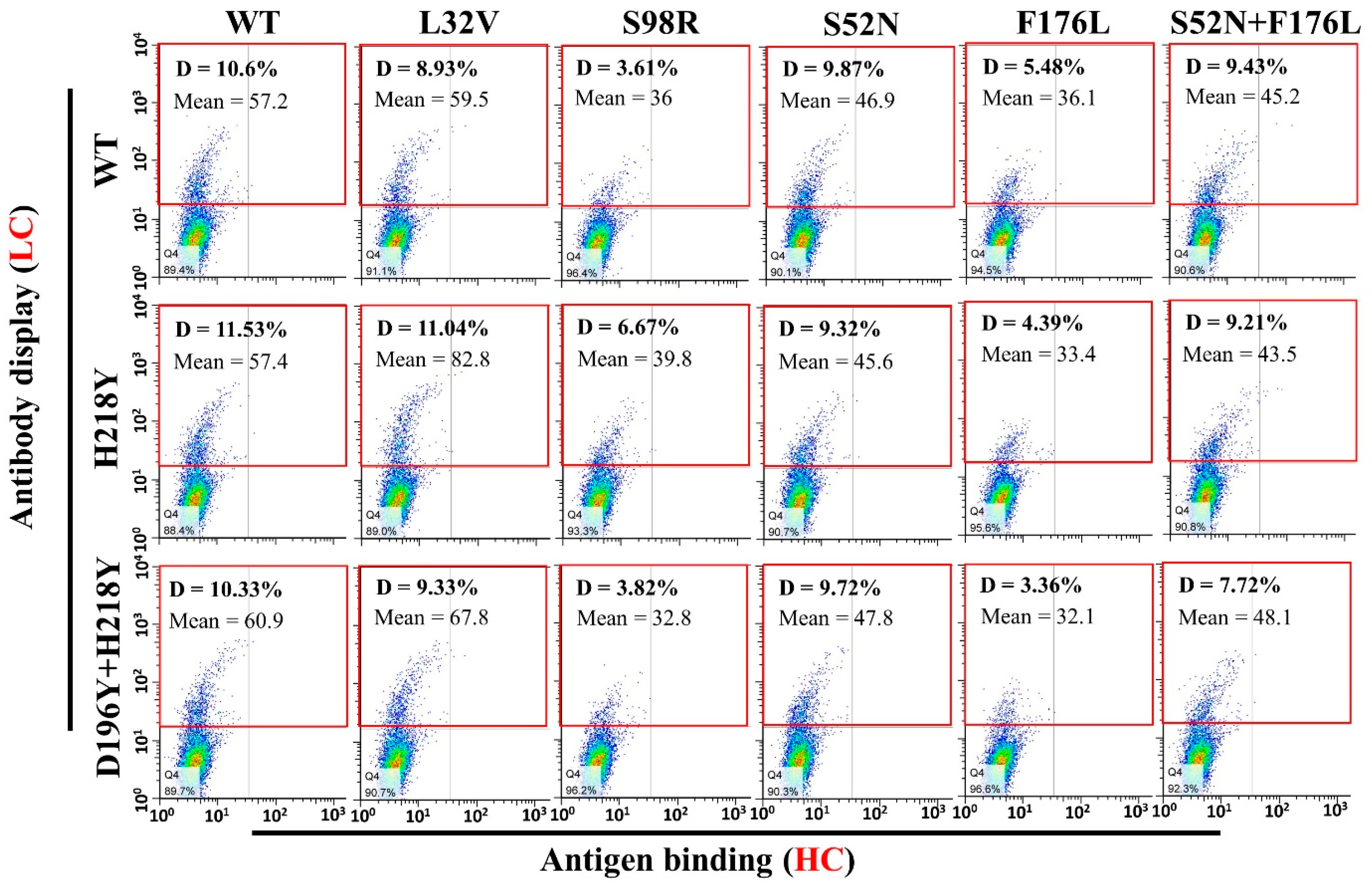
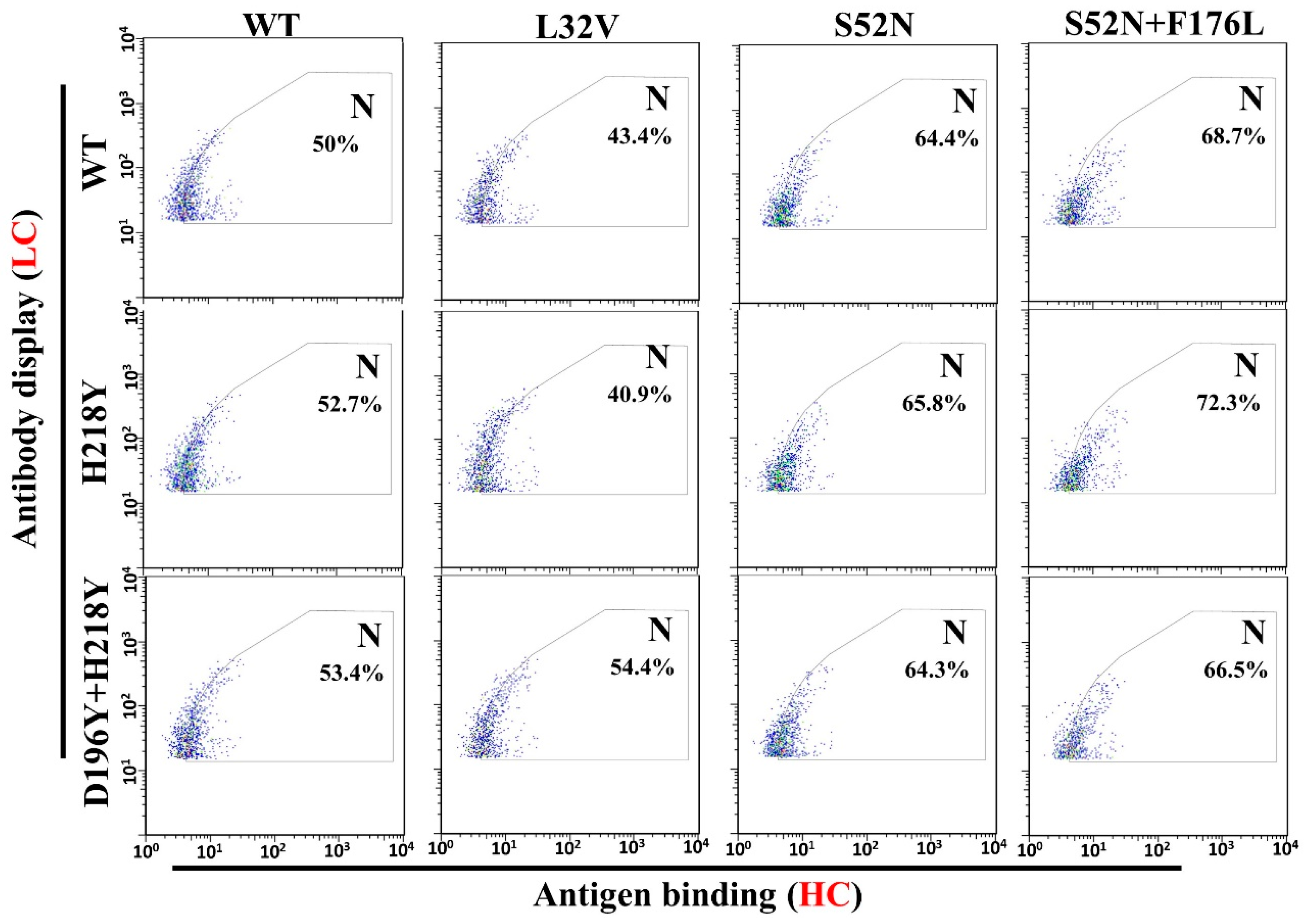
3.4. Antigen Binding and Affinity Determination
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Leickt, L.; Grubb, A.; Ohlson, S. Affinity screening for weak monoclonal antibodies. J. Immunol. Methods 1998, 220, 19–24. [Google Scholar] [CrossRef]
- Chappey, O.N.; Sandouk, P.; Scherrmann, J.M.G. Monoclonal-antibodies in hapten immunoassays. Pharmaceut. Res. 1992, 9, 1375–1379. [Google Scholar] [CrossRef]
- Yuan, B.; Schulz, P.; Liu, R.; Sierks, M.R. Improved affinity selection using phage display technology and off-rate based selection. Electron. J. Biotechn. 2006, 9, 171–175. [Google Scholar] [CrossRef]
- Lowman, H.B.; Wells, J.A. Affinity maturation of human growth-hormone by monovalent phage display. J. Mol. Biol. 1993, 234, 564–578. [Google Scholar] [CrossRef]
- Sun, S.; Yang, X.; Wang, H.F.; Zhao, Y.; Lin, Y.; Ye, C.; Fang, X.D.; Hang, H.Y. Antibody affinity maturation through combining display of two-chain paired antibody and precision flow cytometric sorting. Appl. Microbiol. Biot. 2016, 100, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Chen, G.; Olsen, M.J.; Iverson, B.L.; Georgiou, G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998, 11, 825–832. [Google Scholar] [CrossRef]
- Lofblom, J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011, 6, 1115–1129. [Google Scholar] [CrossRef]
- Tillotson, B.J.; de Larrinoa, I.F.; Skinner, C.A.; Klavas, D.M.; Shusta, E.V. Antibody affinity maturation using yeast display with detergent-solubilized membrane proteins as antigen sources. Protein Eng. Des. Sel. 2013, 26, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Keck, Z.Y.; Saha, A.; Xia, J.M.; Conrad, F.; Lou, J.L.; Eckart, M.; Marks, J.D.; Foung, S.K.H. Affinity maturation to improve human monoclonal antibody neutralization potency and breadth against hepatitis c virus. J. Biol. Chem. 2011, 286, 44218–44233. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Pakabunto, K.; Xu, Z.H.; Zhang, Y.; Tsurushita, N. Whole igg surface display on mammalian cells: Application to isolation of neutralizing chicken monoclonal anti-il-12 antibodies. J. Immunol. Methods 2007, 327, 40–52. [Google Scholar] [CrossRef]
- Zhou, C.; Jacobsen, F.W.; Cai, L.; Chen, Q.; Shen, W.D. Development of a novel mammalian cell surface antibody display platform. Mabs-Austin 2010, 2, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.P.; Qiu, J.K.; Chen, C.; Liu, C.C.; Liu, Y.H.; An, L.L.; Jia, J.Y.; Tang, J.; Wu, L.J.; Hang, H.Y. Affinity maturation of anti-tnf-alpha scfv with somatic hypermutation in non-b cells. Protein Cell 2012, 3, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, N.; Zhao, Y.; Hang, H.Y. Coupling recombinase-mediated cassette exchange with somatic hypermutation for antibody affinity maturation in cho cells. Biotechnol. Bioeng. 2016, 113, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.D.; Chernukhin, I.V.; Bigas, A.; Klenova, E.M.; Leon, J. Differential expression and phosphorylation of ctcf, a c-myc transcriptional regulator, during differentiation of human myeloid cells. Febs. Lett. 1999, 444, 5–10. [Google Scholar] [CrossRef]
- Suriano, R.; Ghosh, S.K.; Ashok, B.T.; Mittelman, A.; Chen, Y.G.; Banerjee, A.; Tiwari, R.K. Differences in glycosylation patterns of heat shock protein, gp96: Implications for prostate cancer prevention. Cancer Res. 2005, 65, 6466–6475. [Google Scholar] [CrossRef] [PubMed]
- Bowers, P.M.; Horlick, R.A.; Neben, T.Y.; Toobian, R.M.; Tomlinson, G.L.; Dalton, J.L.; Jones, H.A.; Chen, A.; Altobell, L.; Zhang, X.; et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 20455–20460. [Google Scholar] [CrossRef]
- Li, F.; Liu, Y.H.; Li, Y.W.; Ju, Q.; Chen, L.; Xie, P.L.; Li, Y.H.; Li, G.C. Human anti-egfl7 recombinant full-length antibodies selected from a mammalian cell-based antibody display library. Mol. Cell. Biochem. 2012, 365, 77–84. [Google Scholar] [CrossRef]
- Bowers, P.M.; Horlick, R.A.; Kehry, M.R.; Neben, T.Y.; Tomlinson, G.L.; Altobell, L.; Zhang, X.; Macomber, J.L.; Krapf, I.P.; Wu, B.F.; et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods 2014, 65, 44–56. [Google Scholar] [CrossRef]
- Li, Z.J.; Michael, I.P.; Zhou, D.X.; Nagy, A.; Rini, J.M. Simple piggybac transposon-based mammalian cell expression system for inducible protein production. Proc. Natl. Acad. Sci. USA 2013, 110, 5004–5009. [Google Scholar] [CrossRef]
- Cohen, R.N.; van der Aa, M.A.E.M.; Macaraeg, N.; Lee, A.P.; Szoka, F.C. Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection. J. Control. Release 2009, 138, 166–174. [Google Scholar] [CrossRef]
- Breous-Nystrom, E.; Schultze, K.; Meier, M.; Flueck, L.; Holzer, C.; Boll, M.; Seibert, V.; Schuster, A.; Blanusa, M.; Schaefer, V.; et al. Retrocyte display (r) technology: Generation and screening of a high diversity cellular antibody library. Methods 2014, 65, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Horlick, R.A.; Schilling, A.E.; Samama, P.; Swanson, R.N.; Fitzpatrick, V.D.; Robbins, A.K.; Damaj, B. Combinatorial gene expression using multiple episomal vectors. Gene 2000, 243, 187–194. [Google Scholar] [CrossRef]
- Martin, A.; Scharff, M.D. Somatic hypermutation of the aid transgene in b and non-b cells. Proc. Natl. Acad. Sci. USA 2002, 99, 12304–12308. [Google Scholar] [CrossRef] [PubMed]
- Maul, R.W.; Gearhart, P.J. Aid and somatic hypermutation. Adv. Immunol. 2010, 105, 159–191. [Google Scholar] [PubMed]
- Seo, H.; Hashimoto, S.; Tsuchiya, K.; Lin, W.; Shibata, T.; Ohta, K. An ex vivo method for rapid generation of monoclonal antibodies (adlib system). Nat. Protocols 2006, 1, 1502–1506. [Google Scholar] [CrossRef]
- Todo, K.; Miyake, K.; Magari, M.; Kanayama, N.; Ohmori, H. Novel in vitro screening system for monoclonal antibodies using hypermutating chicken b cell library. J. Biosci. Bioeng. 2006, 102, 478–481. [Google Scholar] [CrossRef]
- Cumbers, S.J.; Williams, G.T.; Davies, S.L.; Grenfell, R.L.; Takeda, S.; Batista, F.D.; Sale, J.E.; Neuberger, M.S. Generation and iterative affinity maturation of antibodies in vitro using hypermutating b-cell lines. Nat. Biotechnol. 2002, 20, 1129–1134. [Google Scholar] [CrossRef]
- McConnell, A.D.; Do, M.; Neben, T.Y.; Spasojevic, V.; MacLaren, J.; Chen, A.P.; Altobell, L.; Macomber, J.L.; Berkebile, A.D.; Horlick, R.A.; et al. High affinity humanized antibodies without making hybridomas; immunization paired with mammalian cell display and in vitro somatic hypermutation. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Horlick, R.A.; Macomber, J.L.; Bowers, P.M.; Neben, T.Y.; Tomlinson, G.L.; Krapf, I.P.; Dalton, J.L.; Verdino, P.; King, D.J. Simultaneous surface display and secretion of proteins from mammalian cells facilitate efficient in vitro selection and maturation of antibodies. J. Biol. Chem. 2013, 288, 19861–19869. [Google Scholar] [CrossRef]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from epstein-barr virus in various mammalian-cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef]
- Bachl, J.; Carlson, C.; Gray-Schopfer, V.; Dessing, M.; Olsson, C. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol. 2001, 166, 5051–5057. [Google Scholar] [CrossRef] [PubMed]
- Kulaeva, O.I.; Nizovtseva, E.V.; Polikanov, Y.S.; Ulianov, S.V.; Studitsky, V.M. Distant activation of transcription: Mechanisms of enhancer action. Mol. Cell. Biol. 2012, 32, 4892–4897. [Google Scholar] [CrossRef]
- Tippens, N.D.; Vihervaara, A.; Lis, J.T. Enhancer transcription: What, where, when, and why? Gene Dev. 2018, 32, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Shen, H.M.; Ratnam, S.; Kodgire, P.; Storb, U. Attracting aid to targets of somatic hypermutation. J. Exp. Med. 2010, 207, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Inlay, M.A.; Gao, H.H.; Odegard, V.H.; Lin, T.X.; Schatz, D.G.; Xu, Y. Roles of the ig kappa light chain intronic and 3′ enhancers in igk somatic hypermutation. J. Immunol. 2006, 177, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Bachl, J.; Wabl, M. Enhancers of hypermutation. Immunogenetics 1996, 45, 59–64. [Google Scholar] [CrossRef]
- Yang, S.Y.; Fugmann, S.D.; Schatz, D.G. Control of gene conversion and somatic hypermutation by immunoglobulin promoter and enhancer sequences. J. Exp. Med. 2006, 203, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Bachl, J.; Olsson, C.; Chitkara, N.; Wabl, M. The ig mutator is dependent on the presence, position, and orientation of the large intron enhancer. Proc. Natl. Acad. Sci. USA 1998, 95, 2396–2399. [Google Scholar] [CrossRef] [PubMed]
- Banerji, J.; Olson, L.; Schaffner, W. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy-chain genes. Cell 1983, 33, 729–740. [Google Scholar] [CrossRef]
- Meyer, K.B.; Neuberger, M.S. The immunoglobulin kappa-locus contains a second, stronger b-cell-specific enhancer which is located downstream of the constant region. EMBO J. 1989, 8, 1959–1964. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.C.; Zhang, Q.H.; Hou, W.; Wu, S.; Cao, L.Z.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.W.; et al. Hmgb1 in health and disease. Mol. Aspects Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Rauvala, H.; Rouhiainen, A. Rage as a receptor of hmgb1 (amphoterin): Roles in health and disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.A.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.C.; Ochani, M.; Li, J.H.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for hmgb1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tracey, K.J. Targeting hmgb1 in inflammation. Bba-Gene Regul. Mech. 2010, 1799, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; DeMarco, R.A. Dealing with death: Hmgb1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs 2003, 4, 1405–1409. [Google Scholar] [PubMed]
- Zhou, H.Z.; Wang, Y.B.; Wang, W.; Jia, J.Y.; Li, Y.; Wang, Q.Y.; Wu, Y.F.; Tang, J. Generation of monoclonal antibodies against highly conserved antigens. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Okazaki, I.M.; Eto, T.; Kinoshita, K.; Muramatsu, M.; Nagaoka, H.; Honjo, T. Aid enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science 2002, 296, 2033–2036. [Google Scholar] [CrossRef]
- Wang, C.L.; Harper, R.A.; Wabl, M. Genome-wide somatic hypermutation. Proc. Natl. Acad. Sci. USA 2004, 101, 7352–7356. [Google Scholar] [CrossRef]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-cd22 fv with high affinity by fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K.; Vanhoenacker, P.; Haegeman, G. Episomal vectors for gene expression in mammalian cells. Eur. J. Biochem. 2000, 267, 5665–5678. [Google Scholar] [CrossRef]
- Einav, Y.; Shistik, E.; Shenfeld, M.; Simons, A.H.; Melton, D.W.; Canaani, D. Replication and episomal maintenance of epstein-barr virus-based vectors in mouse embryonal fibroblasts enable synthetic lethality screens. Mol. Cancer Ther. 2003, 2, 1121–1128. [Google Scholar] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. Mabs-Austin 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Zan, H.; Casali, P. Regulation of aicda expression and aid activity. Autoimmunity 2013, 46, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.M.; Pone, E.J.; Al-Qahtani, A.; Park, S.R.; Zan, H.; Casali, P. Regulation of aicda expression and aid activity: Relevance to somatic hypermutation and class switch DNA recombination. Crit. Rev. Immunol. 2007, 27, 367–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Rada, C.; Neuberger, M.S. Altering the spectrum of immunoglobulin v gene somatic hypermutation by modifying the active site of aid. J. Exp.Med. 2010, 207, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Z.Z.; Rada, C.; Neuberger, M.S. Aid upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat. Struct. Mol. Biol. 2009, 16, 769–U797. [Google Scholar] [CrossRef] [PubMed]
- Barreto, V.; Reina-San-Martin, B.; Ramiro, A.R.; McBride, K.M.; Nussenzweig, M.C. C-terminal deletion of aid uncouples class switch recombination from somatic hypermutation and gene conversion. Mol. Cell 2003, 12, 501–508. [Google Scholar] [CrossRef]
- Shinkura, R.; Ito, S.; Begum, N.A.; Nagaoka, H.; Muramatsu, M.; Kinoshita, K.; Sakakibara, Y.; Hijikata, H.; Honjo, T. Separate domains of aid are required for somatic hypermutation and class-switch recombination. Nat. Immunol. 2004, 5, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Luginbuhl, B.; Kanyo, Z.; Jones, R.M.; Fletterick, R.J.; Prusiner, S.B.; Cohen, F.E.; Williamson, R.A.; Burton, D.R.; Pluckthun, A. Directed evolution of an anti-prion protein scfv fragment to an affinity of 1 pm and its structural interpretation. J. Mol. Biol. 2006, 363, 75–97. [Google Scholar] [CrossRef]
- Shafikhani, S.; Siegel, R.A.; Ferrari, E.; Schellenberger, V. Generation of large libraries of random mutants in bacillus subtilis by pcr-based plasmid multimerization. Biotechniques 1997, 23, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Iverson, B.L.; Georgiou, G.; Arnold, F.H. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J. Mol. Biol. 2005, 350, 806–816. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants Rounds | Light Chain (Mut/Seq) 1 | Heavy Chain (Mut/Seq) |
|---|---|---|
| Round 6 | H218Y (3/46) | S98R (1/41) L32V (1/41) |
| Round 8 | H218Y (9/39) | F176L (2/43) S52N + F176L (24/43) |
| Round 10 | H218Y (14/51) D196Y + H218Y (1/51) | S52N (1/55) S52N + F176L (54/55) |
| Mutants (LC/HC 1) | Kon (1/Ms) | Koff (1/s) | KD (M) |
|---|---|---|---|
| WT/WT | 5.18 × 104 | 2.32 × 10−3 | 4.47 × 10−8 |
| WT/S52N | 3.90 × 105 | 8.96 × 10−4 | 2.30 × 10−9 |
| WT/S52N + F176L | 1.72 × 105 | 8.81 × 10−4 | 5.11 × 10−9 |
| H218Y/WT | 1.09 × 105 | 1.36 × 10−3 | 1.24 × 10−8 |
| H218Y/S52N | 1.55 × 105 | 1.33 × 10−3 | 8.56 × 10−9 |
| H218Y/S52N + F176L | 2.08 × 105 | 9.55 × 10−4 | 4.59 × 10−9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Wang, J.; Zhao, Y.; Chen, S.; Hu, Z.; Chen, L.; Hang, H. Enhancers Improve the AID-Induced Hypermutation in Episomal Vector for Antibody Affinity Maturation in Mammalian Cell Display. Antibodies 2018, 7, 42. https://doi.org/10.3390/antib7040042
Chen C, Wang J, Zhao Y, Chen S, Hu Z, Chen L, Hang H. Enhancers Improve the AID-Induced Hypermutation in Episomal Vector for Antibody Affinity Maturation in Mammalian Cell Display. Antibodies. 2018; 7(4):42. https://doi.org/10.3390/antib7040042
Chicago/Turabian StyleChen, Chuan, Jie Wang, Yun Zhao, Shaopeng Chen, Zhishang Hu, Long Chen, and Haiying Hang. 2018. "Enhancers Improve the AID-Induced Hypermutation in Episomal Vector for Antibody Affinity Maturation in Mammalian Cell Display" Antibodies 7, no. 4: 42. https://doi.org/10.3390/antib7040042
APA StyleChen, C., Wang, J., Zhao, Y., Chen, S., Hu, Z., Chen, L., & Hang, H. (2018). Enhancers Improve the AID-Induced Hypermutation in Episomal Vector for Antibody Affinity Maturation in Mammalian Cell Display. Antibodies, 7(4), 42. https://doi.org/10.3390/antib7040042