
Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders

Abstract
:1. Introduction
2. TNF-α Receptor Signaling Pathways
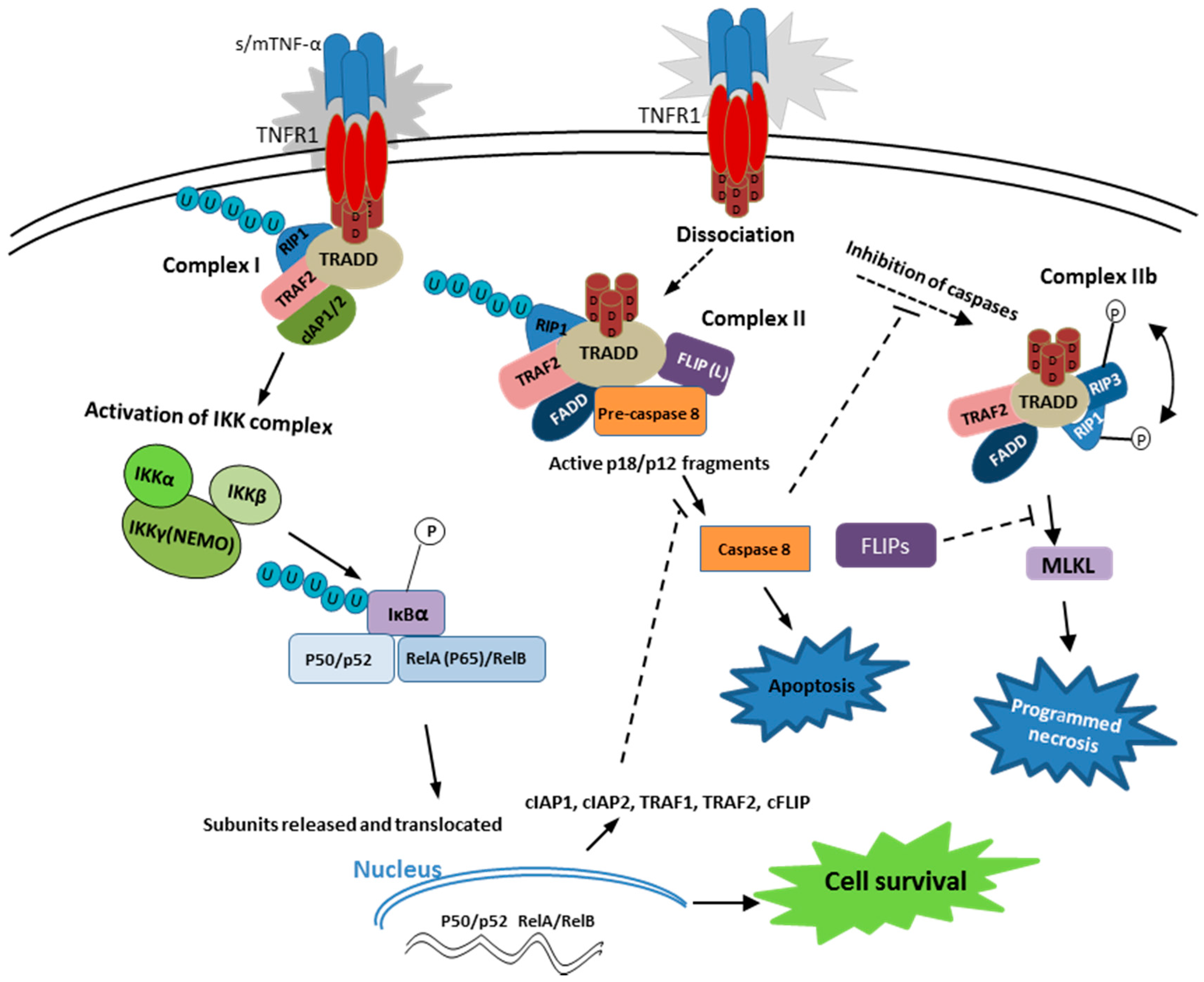
2.1. TNF-α Receptor 1 Signaling
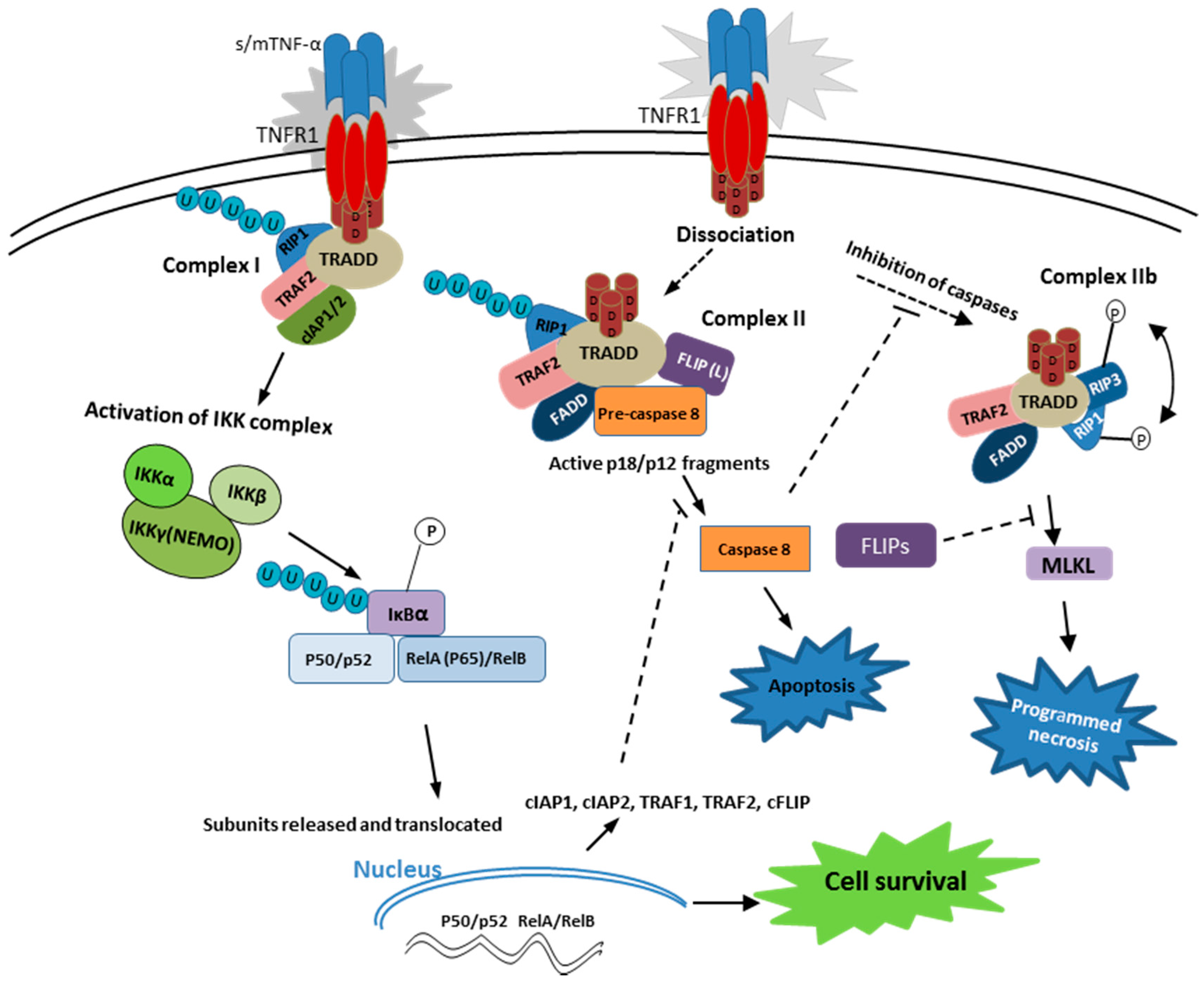
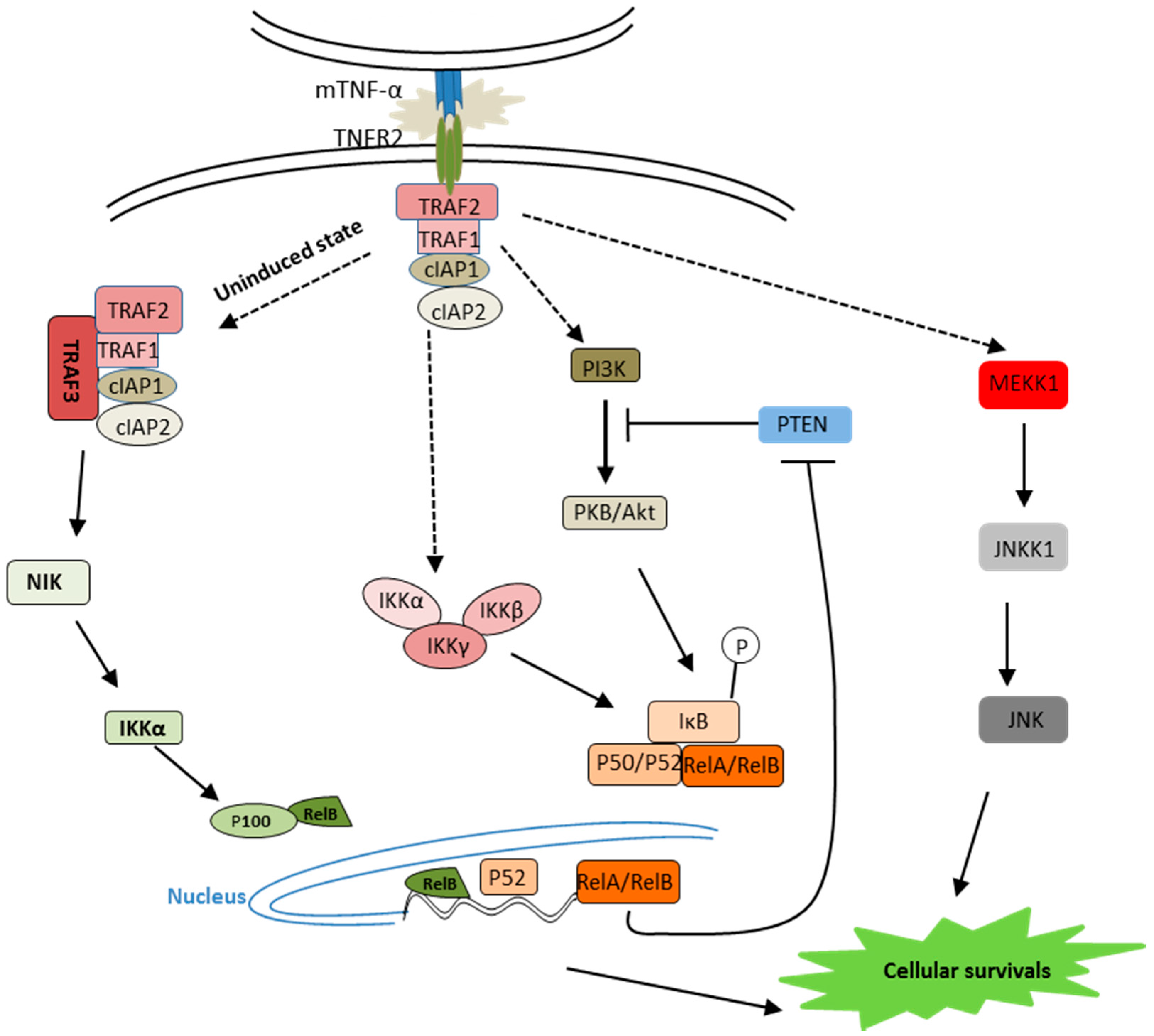
2.2. TNF-α Receptor 2 Signaling
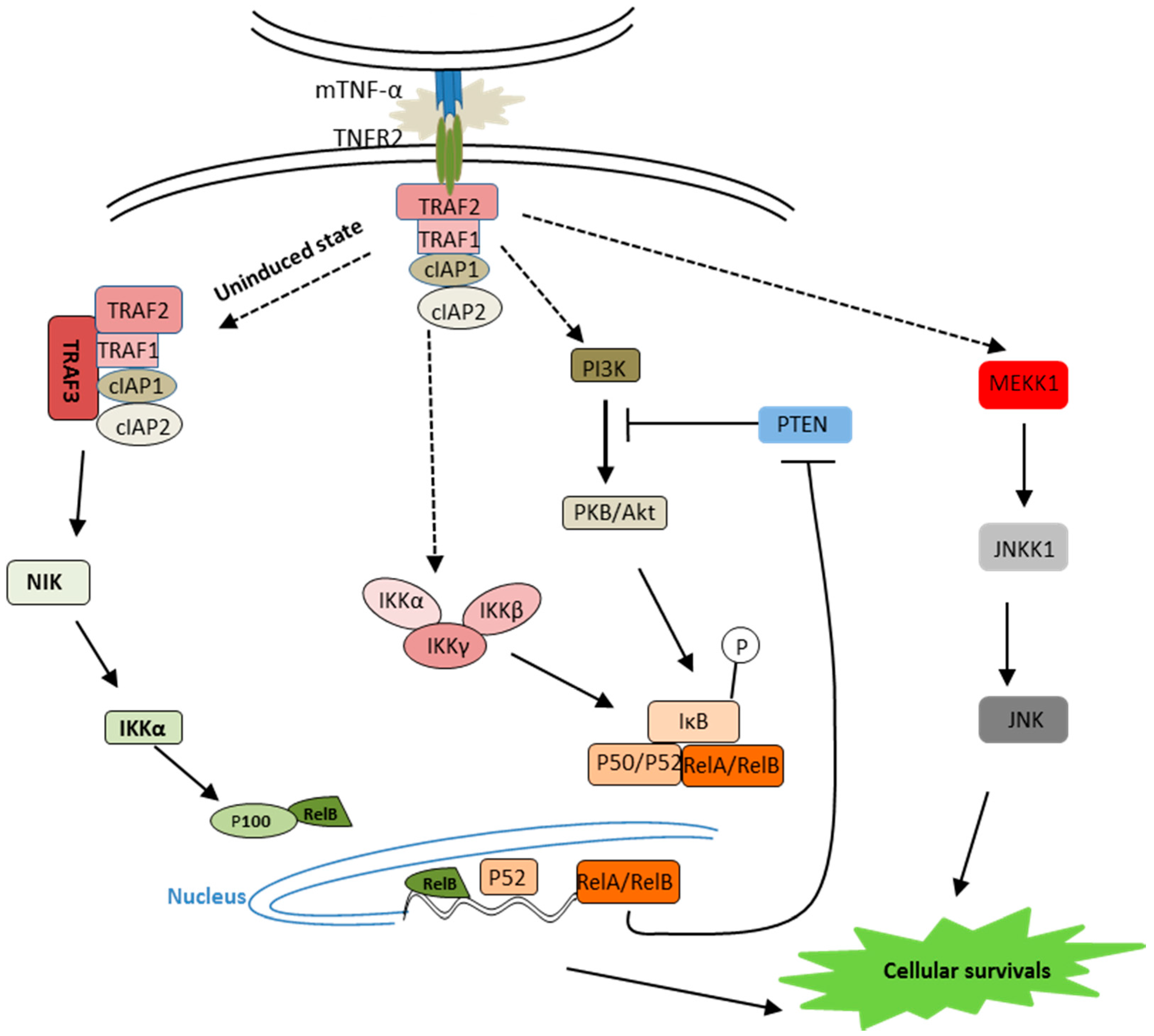
3. TNF and Its Receptors—Involvement in Neurodegenerative Disorders
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Ischemic Stroke
3.4. Multiple Sclerosis

{kind=link}
{kind=link}
{kind=link}
| Condition | TNF-α family member | Tissue | Finding | Model | Ref. |
|---|---|---|---|---|---|
| Alzheimer’s disease (AD) | TNF-α | CNS | TNF-α protein levels are increased in AD brain tissue. | Human AD patients. | [86,87,88] |
| Plasma and serum | TNF-α protein levels are increased in AD plasma and serum. | Human AD patients. | [83,84,85] | ||
| TNFR1 and TNFR2 | CNS | TNFR1 protein levels are increased, TNFR2 protein levels are decreased. | Human AD patients. | [88,93] | |
| Deletion of both TNFRs exacerbates AD pathology. | 3xTg-AD mouse model. | [96] | |||
| Silencing or deletion of TNFR2 aggravates AD pathology. TNFR2 overexpression reverses these effects. | 3xTg-AD mouse model and APP23 mouse model. | [97] | |||
| In vitro, TNFR2 silencing promotes Aβ neurotoxic effects. | SH-SY5Y cell line. | [100] | |||
| Deletion of TNFR1 diminishes AD pathology. | 3xTg-AD mouse model and APP23 mouse model. | [91] | |||
| sTNF-α inhibitors diminish AD pathology. | 3xTg-AD mouse model. | [99] | |||
| sTNFR1 and sTNFR2 | CSF, serum and plasma | sTNFR1 levels are increased. sTNFR2 levels are unchanged or decreased. | Human control and MCI patients. | [159,160,161,162,163] | |
| Higher sTNFR1 serum levels can predict conversion from MCI to AD. | Human control and MCI patients. | [161] | |||
| sTNFR1 and sTNFR2 levels correlate with BACE1 activity and Aβ40 levels, as well as with tau CSF levels. | Human control and MCI patients. | [164] | |||
| Parkinson’s disease (PD) | TNF-α | CNS and CSF | TNF-α levels are increased in brain and CSF. | Human control and PD patients. | [96,98] |
| TNF-α levels are increased in brain. | α-Synuclein overexpression cell line and mouse models. | [106] | |||
| TNFR1 | CNS | TNFR1 levels are increased in the substantia nigra. | Human control and PD patients. | [105] | |
| sTNF-α inhibitors reduce cell death of dopamine neurons. | Rat 6-OHDA toxicity model. | [111,112,113,114] | |||
| TNFR2 | CNS | Selective activation of TNFR2 protects dopaminergic neurons. | Neuronal culture, 6-OHDA toxicity model. | [110] | |
| TNFR1 and TNFR2 | CNS | Deletion of both TNFRs protects from dopaminergic toxicity, while lack of either TNFRs alone is not protective. | Mouse, MPTP toxicity model. | [116] | |
| sTNFR1 | Serum and plasma | Serum sTNFR1 levels are increased. | Human control and PD patients. | [102,104,165] | |
| Higher serum sTNFR1 correlate with a later onset of sporadic PD. | Human control and PD patients. | [165] | |||
| Elevated plasma sTNFR1 levels predict poorer executive functioning in PD. | Human control and PD patients. | [166] | |||
| Ischemic stroke | TNF-α | CNS | TNF-α production is increased around the lesion site. | Human brain tissue and animal models of stroke. | [117,118,119,120] |
| Inhibition of TNF-α reduces infarct size and neuroinflammation. | Stroke mouse models. | [122,123,124,125] | |||
| TNFR1 | CNS | TNFR1 knockout mice have larger infarct sizes compared to wild-type and TNFR2 knockout mice. | Stroke mouse model. | [134,135] | |
| TNFR1 is responsible for expression of neuroprotective factors upon ischemia. | Stroke mouse model. | [135,136] | |||
| Absence of TNFR1 reduces retinal ischemia-reperfusion damage. | Mouse retinal ischemia-reperfusion model. | [16] | |||
| TNFR1 signaling causes neuroinflammation and neurovascular damage in the immature brain. | LPS-sensitized hypoxic-ischemia mouse model. | [138] | |||
| TNFR2 | CNS | Absence of TNFR2 aggravates retinal ischemia-reperfusion damage. | Mouse retinal ischemia-reperfusion model. | [16] | |
| TNFR2 silencing increases cell injury upon hypoxic conditions. | SH-SY5Y cell line. | [100] | |||
| TNFR2 signaling can result in inflammatory ischemia. | Stroke mouse model. | [137] | |||
| TNFR1 and TNFR2 | CNS | Deletion of both TNFRs aggravates neuronal damage. | Stroke mouse model. | [5] | |
| Multiple sclerosis (MS) | TNF-α | CNS | TNF-α levels are increased in MS lesions. | Human MS brain tissue. | [140,141] |
| Constitutive TNF-α overexpression can cause a spontaneous inflammatory demyelinating disorder. | TNF-overexpressing mouse model. | [142] | |||
| TNF-α knockout increases demyelination and inflammation. | EAE mouse model. | [144,145] | |||
| TNF-α knockout delays both demyelination and remyelination. | Cuprizone mouse model. | [146] | |||
| General blockage of TNF-α by etanercept is linked to onset of MS in human case reports. | Human case reports. | [157,158] | |||
| General blockage of TNF-α by lenercept may exacerbate symptoms in human MS patients. | Human MS patients. | [156] | |||
| TNFR1 | CNS | TNFR1 knockout mice do not develop EAE or have a less severe disease course. | EAE mouse model. | [18,152,153,154] | |
| TNFR1 signaling induces oligodendrocyte apoptosis and primary demyelination. | TNF-transgenic mice. | [147] | |||
| TNFR1 may contribute to inflammatory infiltration of the spinal cord. | EAE mouse model. | [148] | |||
| Selective inhibition of TNFR1 signaling ameliorates EAE-induced pathology. | EAE mouse model. | [18,149] | |||
| sTNF-α inhibition protects against EAE symptoms. | EAE mouse model. | [150,151] | |||
| TNFR2 | CNS | TNFR2 knockout mice show aggravated demyelination and disease symptoms. | EAE mouse model. | [18,152,153,154] | |
| TNFR2 signaling mediates remyelination and oligodendrocyte precursor cell proliferation. | Cuprizone mouse model. | [146] | |||
| Selective stimulation of TNFR2 protects primary oligodendrocytes from oxidative stress. | Primary oligodendrocyte cell culture. | [155] |
3.5. Other Neurodegenerative Disorders
4. TNFR1- and TNFR2-Mediated Signaling in Neurodegeneration
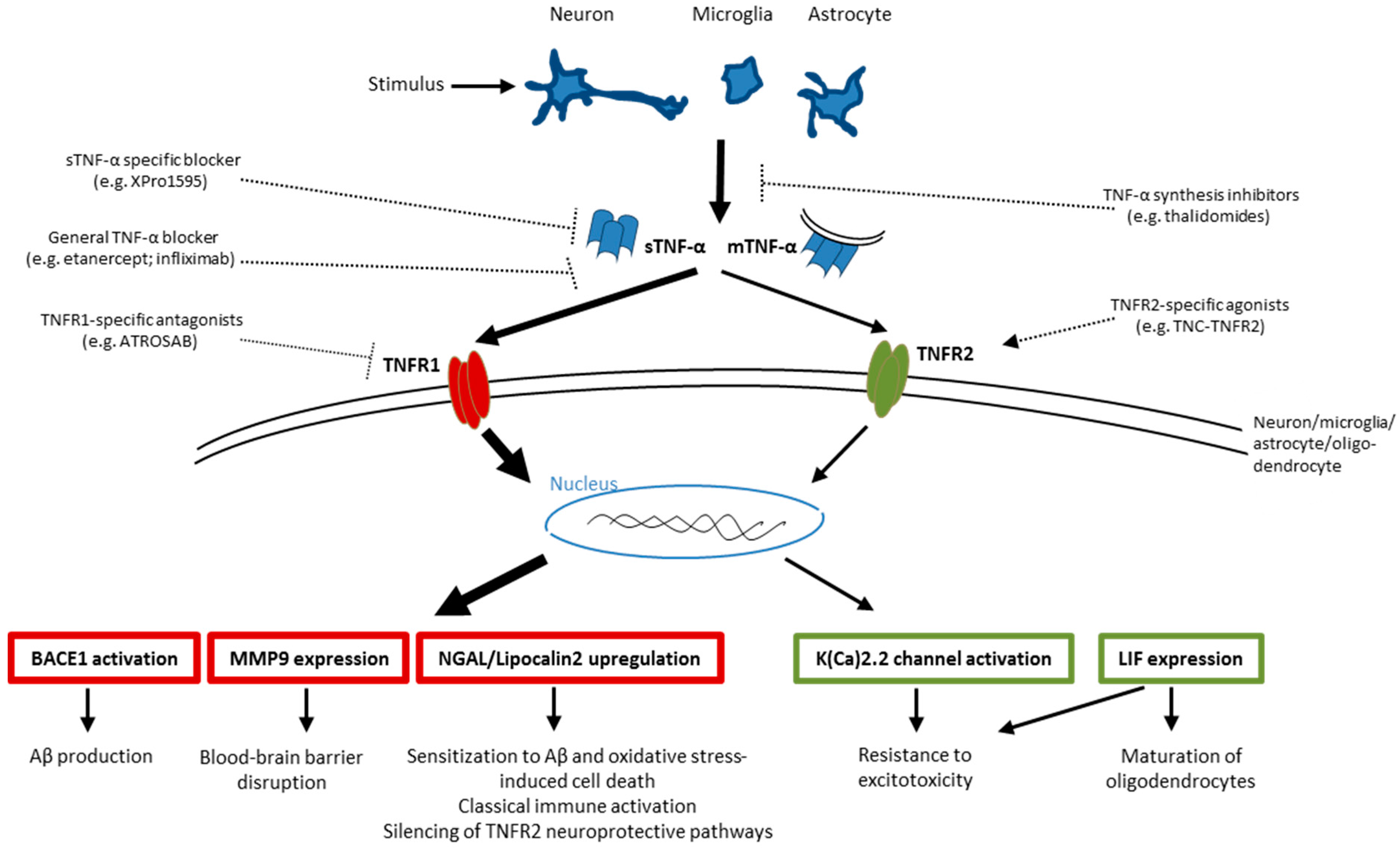
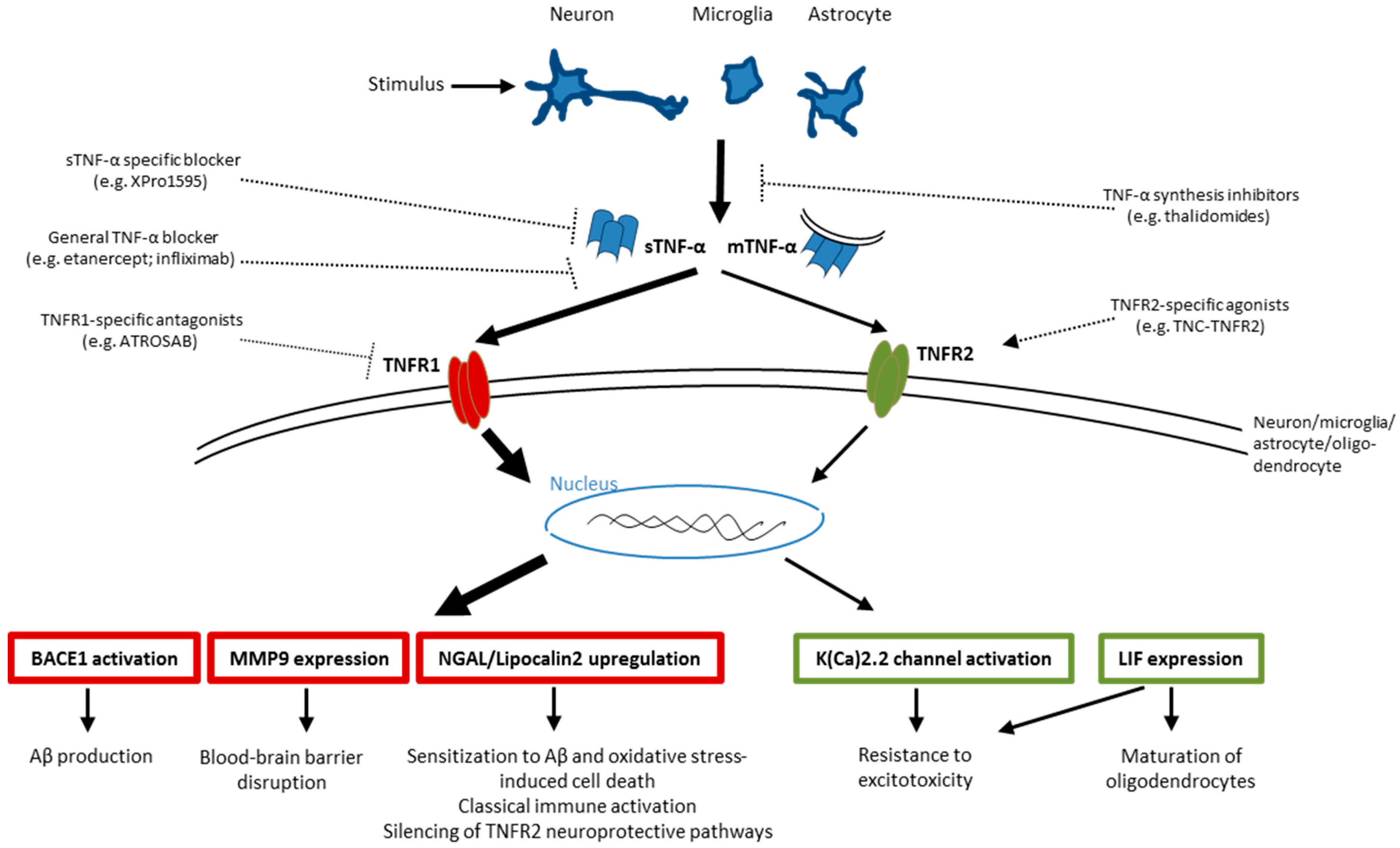
4.1. TNFR1—Possible Downstream Targets in Neurodegeneration
4.2. TNFR2—Possible Downstream Targets in Neurodegeneration
5. Complex matters: TNFR1 Signaling Is Primarily Damaging and TNFR2 Beneficial?
5.1. Selective Harmful Downstream Targets of TNFR1 and Beneficial Downstream Targets of TNFR2?
5.2. Soluble TNF Receptors
5.3. Interaction between TNFR2 and Interleukin-17 Receptor D
6. Targeting TNF-α Signaling: An Opportunity for Treatment of Neurodegenerative Disorders?
6.1. Targeting TNF-α as Treatment for Neurodegenerative Disorders
6.2. Targeting TNFRs as Treatment for Neurodegenerative Disorders
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K.M. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [PubMed]
- Siebert, S.; Tsoukas, A.; Robertson, J.; McInnes, I. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol. Rev. 2015, 67, 280–309. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Franciotta, D.M.; Grimaldi, L.M.; Martino, G.V.; Piccolo, G.; Bergamaschi, R.; Citterio, A.; Melzi d’Eril, G.V. Tumor necrosis factor in serum and cerebrospinal fluid of patients with multiple sclerosis. Ann. Neurol. 1989, 26, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.J.; Boling, W.; Kindy, M.S.; Peschon, J.; Kraemer, P.J.; Carpenter, M.K.; Holtsberg, F.W.; Mattson, M.P. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996, 2, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, H.; Pan, Y.C.; Lahm, H.W.; Gentz, R.; Brockhaus, M.; Tabuchi, H.; Lesslauer, W. Molecular cloning and expression of the human 55 kd tumor necrosis factor receptor. Cell 1990, 61, 351–359. [Google Scholar] [CrossRef]
- Schall, T.J.; Lewis, M.; Koller, K.J.; Lee, A.; Rice, G.C.; Wong, G.H.; Gatanaga, T.; Granger, G.A.; Lentz, R.; Raab, H. Molecular cloning and expression of a receptor for human tumor necrosis factor. Cell 1990, 61, 361–370. [Google Scholar] [CrossRef]
- Smith, C.A.; Davis, T.; Anderson, D.; Solam, L.; Beckmann, M.P.; Jerzy, R.; Dower, S.K.; Cosman, D.; Goodwin, R.G. A receptor for tumor necrosis factor defines an unusual family of cellular and viral proteins. Science 1990, 248, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Hierro, L.; Lazo, P.S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 2012, 24, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Fiers, W.; Beyaert, R.; Boone, E.; Cornelis, S.; Declercq, W.; Decoster, E.; Denecker, G.; Depuydt, B.; De Valck, D.; De Wilde, G.; et al. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J. Inflamm. 1995, 47, 67–75. [Google Scholar] [PubMed]
- Weiss, T.; Grell, M.; Hessabi, B.; Bourteele, S.; Müller, G.; Scheurich, P.; Wajant, H. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: Requirement of the TNF receptor-associated factor-2 binding site. J. Immunol. 1997, 158, 2398–2404. [Google Scholar] [PubMed]
- Weiss, T.; Grell, M.; Siemienski, K.; Mühlenbeck, F.; Dürkop, H.; Pfizenmaier, K.; Scheurich, P.; Wajant, H. TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J. Immunol. 1998, 161, 3136–3142. [Google Scholar] [PubMed]
- Van Herreweghe, F.; Festjens, N.; Declercq, W.; Vandenabeele, P. Tumor necrosis factor-mediated cell death: To break or to burst, that’s the question. Cell. Mol. Life Sci. 2010, 67, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Kimura, M.; Watanabe, N.; Ogihara, M. Tumor necrosis factor (TNF) receptor-2-mediated DNA synthesis and proliferation in primary cultures of adult rat hepatocytes: The involvement of endogenous transforming growth factor-alpha. Eur. J. Pharmacol. 2009, 604, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [PubMed]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014, 20, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, e90117. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.; Tartaglia, L.A.; Lee, A.; Bennett, G.L.; Rice, G.C.; Wong, G.H.; Chen, E.Y.; Goeddel, D.V. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA 1991, 88, 2830–2834. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74, 845–853. [Google Scholar] [CrossRef]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Chen, N.-J.; Chio, I.I.C.; Lin, W.-J.; Duncan, G.; Chau, H.; Katz, D.; Huang, H.-L.; Pike, K.A.; Hao, Z.; Su, Y.-W.; et al. Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 12429–12434. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr. Biol. 2007, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 2004, 5, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Woronicz, J.D.; Liu, W.; Goeddel, D.V. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science 1999, 283, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Juo, P.; Kuo, C.J.; Yuan, J.; Blenis, J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998, 8, 1001–1008. [Google Scholar] [CrossRef]
- Yeh, W.C.; de la Pompa, J.L.; McCurrach, M.E.; Shu, H.B.; Elia, A.J.; Shahinian, A.; Ng, M.; Wakeham, A.; Khoo, W.; Mitchell, K.; et al. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998, 279, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Choksi, S.; Shen, H.-M.; Yang, Q.-F.; Hur, G.M.; Kim, Y.S.; Tran, J.H.; Nedospasov, S.A.; Liu, Z. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 2004, 279, 10822–10828. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, J.; Li, L.; Zhang, Z.; Ren, J.; Liang, Y.; Chen, F.; Yang, C.; Zhou, Z.; Su, S.S.; et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat. Cell Biol. 2015, 17, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Fernandes, J.; Carreto, L.; Anuncibay-Soto, B.; Santos, M.; Han, J.; Fernández-López, A.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol. Dis. 2014, 68, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, X.; Li, Y.; Xu, L.; Yu, X.; Ge, L.; Li, J.; Zhu, Y.; He, S. Necroptosis mediates TNF-induced toxicity of hippocampal neurons. BioMed Res. Int. 2014, 2014, 290182. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Vilalta, A.; Tolkovsky, A.M.; Brown, G.C. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J. Biol. Chem. 2013, 288, 9145–9152. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Rothe, M.; Pan, M.G.; Henzel, W.J.; Ayres, T.M.; Goeddel, D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252. [Google Scholar] [CrossRef]
- Cabal-Hierro, L.; Rodríguez, M.; Artime, N.; Iglesias, J.; Ugarte, L.; Prado, M.A.; Lazo, P.S. TRAF-mediated modulation of NF-kB AND JNK Activation by TNFR2. Cell. Signal. 2014, 26, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Ley, S.C. New insights into NF-kappaB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.; Cabal-Hierro, L.; Carcedo, M.T.; Iglesias, J.M.; Artime, N.; Darnay, B.G.; Lazo, P.S. NF-kappaB signal triggering and termination by tumor necrosis factor receptor 2. J. Biol. Chem. 2011, 286, 22814–22824. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L.M. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.A.; Ozes, O.N.; Akca, H.; Pincheira, R.; Mayo, L.D.; Li, Q.; Guzman, J.R.; Korgaonkar, C.K.; Donner, D.B. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation. J. Biol. Chem. 2004, 279, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Eisel, U.L.M.; Biber, K.; Luiten, P.G.M. Life and Death of Nerve Cells: Therapeutic Cytokine Signaling Pathways. Curr. Signal Transduct. Ther. 2006, 1, 133–146. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Tseng, P.-H.; Vallabhapurapu, S.; Luo, J.-L.; Zhang, W.; Wang, H.; Vignali, D.A.A.; Gallagher, E.; Karin, M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 2008, 321, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Ruspi, G.; Schmidt, E.M.; McCann, F.; Feldmann, M.; Williams, R.O.; Stoop, A.A.; Dean, J.L.E. TNFR2 increases the sensitivity of ligand-induced activation of the p38 MAPK and NF-κB pathways and signals TRAF2 protein degradation in macrophages. Cell. Signal. 2014, 26, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Liu, Z.G.; Bennett, B.; Suzuki, N.; Xia, Y.; Karin, M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999, 13, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Tan, T.H. The c-Jun N-terminal kinase pathway and apoptotic signaling (review). Int. J. Oncol. 2000, 16, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Li, Y.; Wan, T.; Wang, J.; Zhang, H.; Chen, H.; Min, W. Both internalization and AIP1 association are required for tumor necrosis factor receptor 2-mediated JNK signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Choi, Y.; Watts, T.H. Role of TNF Receptor-Associated Factor 2 and p38 Mitogen-Activated Protein Kinase Activation During 4-1BB-Dependent Immune Response. J. Immunol. 2000, 165, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Naudé, P.J.W.; den Boer, J.A.; Luiten, P.G.M.; Eisel, U.L.M. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M.; Henkler, F.; Samel, D.; Reichwein, M.; Hausser, A.; Parmryd, I.; Scheurich, P.; Schmid, J.A.; Wajant, H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 2002, 115, 2757–2770. [Google Scholar] [PubMed]
- Li, X.; Yang, Y.; Ashwell, J.D. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 2002, 416, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [PubMed]
- McAlpine, F.E.; Tansey, M.G. Neuroinflammation and tumor necrosis factor signaling in the pathophysiology of Alzheimer’s disease. J. Inflamm. Res. 2008, 1, 29–39. [Google Scholar] [PubMed]
- Di Bona, D.; Candore, G.; Franceschi, C.; Licastro, F.; Colonna-Romano, G.; Cammà, C.; Lio, D.; Caruso, C. Systematic review by meta-analyses on the possible role of TNF-alpha polymorphisms in association with Alzheimer’s disease. Brain Res. Rev. 2009, 61, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-J.; Kim, J.-M.; Kim, S.-W.; Shin, I.-S.; Park, S.-W.; Kim, Y.-H.; Yoon, J.-S. Associations of cytokine genes with Alzheimer’s disease and depression in an elderly Korean population. J. Neurol. Neurosurg. Psychiatry 2014, 86, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. Association between TNF-α promoter -308 A/G polymorphism and Alzheimer’s disease: A meta-analysis. Neurol. Sci. 2015, 36, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. TNF-alpha G308A Polymorphism and the Susceptibility to Alzheimer’s Disease: An Updated Meta-analysis. Arch. Med. Res. 2015, 46, 24–30.e1. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Cacabelos, R.; Sanpedro, C.; García-Fantini, M.; Aleixandre, M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 2007, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Jeune, B.; Pedersen, A.N.; Skinhøj, P.; Pedersen, B.K. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J. Gerontol. A-Biol. 1999, 54, M357–M364. [Google Scholar] [CrossRef]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef]
- McGeer, E.G.; McGeer, P.L. Inflammatory processes in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 741–749. [Google Scholar] [CrossRef]
- Zhao, M.; Cribbs, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiyota, T.; Walsh, S.M.; Liu, J.; Kipnis, J.; Ikezu, T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J. Immunol. 2008, 181, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-F.; Wang, B.-J.; Cheng, H.-T.; Kuo, L.-H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimers Dis. 2010, 19, 621–630. [Google Scholar] [PubMed]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimers Dis. 2010, 19, 621–630. [Google Scholar] [PubMed]
- Perry, R.T.; Collins, J.S.; Wiener, H.; Acton, R.; Go, R.C. The role of TNF and its receptors in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 873–883. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Mastrangelo, M.A.; Habib, D.; Narrow, W.C.; Knowlden, S.A.; Wright, T.W.; Bowers, W.J. Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: Implications for chronic pan-TNF-α suppressive therapeutic strategies in the brain. Am. J. Pathol. 2011, 179, 2053–2070. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.L.; Narrow, W.C.; Mastrangelo, M.A.; Olschowka, J.A.; O’Banion, M.K.; Bowers, W.J. Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic Alzheimer disease mice leads to significant modulation of amyloid- and Tau-related pathologies. Am. J. Pathol. 2013, 182, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, P.; Xie, J.; Staufenbiel, M.; Li, R.; Shen, Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα. Hum. Mol. Genet. 2014, 23, 4906–4918. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.E.; Lee, J.-K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, R.; Shiosaki, K. Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and beta-amyloid toxicity in human neuronal cell line. J. Biol. Chem. 1997, 272, 3550–3553. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Rojanathammanee, L.; Murphy, E.J.; Combs, C.K. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J. Neuroinflamm. 2011, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Federoff, H.J.; Maguire-Zeiss, K.A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 2009, 16, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Park. Dis. 2014, 4, 349–360. [Google Scholar]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Martinez, T.N.; McAlpine, F.E.; Blesch, A.; Tansey, M.G. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol. Ther. 2008, 16, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Barnum, C.J.; Ruhn, K.A.; Varghese, S.; Treviño, I.; Blesch, A.; Tansey, M.G. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson’s disease. Mol. Ther. 2011, 19, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Dziewulska, D.; Mossakowski, M.J. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin. Neuropathol. 2003, 22, 35–40. [Google Scholar] [PubMed]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, T.; Carpén, O.; Karjalainen-Lindsberg, M.L.; Paetau, A.; Turpeinen, U.; Kaste, M.; Lindsberg, P.J. Evolution of cerebral tumor necrosis factor-alpha production during human ischemic stroke. Stroke 2001, 32, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; di Sciacca, R.; Pinto, A.; Licata, G. Inflammatory cytokines in acute ischemic stroke. Curr. Pharm. Des. 2008, 14, 3574–3589. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Kastin, A.J. Tumor necrosis factor and stroke: Role of the blood-brain barrier. Prog. Neurobiol. 2007, 83, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, R.K.; Boado, R.J.; Pardridge, W.M. Brain protection from stroke with intravenous TNFα decoy receptor-Trojan horse fusion protein. J. Cereb. Blood Flow Metab. 2012, 32, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Kim, N.M.; Reyzin, G.; Rodriguez-Romanacce, H.; DePuy, V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 2012, 26, 1051–1070. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-H.; Huang, C.-C.; Chio, C.-C.; Tsai, K.-J.; Chang, C.-P.; Lin, N.-K.; Lin, M.-T. Inhibition of Peripheral TNF-α and Downregulation of Microglial Activation by Alpha-Lipoic Acid and Etanercept Protect Rat Brain Against Ischemic Stroke. Mol. Neurobiol. 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Arango-Dávila, C.A.; Vera, A.; Londoño, A.C.; Echeverri, A.F.; Cañas, F.; Cardozo, C.F.; Orozco, J.L.; Rengifo, J.; Cañas, C.A. Soluble or soluble/membrane TNF-α inhibitors protect the brain from focal ischemic injury in rats. Int. J. Neurosci. 2014, 125, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-H.; Mrizek, M.; Lai, Q.; Wu, Y.; Reyes, R.; Li, J.; Davis, W.W.; Ding, Y. Exercise preconditioning reduces brain damage and inhibits TNF-alpha receptor expression after hypoxia/reoxygenation: An in vivo and in vitro study. Curr. Neurovasc. Res. 2006, 3, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Tasaki, K.; Ruetzler, C.A.; Hallenbeck, J.M. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997, 17, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Botchkina, G.I.; Meistrell, M.E.; Botchkina, I.L.; Tracey, K.J. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol. Med. Camb. Mass 1997, 3, 765–781. [Google Scholar] [PubMed]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.M.; Eisel, U.L.M. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alzheimers Dis. 2008, 13, 111–122. [Google Scholar] [PubMed]
- Rasmussen, L.M.; Hansen, P.R.; Nabipour, M.T.; Olesen, P.; Kristiansen, M.T.; Ledet, T. Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells. Biochem. J. 2001, 360, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Santee, S.M.; Owen-Schaub, L.B. Human tumor necrosis factor receptor p75/80 (CD120b) gene structure and promoter characterization. J. Biol. Chem. 1996, 271, 21151–21159. [Google Scholar] [PubMed]
- Gary, D.S.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998, 18, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Pradillo, J.M.; Romera, C.; Hurtado, O.; Cárdenas, A.; Moro, M.A.; Leza, J.C.; Dávalos, A.; Castillo, J.; Lorenzo, P.; Lizasoain, I. TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J. Cereb. Blood Flow Metab. 2005, 25, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Valable, S.; Müller, G.J.; Roberts, M.L.; Divoux, D.; Tinel, A.; Voulgari-Kokota, A.; Tseveleki, V.; Altruda, F.; Lassmann, H.; et al. FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J. Neurosci. 2007, 27, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Akassoglou, K.; Douni, E.; Bauer, J.; Lassmann, H.; Kollias, G.; Probert, L. Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-W.; Chang, Y.-C.; Chen, S.-J.; Tseng, C.-H.; Tu, Y.-F.; Liao, N.-S.; Huang, C.-C.; Ho, C.-J. TNFR1-JNK signaling is the shared pathway of neuroinflammation and neurovascular damage after LPS-sensitized hypoxic-ischemic injury in the immature brain. J. Neuroinflamm. 2014, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Cudrici, C.; Niculescu, T.; Niculescu, F.; Shin, M.L.; Rus, H. Oligodendrocyte cell death in pathogenesis of multiple sclerosis: Protection of oligodendrocytes from apoptosis by complement. J. Rehabil. Res. Dev. 2006, 43, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; Merrill, J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989, 170, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Raine, C.S.; Cannella, B.; Brosnan, C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Invest. 1991, 87, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Parkhurst, C.N.; Hayes, S.; Gan, W.-B. Peripheral elevation of TNF-α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2013, 110, 10306–10311. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- Gimenez, M.A.; Sim, J.; Archambault, A.S.; Klein, R.S.; Russell, J.H. A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am. J. Pathol. 2006, 168, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; Kawara, T.; Arita, S.; Furuya, T.; Yamashita, T.; et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J. Control. Release 2011, 149, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain J. Neurol. 2011, 134, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain J. Neurol. 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Bachmann, R.; Bluethmann, H.; Lassmann, H.; Fontana, A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 1999, 29, 626–632. [Google Scholar] [CrossRef]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Kollias, G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: Implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 2001, 193, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Fischer, R.; Agresti, C.; Pfizenmaier, K. TNF receptor 2 protects oligodendrocyte progenitor cells against oxidative stress. Biochem. Biophys. Res. Commun. 2013, 440, 336–341. [Google Scholar] [CrossRef] [PubMed]
- The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar]
- Pfueller, C.F.; Seipelt, E.; Zipp, F.; Paul, F. Multiple sclerosis following etanercept treatment for ankylosing spondylitis. Scand. J. Rheumatol. 2008, 37, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Sicotte, N.L.; Voskuhl, R.R. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology 2001, 57, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hampel, H.; Prvulovic, D.; Wallin, A.; Blennow, K.; Li, R.; Shen, Y. Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Song, N.; Yu, J.; Tan, L.; Shen, Y.; Xie, J.; Jiang, H. Elevated plasma levels of soluble TNFRs and TACE activity in Alzheimer’s disease patients of Northern Han Chinese descent. Curr. Alzheimer Res. 2013, 10, 57–62. [Google Scholar] [PubMed]
- Diniz, B.S.; Teixeira, A.L.; Ojopi, E.B.; Talib, L.L.; Mendonça, V.A.; Gattaz, W.F.; Forlenza, O.V. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1305–1311. [Google Scholar] [PubMed]
- Faria, M.C.; Gonçalves, G.S.; Rocha, N.P.; Moraes, E.N.; Bicalho, M.A.; Gualberto Cintra, M.T.; Jardim de Paula, J.; José Ravic de Miranda, L.F.; Clayton de Souza Ferreira, A.; Teixeira, A.L.; et al. Increased plasma levels of BDNF and inflammatory markers in Alzheimer’s disease. J. Psychiatr. Res. 2014, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hampel, H.; Blennow, K.; Lista, S.; Levey, A.; Tang, B.; Li, R.; Shen, Y. Increased plasma TACE activity in subjects with mild cognitive impairment and patients with Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 877–886. [Google Scholar] [PubMed]
- Buchhave, P.; Zetterberg, H.; Blennow, K.; Minthon, L.; Janciauskiene, S.; Hansson, O. Soluble TNF receptors are associated with Aβ metabolism and conversion to dementia in subjects with mild cognitive impairment. Neurobiol. Aging 2010, 31, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Increased serum levels of soluble tumor necrosis factor-alpha receptor-1 in patients with Parkinson’s disease. J. Neuroimmunol. 2009, 216, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.P.; Teixeira, A.L.; Scalzo, P.L.; Barbosa, I.G.; de Sousa, M.S.; Morato, I.B.; Vieira, E.L.M.; Christo, P.P.; Palotás, A.; Reis, H.J. Plasma levels of soluble tumor necrosis factor receptors are associated with cognitive performance in Parkinson’s disease. Mov. Disord. 2014, 29, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.; Morganti-Kossmann, C. The role of markers of inflammation in traumatic brain injury. Neurotrauma 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bauer, S.; Nowak, M.; Norwood, B.; Tackenberg, B.; Rosenow, F.; Knake, S.; Oertel, W.H.; Hamer, H.M. Cytokines and epilepsy. Seizure 2011, 20, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A.; Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The Role of the Immune System in Huntington's Disease. J. Immunol. Res. 2013, 2013, e541259. [Google Scholar] [CrossRef] [PubMed]
- Alto, L.T.; Chen, X.; Ruhn, K.A.; Treviño, I.; Tansey, M.G. AAV-Dominant Negative Tumor Necrosis Factor (DN-TNF) Gene Transfer to the Striatum Does Not Rescue Medium Spiny Neurons in the YAC128 Mouse Model of Huntington’s Disease. PLoS ONE 2014, 9, e96544. [Google Scholar] [CrossRef] [PubMed]
- Longhi, L.; Ortolano, F.; Zanier, E.R.; Perego, C.; Stocchetti, N.; De Simoni, M.G. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir. Suppl. 2008, 102, 409–413. [Google Scholar] [PubMed]
- Longhi, L.; Perego, C.; Ortolano, F.; Aresi, S.; Fumagalli, S.; Zanier, E.R.; Stocchetti, N.; De Simoni, M.-G. Tumor necrosis factor in traumatic brain injury: Effects of genetic deletion of p55 or p75 receptor. J. Cereb. Blood Flow Metab. 2013, 33, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.J.; Ashley, M.D.; Stöhr, S.; Schindler, C.; Li, M.; McCarthy-Culpepper, K.A.; Pearson, A.N.; Xiong, Z.-G.; Simon, R.P.; Henshall, D.C.; et al. Suppression of TNF receptor-1 signaling in an in vitro model of epileptic tolerance. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 120–132. [Google Scholar] [PubMed]
- Balosso, S.; Ravizza, T.; Perego, C.; Peschon, J.; Campbell, I.L.; De Simoni, M.G.; Vezzani, A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann. Neurol. 2005, 57, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.-Y.; Chiu, F.-L.; Chen, C.-M.; Wu, Y.-R.; Chen, H.-M.; Chen, Y.-C.; Kuo, H.-C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- German, D.C.; Liang, C.-L.; Song, T.; Yazdani, U.; Xie, C.; Dietschy, J.M. Neurodegeneration in the Niemann–Pick C mouse: Glial involvement. Neuroscience 2002, 109, 437–450. [Google Scholar] [CrossRef]
- Patel, S.C.; Suresh, S.; Kumar, U.; Hu, C.Y.; Cooney, A.; Blanchette-Mackie, E.J.; Neufeld, E.B.; Patel, R.C.; Brady, R.O.; Patel, Y.C.; et al. Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann- Pick type C disease. Proc. Natl. Acad. Sci. USA 1999, 96, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-P.; Mizukami, H.; Matsuda, J.; Saito, Y.; Proia, R.L.; Suzuki, K. Apoptosis accompanied by up-regulation of TNF-alpha death pathway genes in the brain of Niemann-Pick type C disease. Mol. Genet. Metab. 2005, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kim, E.Y.; Jung, S.-C. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar] [PubMed]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain J. Neurol. 2012, 135, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Naudé, P.J.W.; Nyakas, C.; Eiden, L.E.; Ait-Ali, D.; van der Heide, R.; Engelborghs, S.; Luiten, P.G.M.; De Deyn, P.P.; den Boer, J.A.; Eisel, U.L.M. Lipocalin 2: Novel component of proinflammatory signaling in Alzheimer’s disease. FASEB J. 2012, 26, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Marques, F.; Mesquita, S.D.; Sousa, J.C.; Coppola, G.; Gao, F.; Geschwind, D.H.; Columba-Cabezas, S.; Aloisi, F.; Degn, M.; Cerqueira, J.J.; et al. Lipocalin 2 is present in the EAE brain and is modulated by natalizumab. Front. Cell. Neurosci. 2012, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Weng, Y.-C.; Han, X.; Whaley, J.D.; McCrae, K.R.; Chou, W.-H. Lipocalin-2 released in response to cerebral ischaemia mediates reperfusion injury in mice. J. Cell. Mol. Med. 2015, 19, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Huang, C.; Tong, J.; Qiu, G.; Huang, B.; Wu, Q.; Li, F.; Xu, Z.; Bowser, R.; Xia, X.-G.; et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. USA 2013, 110, 4069–4074. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, W.-H.; Lee, M.-S.; Mori, K.; Suk, K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J. Neurosci. Res. 2012, 90, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, S.D.; Ferreira, A.C.; Falcao, A.M.; Sousa, J.C.; Oliveira, T.G.; Correia-Neves, M.; Sousa, N.; Marques, F.; Palha, J.A. Lipocalin 2 modulates the cellular response to amyloid beta. Cell Death Differ. 2014, 21, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.; Kim, J.-H.; Lee, S.; Kim, J.-H.; Seo, J.-W.; Jin, M.; Lee, M.-G.; Jang, I.-S.; Lee, W.-H.; Suk, K. Phenotypic polarization of activated astrocytes: The critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 2013, 191, 5204–5219. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.; Lee, S.; Kim, J.-H.; Kim, J.-H.; Seo, J.-W.; Lee, W.-H.; Mori, K.; Nakao, K.; Suk, K. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 2013, 27, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-T.; Lin, C.-C.; Wu, Y.-C.; Yang, C.-M. TNF-alpha induces matrix metalloproteinase-9 expression in A549 cells: role of TNFR1/TRAF2/PKCalpha-dependent signaling pathways. J. Cell. Physiol. 2010, 224, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Chen, W.-C.; Hsieh, H.-L.; Chi, P.-L.; Hsiao, L.-D.; Yang, C.-M. TNF-α induces matrix metalloproteinase-9-dependent soluble intercellular adhesion molecule-1 release via TRAF2-mediated MAPKs and NF-κB activation in osteoblast-like MC3T3-E1 cells. J. Biomed. Sci. 2014, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Tseng, H.-W.; Hsieh, H.-L.; Lee, C.-W.; Wu, C.-Y.; Cheng, C.-Y.; Yang, C.-M. Tumor necrosis factor-alpha induces MMP-9 expression via p42/p44 MAPK, JNK, and nuclear factor-kappaB in A549 cells. Toxicol. Appl. Pharmacol. 2008, 229, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A.; Tepper, D.; Leonard, A. Matrix Metalloproteinases and Blood-Brain Barrier Disruption in Acute Ischemic Stroke. Front. Neurol. 2013, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, H.; Takuma, K.; Fukuzaki, E.; Ibi, D.; Someya, E.; Akazawa, K.; Alkam, T.; Tsunekawa, H.; Mouri, A.; Noda, Y.; et al. Matrix metalloprotease-9 inhibition improves amyloid beta-mediated cognitive impairment and neurotoxicity in mice. J. Pharmacol. Exp. Ther. 2009, 331, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Takahashi, H.; Machida, T.; Wakigawa, T.; Harada, E.; Miyaji, H.; Koga, M.; Nishioku, T.; et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflamm. 2011, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J. Biol. Chem. 2001, 276, 37258–37265. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Liu, Q.; Wu, J.; Shen, Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. 2012, 26, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Terpolilli, N.; Kepura, F.; Nijholt, I.M.; Knaus, H.-G.; D’Orsi, B.; Prehn, J.H.M.; Eisel, U.L.M.; Plant, T.; Plesnila, N.; et al. KCa2 channels activation prevents [Ca2+]i deregulation and reduces neuronal death following glutamate toxicity and cerebral ischemia. Cell Death Dis. 2011, 2, e147. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, E.F.E.; Nelemans, A.; Luiten, P.; Nijholt, I.; Dolga, A.; Eisel, U. K(Ca)2 and k(ca)3 channels in learning and memory processes, and neurodegeneration. Front. Pharmacol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.; Bond, C.T.; Luján, R.; Ballesteros-Merino, C.; Lin, M.T.; Wang, K.; Klett, N.; Watanabe, M.; Shigemoto, R.; Stackman, R.W.; et al. The SK2-long isoform directs synaptic localization and function of SK2-containing channels. Nat. Neurosci. 2011, 14, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Moidunny, S.; Vinet, J.; Wesseling, E.; Bijzet, J.; Shieh, C.-H.; van Ijzendoorn, S.C.D.; Bezzi, P.; Boddeke, H.W.G.M.; Biber, K. Adenosine A2B receptor-mediated leukemia inhibitory factor release from astrocytes protects cortical neurons against excitotoxicity. J. Neuroinflamm. 2012, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresle, M.M.; Alexandrou, E.; Wu, Q.; Egan, G.; Jokubaitis, V.; Ayers, M.; Jonas, A.; Doherty, W.; Friedhuber, A.; Shaw, G.; et al. Leukemia inhibitory factor protects axons in experimental autoimmune encephalomyelitis via an oligodendrocyte-independent mechanism. PLoS ONE 2012, 7, e47379. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Parachikova, A.; Cotman, C.W. Reduced CXCL12/CXCR4 results in impaired learning and is downregulated in a mouse model of Alzheimer disease. Neurobiol. Dis. 2007, 28, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Aricò, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Maier, O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies 2015, 4, 48–70. [Google Scholar] [CrossRef]
- Rodriguez, M.; Zoecklein, L.; Papke, L.; Gamez, J.; Denic, A.; Macura, S.; Howe, C. Tumor necrosis factor alpha is reparative via TNFR2 [corrected] in the hippocampus and via TNFR1 [corrected] in the striatum after virus-induced encephalitis. Brain Pathol. 2009, 19, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Wilde, G.J.; Pringle, A.K.; Sundstrom, L.E.; Mann, D.A.; Iannotti, F. Attenuation and augmentation of ischaemia-related neuronal death by tumour necrosis factor-alpha in vitro. Eur. J. Neurosci. 2000, 12, 3863–3870. [Google Scholar] [CrossRef] [PubMed]
- Kuno, R.; Yoshida, Y.; Nitta, A.; Nabeshima, T.; Wang, J.; Sonobe, Y.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res. 2006, 1116, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Lou, J.; Morel, D.R.; Ricou, B.; Suter, P.M.; Grau, G.E. TNF receptors in the microvascular pathology of acute respiratory distress syndrome and cerebral malaria. J. Leuk. Biol. 1997, 61, 551–558. [Google Scholar] [CrossRef]
- Pola, R.; Flex, A.; Gaetani, E.; Santoliquido, A.; Serricchio, M.; Pola, P.; Bernabei, R. Intercellular adhesion molecule-1 K469E gene polymorphism and Alzheimer’s disease. Neurobiol. Aging 2003, 24, 385–387. [Google Scholar] [CrossRef]
- Hattori, A.; Hayashi, K.; Kohno, M. Tumor necrosis factor (TNF) stimulates the production of nerve growth factor in fibroblasts via the 55-kDa type 1 TNF receptor. FEBS Lett. 1996, 379, 157–160. [Google Scholar] [CrossRef]
- Dopp, J.M.; Mackenzie-Graham, A.; Otero, G.C.; Merrill, J.E. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J. Neuroimmunol. 1997, 75, 104–112. [Google Scholar] [CrossRef]
- Etemadi, N.; Holien, J.K.; Chau, D.; Dewson, G.; Murphy, J.M.; Alexander, W.S.; Parker, M.W.; Silke, J.; Nachbur, U. Lymphotoxin α induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J. 2013, 280, 5283–5297. [Google Scholar] [CrossRef] [PubMed]
- Hawari, F.I.; Rouhani, F.N.; Cui, X.; Yu, Z.-X.; Buckley, C.; Kaler, M.; Levine, S.J. Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism for generation of soluble cytokine receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Diez-Ruiz, A.; Tilz, G.P.; Zangerle, R.; Baier-Bitterlich, G.; Wachter, H.; Fuchs, D. Soluble receptors for tumour necrosis factor in clinical laboratory diagnosis. Eur. J. Haematol. 1995, 54, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; van der Poll, T.; Levi, M.; ten Cate, H.; Gallati, H.; ten Cate, J.W.; van Deventer, S.J. Inhibition of the release of soluble tumor necrosis factor receptors in experimental endotoxemia by an anti-tumor necrosis factor-alpha antibody. J. Clin. Immunol. 1995, 15, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Eissner, G.; Kolch, W.; Scheurich, P. Ligands working as receptors: Reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004, 15, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Sipos, O.; Török, A.; Kalic, T.; Duda, E.; Filkor, K. Reverse Signaling Contributes to Control of Chronic Inflammation by Anti-TNF Therapeutics. Antibodies 2015, 4, 123–140. [Google Scholar] [CrossRef]
- Waetzig, G.H.; Rosenstiel, P.; Arlt, A.; Till, A.; Bräutigam, K.; Schäfer, H.; Rose-John, S.; Seegert, D.; Schreiber, S. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J. 2005, 19, 91–93. [Google Scholar] [PubMed]
- Kisiswa, L.; Osório, C.; Erice, C.; Vizard, T.; Wyatt, S.; Davies, A.M. TNFα reverse signaling promotes sympathetic axon growth and target innervation. Nat. Neurosci. 2013, 16, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Meusch, U.; Rossol, M.; Baerwald, C.; Hauschildt, S.; Wagner, U. Outside-to-inside signaling through transmembrane tumor necrosis factor reverses pathologic interleukin-1beta production and deficient apoptosis of rheumatoid arthritis monocytes. Arthritis Rheum. 2009, 60, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Holler, E.; Haffner, S.; Andreesen, R.; Eissner, G. Effect of different tumor necrosis factor (TNF) reactive agents on reverse signaling of membrane integrated TNF in monocytes. Cytokine 2004, 28, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hawari, F.; Alsaaty, S.; Lawrence, M.; Combs, C.A.; Geng, W.; Rouhani, F.N.; Miskinis, D.; Levine, S.J. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J. Clin. Invest. 2002, 110, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Adamik, B.; Hawari, F.I.; Ma, G.; Rouhani, F.N.; Zhang, J.; Levine, S.J. Extracellular TNFR1 release requires the calcium-dependent formation of a nucleobindin 2-ARTS-1 complex. J. Biol. Chem. 2006, 281, 6860–6873. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Rouhani, F.N.; Hawari, F.; Levine, S.J. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J. Immunol. 2003, 171, 6814–6819. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, Y.; Mei, K.; Zhang, S.; Sun, X.; Ren, F.; Liu, S.; Yang, Z.; Wang, X.; Qin, Z.; et al. Tumor necrosis factor receptor 2 (TNFR2)·interleukin-17 receptor D (IL-17RD) heteromerization reveals a novel mechanism for NF-κB activation. J. Biol. Chem. 2015, 290, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Mease, P. Psoriatic arthritis: The role of TNF inhibition and the effect of its inhibition with etanercept. Clin. Exp. Rheumatol. 2002, 20, S116–121. [Google Scholar] [PubMed]
- Liu, Y.; Yang, G.; Zhang, J.; Xing, K.; Dai, L.; Cheng, L.; Liu, J.; Deng, J.; Shi, G.; Li, C.; et al. Anti-TNF-α monoclonal antibody reverses psoriasis through dual inhibition of inflammation and angiogenesis. Int. Immunopharmacol. 2015, 28, 731–743. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.E.; Lee, J.-K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.R.; Di Franco, M.; Metere, A.; Conti, F.; Iannuccelli, C.; Agati, L.; Valesini, G. Decrease of asymmetric dimethyl arginine after anti-TNF therapy in patients with rheumatoid arthritis. Drug Dev. Res. 2014, 75 (Suppl. 1), S67–S69. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, C.; Carvalho, D.; Russo, P.; Saiote, J.; Ramos, J. Anti-TNF alpha therapy in Inflammatory Bowel Disease - safety profile in elderly patients. J. Crohns Colitis 2015, 9 (Suppl. 1), S400. [Google Scholar]
- Maini, R.N.; Taylor, P.C. Anti-cytokine therapy for rheumatoid arthritis. Annu. Rev. Med. 2000, 51, 207–229. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Sambamurti, K.; Greig, N.H. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: New drug candidates and targets. Curr. Alzheimer Res. 2007, 4, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Mattson, M.P.; Perry, T.; Chan, S.L.; Giordano, T.; Sambamurti, K.; Rogers, J.T.; Ovadia, H.; Lahiri, D.K. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann. NY Acad. Sci. 2004, 1035, 290–315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Giordano, T.; Yu, Q.-S.; Holloway, H.W.; Perry, T.A.; Lahiri, D.K.; Brossi, A.; Greig, N.H. Thiothalidomides: Novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J. Med. Chem. 2003, 46, 5222–5229. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P. The first decade of biologic TNF antagonists in clinical practice: Lessons learned, unresolved issues and future directions. Curr. Dir. Autoimmun. 2010, 11, 180–210. [Google Scholar] [PubMed]
- Hyrich, K.L.; Lunt, M.; Watson, K.D.; Symmons, D.P.M.; Silman, A.J. British Society for Rheumatology Biologics Register Outcomes after switching from one anti-tumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum. 2007, 56, 13–20. [Google Scholar] [PubMed]
- Atzeni, F.; Gianturco, L.; Talotta, R.; Varisco, V.; Ditto, M.C.; Turiel, M.; Sarzi-Puttini, P. Investigating the potential side effects of anti-TNF therapy for rheumatoid arthritis: Cause for concern? Immunotherapy 2015, 7, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Beigel, F.; Steinborn, A.; Schnitzler, F.; Tillack, C.; Breiteneicher, S.; John, J.M.; Van Steen, K.; Laubender, R.P.; Göke, B.; Seiderer, J.; et al. Risk of malignancies in patients with inflammatory bowel disease treated with thiopurines or anti-TNF alpha antibodies. Pharmacoepidemiol. Drug Saf. 2014, 23, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Richez, C.; Blanco, P.; Lagueny, A.; Schaeverbeke, T.; Dehais, J. Neuropathy resembling CIDP in patients receiving tumor necrosis factor-alpha blockers. Neurology 2005, 64, 1468–1470. [Google Scholar] [CrossRef] [PubMed]
- Guiddir, T.; Frémond, M.-L.; Triki, T.B.; Candon, S.; Croisille, L.; Leblanc, T.; de Pontual, L. Anti-TNF-α therapy may cause neonatal neutropenia. Pediatrics 2014, 134, e1189–e1193. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; Osvath, S.R.; Xu, X.-M.; Li, G.; Chan, F.K.-M.; Barrett, J.W.; McFadden, G. Poxvirus tumor necrosis factor receptor (TNFR)-like T2 proteins contain a conserved preligand assembly domain that inhibits cellular TNFR1-induced cell death. J. Virol. 2006, 80, 9300–9309. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF receptor one-specific antibody (ATROSAB): receptor binding and in vitro bioactivity. PLoS ONE 2013, 8, e72156. [Google Scholar] [CrossRef] [PubMed]
- Zettlitz, K.A.; Lorenz, V.; Landauer, K.; Münkel, S.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. mAbs 2010, 2, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Novrup, H.G.; Bracchi-Ricard, V.; Ellman, D.G.; Ricard, J.; Jain, A.; Runko, E.; Lyck, L.; Yli-Karjanmaa, M.; Szymkowski, D.E.; Pearse, D.D.; et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J. Neuroinflamm. 2014, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Park. Dis. 2014, 4, 349–360. [Google Scholar]
- Deng, M.; Loughran, P.A.; Zhang, L.; Scott, M.J.; Billiar, T.R. Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci. Signal. 2015, 8, ra11. [Google Scholar] [CrossRef] [PubMed]
- Nübel, T.; Schmitt, S.; Kaina, B.; Fritz, G. Lovastatin stimulates p75 TNF receptor (TNFR2) expression in primary human endothelial cells. Int. J. Mol. Med. 2005, 16, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.M.; Eisel, U.L.M. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alzheimers Dis. 2008, 13, 111–122. [Google Scholar] [PubMed]
- Dolga, A.M.; Granic, I.; Nijholt, I.M.; Nyakas, C.; van der Zee, E.A.; Luiten, P.G.M.; Eisel, U.L.M. Pretreatment with lovastatin prevents N-methyl-D-aspartate-induced neurodegeneration in the magnocellular nucleus basalis and behavioral dysfunction. J. Alzheimers Dis. 2009, 17, 327–336. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Dekens, D.W.; De Deyn, P.P.; Naudé, P.J.W.; Eisel, U.L.M. Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders. Antibodies 2015, 4, 369-408. https://doi.org/10.3390/antib4040369
Dong Y, Dekens DW, De Deyn PP, Naudé PJW, Eisel ULM. Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders. Antibodies. 2015; 4(4):369-408. https://doi.org/10.3390/antib4040369
Chicago/Turabian StyleDong, Yun, Doortje W. Dekens, Peter Paul De Deyn, Petrus J. W. Naudé, and Ulrich L. M. Eisel. 2015. "Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders" Antibodies 4, no. 4: 369-408. https://doi.org/10.3390/antib4040369
APA StyleDong, Y., Dekens, D. W., De Deyn, P. P., Naudé, P. J. W., & Eisel, U. L. M. (2015). Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders. Antibodies, 4(4), 369-408. https://doi.org/10.3390/antib4040369