
Neutrophil Extracellular Traps, Antiphospholipid Antibodies and Treatment

{kind=link}
Abstract
:1. Introduction
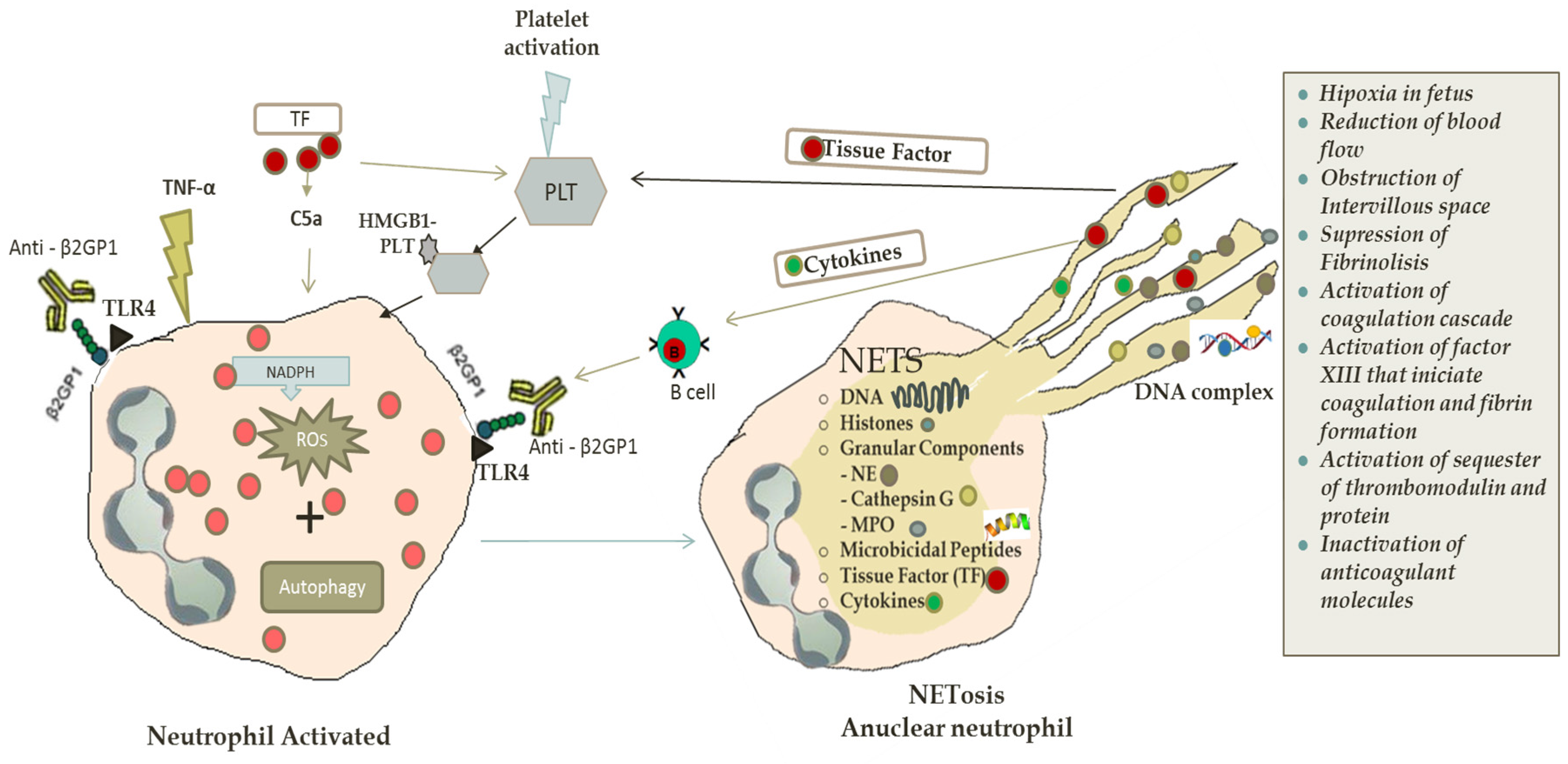
2. NETs Formation
2.1. Autophagy and NETosis
2.2. NETs in Antiphospholipid Syndrome and Thrombosis
3. New Mechanisms of Old Therapeutics Agents
3.1. Acetylsalicylic Acid
3.2. Heparins
3.3. Hydroxychloroquine and Chloroquine
3.4. Vitamin D
3.5. Vitamin C
4. Biologic Anti-Cytokine Therapy
4.1. Statins
4.2. Potential Therapeutic Agents
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grayson, P.C.; Schauer, C.; Herrmann, M.; Kaplan, M.J. Review: Neutrophils as Invigorated Targets in Rheumatic Diseases. Arthritis Rheumatol. 2016, 68, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M. Neutrophil extracelullar traps (NETs): Double-edged swords of innate immunity 1. J. Immunol. 2013, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE 2007, 2007, pe11. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Lysov, Z.; Liaw, P.C. Extracellular DNA and histones: Double-edged swords in immunothrombosis. J. Thromb. Haemost. 2015, 13, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Darrah, E.; Andrade, F. NETs: The missing link between cell death and systemic autoimmune diseases? Front. Immunol. 2013, 3, 428. [Google Scholar] [CrossRef] [PubMed]
- Porto, B.N.; Stein, R.T. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front. Immunol. 2016, 7, 311. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Fortin, P.R. Connective tissue diseases: Mitochondria drive NETosis and inflammation in SLE. Nat. Rev. Rheumatol. 2016, 12, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, D.; Segelmark, M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 2016, 7, 256. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Pratesi, F.; Prediletto, E.; Bombardieri, M.; Migliorini, P. NETosis as Source of Autoantigens in Rheumatoid Arthritis. Front. Immunol. 2016, 7, 485. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. Gout, tophi and the wonders of NETs. Arthritis Res. Ther. 2014, 16, 431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.; Boumpas, D.; Ritis, K. Neutrophil Extracellular Trap Formation Is Associated with IL-1β and Autophagy-Related Signaling in Gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef] [PubMed]
- Apostolidou, E.; Skendros, P.; Kambas, K.; Mitroulis, I.; Konstantinidis, T.; Chrysanthopoulou, A.; Nakos, K.; Tsironidou, V.; Koffa, M.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular traps regulate IL-1β-mediated inflammation in familial Mediterranean fever. Ann. Rheum. Dis. 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The emerging role of NETs in venousthrombosis and immunothrombosis. Front. Immunol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.N.; Kazzaz, N.M.; Knight, J.S. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases? World J. Cardiol. 2015, 7, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Almyroudis, N.G.; Grimm, M.J.; Davidson, B.A.; Röhm, M.; Urban, C.F.; Segal, B.H. NETosis and NADPH oxidase: At the intersection of host defense, inflammation, and injury. Front. Immunol. 2013, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Hashiguchi, N.; Nagaoka, I.; Tabe, Y.; Murai, M. Neutrophil cell death in response to infection and its relation to coagulation. J. Intensive Care 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy : Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Craft, J.E. Dissecting the immune Cell Mayhem that Drives Lupus Pathogenesis. Sci. Transl. Med. 2011, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Björnsdottir, H.; Welin, A.; Michaëlsson, E.; Osla, V.; Berg, S.; Christenson, K.; Sundqvist, M.; Dahlgren, C.; Karlsson, A.; Bylund, J. Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic. Biol. Med. 2015, 89, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed]
- Baregamian, N.; Song, J.; Bailey, C.E.; Papaconstantinou, J.; Evers, B.M.; Chung, D.H. Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid. Med. Cell. Longev. 2009, 2, 297–306. [Google Scholar] [CrossRef]
- Xue, X.; Piao, J.-H.; Nakajima, A.; Sakon-Komazawa, S.; Kojima, Y.; Mori, K.; Yagita, H.; Okumura, K.; Harding, H.; Nakano, H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005, 280, 33917–33925. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Yoshinori Ohsumi: Autophagy from beginning to end. Interview by Caitlin Sedwick. J. Cell Biol. 2012, 197, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Nobel Prize Honors Autophagy Discovery. Cancer Discov. 2016, 6, 1298–1299. [CrossRef]
- Sha, L.-L.; Wang, H.; Wang, C.; Peng, H.-Y.; Chen, M.; Zhao, M.-H. Autophagy is induced by anti-neutrophil cytoplasmic Abs and promotes neutrophil extracellular traps formation. Innate Immun. 2016, 22, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Lee, S.-J.; Smith, A.; Choi, A.M.K. Autophagy in Vascular Disease. Proc. Am. Thorac. Soc. 2010, 7, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ouseph, M.M.; Huang, Y.; Banerjee, M.; Joshi, S.; MacDonald, L.; Zhong, Y.; Liu, H.; Li, X.; Xiang, B.; Zhang, G.; et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 2015, 126, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Rottem, M.; Krause, I.; Fraser, A.; Stojanovich, L.; Rovensky, J.; Shoenfeld, Y. Antiphospholipid syndrome. Lupus 2006, 95, 336–342. [Google Scholar] [CrossRef]
- Conti, F.; Sorice, M.; Circella, A.; Alessandri, C.; Pittoni, V.; Caronti, B.; Calderaro, C.; Griggi, T.; Misasi, R.; Valesini, G. Beta-2-glycoprotein I expression on monocytes is increased in anti-phospholipid antibody syndrome and correlates with tissue factor expression. Clin. Exp. Immunol. 2003, 132, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Buttari, B.; Capozzi, A.; Profumo, E.; Facchiano, F.; Truglia, S.; Recalchi, S.; Alessandri, C.; Conti, F.; Misasi, R.; et al. Antibodies to age-β2glycoprotein I in patients with anti-phospholipid antibody syndrome. Clin. Exp. Immunol. 2016, 184, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; DE Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Mccrae, K.R. The antiphospholipid syndrome: still an enigma. ASH Educ. Progr. B. 2015, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S.; et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Giaglis, S.; Hoesli, I.; Hasler, P. Neutrophil NETs in reproduction: From infertility to preeclampsia and the possibility of fetal loss. Front. Immunol. 2012, 3, 362. [Google Scholar] [CrossRef]
- Leffler, J.; Gullstrand, B.; Jönsen, A.; Nilsson, J.Å.; Martin, M.; Blom, A.M.; Bengtsson, A.A. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2013, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Stojanovich, L.; Shoenfeld, Y.; Bogdanovic, G.; Hesselstrand, R.; Blom, A.M. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin. Exp. Rheumatol. 2014, 32, 66–70. [Google Scholar] [PubMed]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE 2012, 7, e45427. [Google Scholar] [CrossRef] [PubMed]
- Kambas, K.; Mitroulis, I.; Ritis, K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front. Immunol. 2012, 3, 385. [Google Scholar] [CrossRef] [PubMed]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D.; et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Pinegin, B.; Vorobjeva, N.; Pinegin, V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun. Rev. 2015, 14, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Redecha, P.; Tilley, R.; Tencati, M.; Salmon, J.E.; Kirchhofer, D.; Mackman, N.; Girardi, G. Tissue factor: A link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood 2007, 110, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Fay, W.P. Linking inflammation and thrombosis: Role of C-reactive protein. World J. Cardiol. 2010, 2, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D.; et al. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Mackman, N. Chapter 5 Tissue Factor in Antiphospholipid Antibody-induced Pregnancy Loss: Thrombosis versus Inflammation. Handb. Syst. Autoimmune Dis. 2009, 10, 69–79. [Google Scholar] [CrossRef]
- Galarza-Maldonado, C.; Kourilovitch, M.R.; Andrade-Sánchez, P.; Durán, M.C.; Asanza, E. Treating obstetric antiphospholipid syndrome. Int. J. Clin. Rheumtol. 2013, 8, 407–414. [Google Scholar] [CrossRef]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2013, 345, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ling, Y.; Huang, M.; Yin, T.; Gou, S.M.; Zhan, N.Y.; Xiong, J.X.; Wu, H.S.; Yang, Z.Y.; Wang, C.Y. Heparin inhibits the inflammatory response induced by LPS and HMGB1 by blocking the binding of HMGB1 to the surface of macrophages. Cytokine 2015, 72, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Rovere-Querini, P.; D’Angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Barnado, A.; Crofford, L.J.; Oates, J.C. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Müller-Calleja, N.; Manukyan, D.; Canisius, A.; Strand, D.; Lackner, K.J. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann. Rheum. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Calton, E.K.; Keane, K.N.; Newsholme, P.; Soares, M.J. The impact of Vitamin D levels on inflammatory status: A systematic review of immune cell studies. PLoS ONE 2015, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Handono, K.; Sidarta, Y.O.; Pradana, B.A.; Nugroho, R.A.; Hartono, I.A.; Kalim, H.; Endharti, A.T. Vitamin D prevents endothelial damage induced by increased neutrophil extracellular traps formation in patients with systemic lupus erythematosus. Acta Med. Indones. 2014, 46, 189–198. [Google Scholar] [PubMed]
- Mohammed, B.M.; Fisher, B.J.; Kraskauskas, D.; Farkas, D.; Brophy, D.F.; Fowler, A.A., 3rd; Natarajan, R. Vitamin C: A novel regulator of neutrophil extracellular trap formation. Nutrients 2013, 5, 3131–3151. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar] [CrossRef]
- Gluud, L.L.; Simonetti, R.G. Mortality in Randomized Trials of Antioxidant Supplements for. J. Am. Med. Assoc. 2007, 297, 844–857. [Google Scholar] [CrossRef]
- Sesso, H.D. Vitamins E and C in the Prevention of Cardiovascular Disease in Men. JAMA 2008, 300, 2123. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Ferrer-Oliveras, R.; Llurba, E.; Gris, J.M. Tumor Necrosis Factor-Alpha and Pregnancy: Focus on Biologics. An Updated and Comprehensive Review. Clin. Rev. Allergy Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Puerta, J.A.; Cervera, R. Are there additional options for the treatment of refractory obstetric antiphospholipid syndrome? Lupus 2013, 22, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, U.; Wolf, D.C.; Dubinsky, M.; Cortot, A.; Lee, S.D.; Siegel, C.A.; Ullman, T.; Glover, S.; Valentine, J.F.; Rubin, D.T.; et al. Placental Transfer of Anti–Tumor Necrosis Factor Agents in Pregnant Patients With Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2013, 11, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Danesh, F.R.; Anel, R.L.; Zeng, L.; Lomasney, J.; Sahai, A.; Kanwar, Y.S. Immunomodulatory effects of HMG-CoA reductase inhibitors. Arch. Immunol. Ther. Exp. 2003, 51, 139–148. [Google Scholar]
- Ferrara, D.E.; Swerlick, R.; Casper, K.; Meroni, P.L.; Vega-Ostertag, M.E.; Harris, E.N.; Pierangeli, S.S. Fluvastatin inhibits up-regulation of tissue factor expression by antiphospholipid antibodies on endothelial cells. J. Thromb. Haemost. 2004, 2, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Raschi, E.; Testoni, C.; Tincani, A.; Balestrieri, G.; Molteni, R.; Khamashta, M.A.; Tremoli, E.; Camera, M. Statins prevent endothelial cell activation induced by antiphospholipid (anti-beta2-glycoprotein I) antibodies: Effect on the proadhesive and proinflammatory phenotype. Arthritis Rheum. 2001, 44, 2870–2878. [Google Scholar] [CrossRef]
- López-Pedrera, C.; Ruiz-Limón, P.; Aguirre, M.Á.; Barbarroja, N.; Pérez-Sánchez, C.; Buendía, P.; Rodriguez-García, I.C.; Rodriguez-Ariza, A.; Collantes-Estevez, E.; Velasco, F.; et al. Global effects of fluvastatin on the prothrombotic status of patients with antiphospholipid syndrome. Ann. Rheum. Dis. 2011, 70, 675–682. [Google Scholar] [CrossRef]
- Chow, O.A.; von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Zeiher, A.M.; Dimmeler, S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: A novel vasculoprotective function of statins. Circulation 2004, 110, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, H.; Patel, J.; Mahida, R.; Wang, Q.; Parekh, D.; Dancer, R.C.; Khiroya, H.; Sapey, E.; Thickett, D.R. Simvastatin to modify neutrophil function in older patients with septic pneumonia (SNOOPI): Study protocol for a randomised placebo-controlled trial. Trials 2014, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- John, S.G.; Thorn, J.; Sobonya, R. Statins as a Potential Risk Factor for Autoimmune Diseases. Am. J. Ther. 2014, 21, e94–e96. [Google Scholar] [CrossRef] [PubMed]
- Mohassel, P.; Mammen, A.L. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve 2013, 48, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Musset, L.; Allenbach, Y.; Benveniste, O.; Boyer, O.; Bossuyt, X.; Bentow, C.; Phillips, J.; Mammen, A.; van Damme, P.; Westhovens, R.; et al. Anti-HMGCR antibodies as a biomarker for immune-mediated necrotizing myopathies: A history of statins and experience from a large international multi-center study. Autoimmun. Rev. 2016, 15, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Zapantis, E.; Furie, R.; Horowitz, D. THU0400 Response to Eculizumab in the Antiphospholipid Antibody Syndrome. Ann. Rheum. Dis. 2015, 74. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo-Barrera, J.; Kourilovitch, M.; Galarza-Maldonado, C. Neutrophil Extracellular Traps, Antiphospholipid Antibodies and Treatment. Antibodies 2017, 6, 4. https://doi.org/10.3390/antib6010004
Bravo-Barrera J, Kourilovitch M, Galarza-Maldonado C. Neutrophil Extracellular Traps, Antiphospholipid Antibodies and Treatment. Antibodies. 2017; 6(1):4. https://doi.org/10.3390/antib6010004
Chicago/Turabian StyleBravo-Barrera, Jessica, Maria Kourilovitch, and Claudio Galarza-Maldonado. 2017. "Neutrophil Extracellular Traps, Antiphospholipid Antibodies and Treatment" Antibodies 6, no. 1: 4. https://doi.org/10.3390/antib6010004
APA StyleBravo-Barrera, J., Kourilovitch, M., & Galarza-Maldonado, C. (2017). Neutrophil Extracellular Traps, Antiphospholipid Antibodies and Treatment. Antibodies, 6(1), 4. https://doi.org/10.3390/antib6010004