Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H



Abstract
:1. Introduction: Monoclonal Antibodies and Hypersensitivity Reactions
2. The Consequences of Complement Activation for the Activator and the Host
3. Therapeutic mAbs, Complement Activation, and CARPA
4. Potential Role of Factor H in Mitigating Complement Activation
5. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Picard, M.; Galvão, V.R. Current Knowledge and Management of Hypersensitivity Reactions to Monoclonal Antibodies. J. Allergy Clin. Immunol. Pract. 2017, 5, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Adverse events to monoclonal antibodies used for cancer therapy: Focus on hypersensitivity responses. Oncoimmunology 2013, 2, e26333. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.I.; Bankova, L.; Cahill, K.N.; Kyin, T.; Castells, M.C. Allergy to monoclonal antibodies: Cutting-edge desensitization methods for cutting-edge therapies. Expert Rev. Clin. Immunol. 2012, 8, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Mayer, E.L.; Je, Y.; Rosenberg, J.E.; Nguyen, P.L.; Azzi, G.R.; Bellmunt, J.; Burstein, H.J.; Schutz, F.A. Congestive Heart Failure Risk in Patients With Breast Cancer Treated With Bevacizumab. J. Clin. Oncol. 2011, 29, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; Flinn, I.; Jain, V.; Binet, J.L.; Hillmen, P.; Byrd, J.; Albitar, M.; Brettman, L.; Santabarbara, P.; Wacker, B.; et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: Results of a large international study. Blood 2002, 99, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Kimby, E. Tolerability and safety of rituximab (MabThera®). Cancer Treat. Rev. 2005, 31, 456–473. [Google Scholar] [CrossRef] [PubMed]
- Coombs, R.R.A.; Gell, P.G.H. Classification of allergic reactions responsible for drug hypersensitivity reactions. In Clinical Aspects of Immunology; Coombs, R.R.A., Gell, P.G.H., Eds.; Davis: Philadelphia, PA, USA, 1968; pp. 575–596. [Google Scholar]
- Demoly, P.; Lebel, B.; Messaad, D.; Sahla, H.; Rongier, M.; Daurès, J.P.; Godard, P.; Bousquet, J. Predictive capacity of histamine release for the diagnosis of drug allergy. Allergy 1999, 54, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy caused by liposomes, micellar carriers of intravenous drugs, and radiocontrast agents. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 567–606. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Urbanics, R.; Bedocs, P.; Rozsnyay, Z.; Rosivall, L.; Toth, M.; Kissel, T.; Szebeni, J. In vitro and in vivo complement activation and related anaphylactic effects associated with polyethylenimine and polyethylenimine-graft-poly (ethylene glycol) block copolymers. Biomaterials 2011, 32, 4936–4942. [Google Scholar] [CrossRef] [PubMed]
- Weiszhar, Z.; Czúcz, J.; Révész, C.; Rosivall, L.; Szebeni, J.; Rozsnyay, Z. Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur. J. Pharm. Sci. 2012, 45, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Rombach-Riegraf, V.; Karle, A.C.; Wolf, B.; Sordé, L.; Koepke, S.; Gottlieb, S.; Krieg, J.; Djidja, M.C.; Baban, A.; Spindeldreher, S.; et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS ONE 2014, 9, e86322. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.M.; Lindorfer, M.A.; van der Horst, H.; Oostindie, S.; Beurskens, F.J.; Schuurman, J.; Zent, C.S.; Burack, R.; Parren, P.W.; Taylor, R.P. Antibodies That Efficiently Form Hexamers upon Antigen Binding Can Induce Complement-Dependent Cytotoxicity under Complement-Limiting Conditions. J. Immunol. 2016, 197, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Romain, G.; Yan, W.; Watanabe, M.; Charab, W.; Todorova, B.; Lee, J.; Triplett, K.; Donkor, M.; Lungu, O.I.; et al. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat. Immunol. 2017, 18, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Beers, S.A.; French, R.R.; Johnson, P.W.; Glennie, M.J.; Cragg, M.S. Anti-CD20 monoclonal antibodies: Historical and future perspectives. Haematologica 2010, 95, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Leusen, J.H. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar] [PubMed]
- Zhou, X.; Hu, W.; Qin, X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: Implications for therapy. Oncologist 2008, 13, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Van der Kolk, L.E.; Grillo-Lopez, A.J.; Baars, J.W.; Hack, C.E.; van Oers, M.H. Complement activation plays a key role in the side-effects of rituximab treatment. Br. J. Haematol. 2001, 115, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Junnikkala, S.; Jokiranta, T.S.; Friese, M.A.; Jarva, H.; Zipfel, P.F.; Meri, S. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J. Immunol. 2000, 164, 6075–6081. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.; Castaño, Z.; Garayoa, M.; Zudaire, E.; Pajares, M.J.; Martinez, A.; Cuttitta, F.; Montuenga, L.M.; Pio, R. Expression of complement factor H by lung cancer cells: Effects on the activation of the alternative pathway of complement. Cancer Res. 2004, 64, 6310–6318. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.M.; Veeramani, S.; Weiner, G.J. Complement in Monoclonal Antibody Therapy of Cancer. Immunol. Res. 2014, 59, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Descotes, J. Immunotoxicity of monoclonal antibodies. mAbs 2009, 1, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. mAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist 2008, 13, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Strait, R.T.; Morris, S.C.; Yang, M.; Qu, X.W.; Finkelman, F.D. Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 2002, 109, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Smith, R.A. Membrane-targeted complement inhibitors. Mol. Immunol. 2011, 38, 249–255. [Google Scholar] [CrossRef]
- Mészáros, T.; Csincsi, Á.I.; Uzonyi, B.; Hebecker, M.; Fülöp, T.G.; Erdei, A.; Szebeni, J.; Józsi, M. Factor H inhibits complement activation induced by liposomal and micellar drugs and the therapeutic antibody rituximab in vitro. Nanomedicine 2016, 12, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Hebecker, M.; Svobodová, E.; Józsi, M. Factor H: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef] [PubMed]
- Parente, R.; Clark, S.J.; Inforzato, A.; Day, A.J. Complement factor H in host defense and immune evasion. Cell. Mol. Life Sci. 2017, 74, 1605–1624. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, M.; Alba-Domínguez, M.; Roumenina, L.T.; Reuter, S.; Hyvärinen, S.; Dragon-Durey, M.A.; Jokiranta, T.S.; Sánchez-Corral, P.; Józsi, M. An engineered construct combining complement regulatory and surface-recognition domains represents a minimal-size functional factor H. J. Immunol. 2013, 191, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Shimizu, T.; Abu Lila, A.S.; Ishida, T.; Kiwada, H. Relationship between the concentration of anti-polyethylene glycol (PEG) immunoglobulin M (IgM) and the intensity of the accelerated blood clearance (ABC) phenomenon against PEGylated liposomes in mice. Biol. Pharm. Bull. 2015, 38, 417–424. [Google Scholar] [CrossRef] [PubMed]
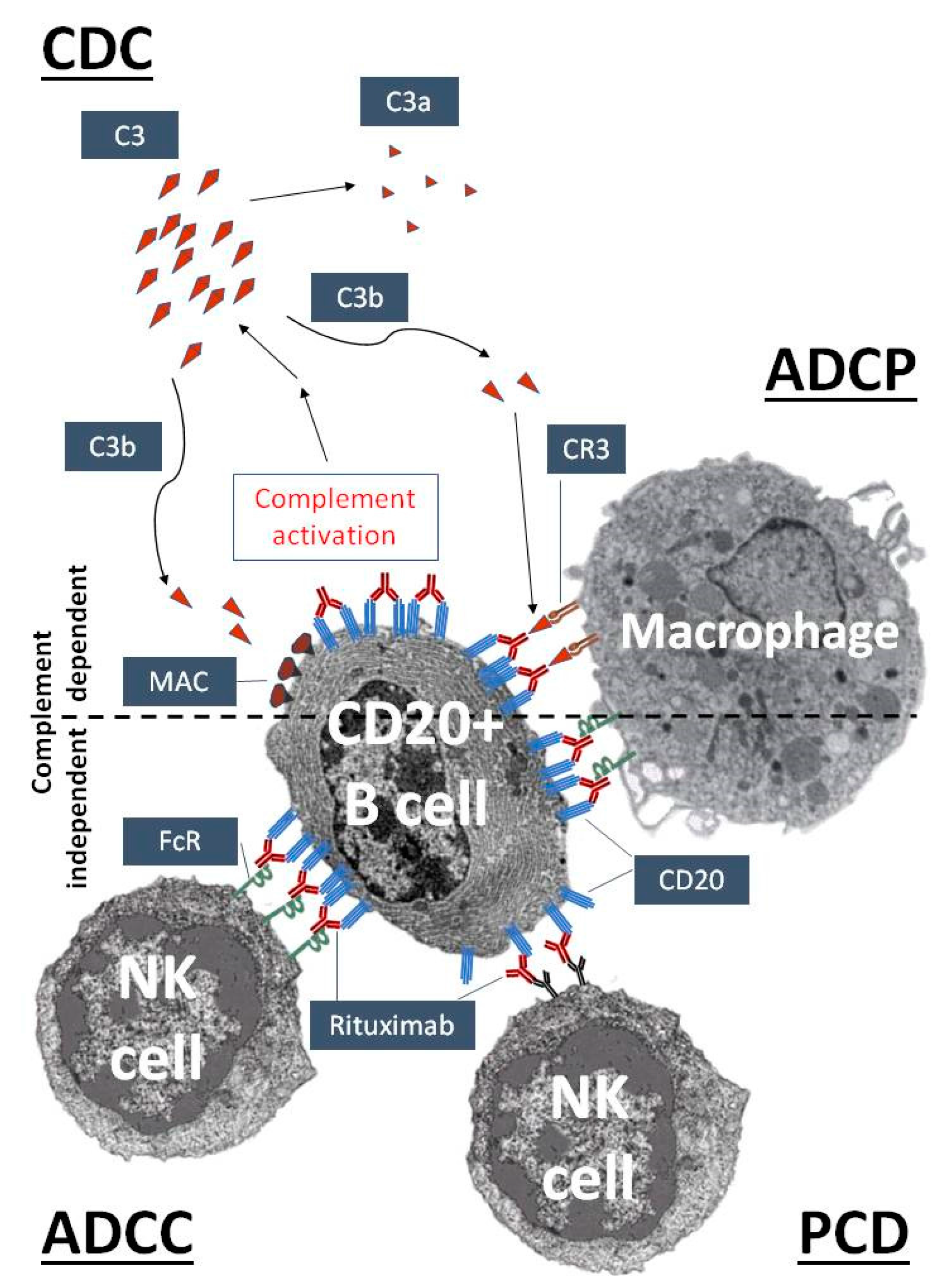

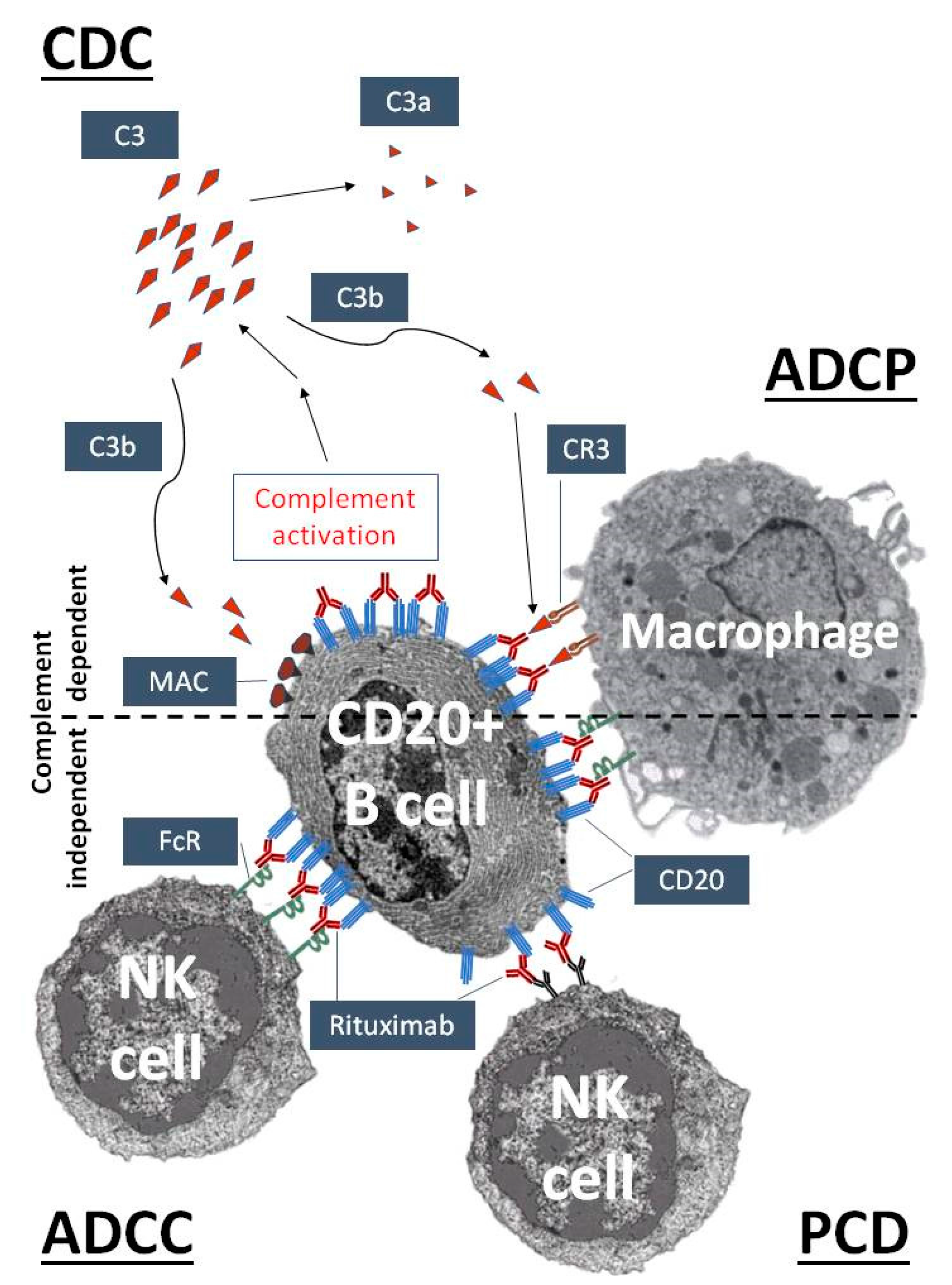{kind=link}
| Brand Name (Manufacturer) | INN, Isotype (Target Antigen) | Indication | Incidence | Symptoms | References |
|---|---|---|---|---|---|
| Anticancer use | |||||
| Avastin (Genentech, San Francisco, CA, USA; Roche, Basel, Switzerland) | bevacizumab, humanized IgG1 (VEGF-A) | combination chemotherapy of metastatic colon, lung, and kidney cancer, and glioblastoma | <3%, severe: 0.2% | chest pain, diaphoresis, headache, hypertension, neurologic signs and symptoms, oxygen desaturation, rigors, wheezing | [2,4] |
| Campath (Genzyme, Cambridge, MA, USA) | alemtuzumab–IH, humanized IgG1κ (CD52 on T and B cells) | B cell chronic lymphocytic leukemia (B-CLL) | 4–7% | bronchospasm, chills, dyspnea, emesis, fever, hypotension, nausea, pyrexia, rash, rigors, tachycardia, urticaria | [2,5] |
| Erbitux (Bristol-Myers Squibb, New York, NY, USA; Eli Lilly, Indianapolis, IN, USA) | cetuximab, chimeric IgG1κ (EGFR) | metastatic colorectal cancer, head and neck cancer, squamous cell carcinomas | <3%, fatal < 0.1% | anaphylaxis, angioedema, bronchospasm, cardiac arrest, chills, dizziness, dyspnea, fever, hoarseness, hypotension, pruritus, rash, rigor, stridor, urticaria, wheezing | [1,2,3] |
| Herceptin (Genentech, San Francisco, CA, USA) | trastuzumab, humanized IgG1κ (EGFR receptor 2, HER2/neu/erbB2) | metastatic breast and gastric cancer | <1% | asthenia, bronchospasm, chills, death within hours, dizziness, dyspnea, further pulmonary complications, headache, hypotension, hypoxia, nausea, pain, rash, severe hypotension, vomiting | [1,2,3] |
| Rituxan (Genentech, San Francisco, CA, USA) | rituximab, chimeric IgG1κ (CD20 on B cells) | B cell leukemias, rheumatoid arthritis and non-Hodgkin’s B-cell lymphoma | >80%, severe: <10% | ARDS, bronchospasm, cardiogenic shock, flushing, hypotension, hypoxia, itching, myocardial infarction, pain (at the site of the tumor), pulmonary infiltrates, runny nose, swelling of the tongue or throat, ventricular fibrillation, vomiting | [1,2,3,6] |
| Anti-inflammatory use | |||||
| Remicade (Janssen Biotech. Inc., Horsham, PA, USA) | infliximab, chimeric IgG1κ (TNF alpha) | Crohn’s disease, rheumatoid arthritis, spondylitis ankylopoetica, arthritis psoriatica, ulcerative colitis | 18% | bronchospasm, laryngeal edema, pharyngeal edema, dyspnea, hypotension, urticaria, serum sickness-like reactions | [3] |
| Xolair (Genentech, San Francisco, CA, USA) | omalizumab, humanized IgG4 (IgE) | atopia, asthma | 39%, Severe: 0.2% | anaphylaxis, bronchospasm, hypotension, syncope, urticaria, and/or angioedema of the throat or tongue, delayed anaphylaxis (with onset two to 24 h or even longer) beyond one year after beginning regularly administered treatment | [1] |
| Cardiovascular | Broncho-Pulmonary | Hematological | Mucocutaneous | Gastrointestinal | Neuro-Psycho-Somatic | Systemic |
|---|---|---|---|---|---|---|
| Angioedema | Apnea | Granulopenia | Cyanosis | Bloating | Back pain | Chills |
| Arrhythmia | Bronchospasm | Leukopenia | Erythmea | Cramping | Chest pain | Diaphoresis |
| Cardiogenic shock | Coughing | Lymphopenia | Flushing | Diarrhea | Chest tightness | Feeling of warmth |
| Edema | Dyspnea | Rebound leukocytosis | Nasal congestion | Metallic taste | Confusion | Fever |
| Hypertension | Hoarsness | Rebound granulocytosis | Rash | Nausea | Dizziness | Loss of consciousness |
| Hypotension | Hyperventillation | Trombocytopenia | Rhinitis | Vomiting | Feeling of imminent death | Rigors |
| Hypoxia | Laryngospasm | Swelling | Fright | Sweating | ||
| Myocardial infarction | Respiratory distress | Tearing | Headache | Wheezing | ||
| Tachycardia | Shortness of breath | Urticaria | Panic | |||
| Ventricular fibrillation | Sneezing | |||||
| Syncope | Stridor |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fülöp, T.; Mészáros, T.; Kozma, G.T.; Szebeni, J.; Józsi, M. Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H. Antibodies 2018, 7, 14. https://doi.org/10.3390/antib7010014
Fülöp T, Mészáros T, Kozma GT, Szebeni J, Józsi M. Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H. Antibodies. 2018; 7(1):14. https://doi.org/10.3390/antib7010014
Chicago/Turabian StyleFülöp, Tamás, Tamás Mészáros, Gergely Tibor Kozma, János Szebeni, and Mihály Józsi. 2018. "Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H" Antibodies 7, no. 1: 14. https://doi.org/10.3390/antib7010014
APA StyleFülöp, T., Mészáros, T., Kozma, G. T., Szebeni, J., & Józsi, M. (2018). Infusion Reactions Associated with the Medical Application of Monoclonal Antibodies: The Role of Complement Activation and Possibility of Inhibition by Factor H. Antibodies, 7(1), 14. https://doi.org/10.3390/antib7010014