
Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies® †

Abstract
:1. Introduction
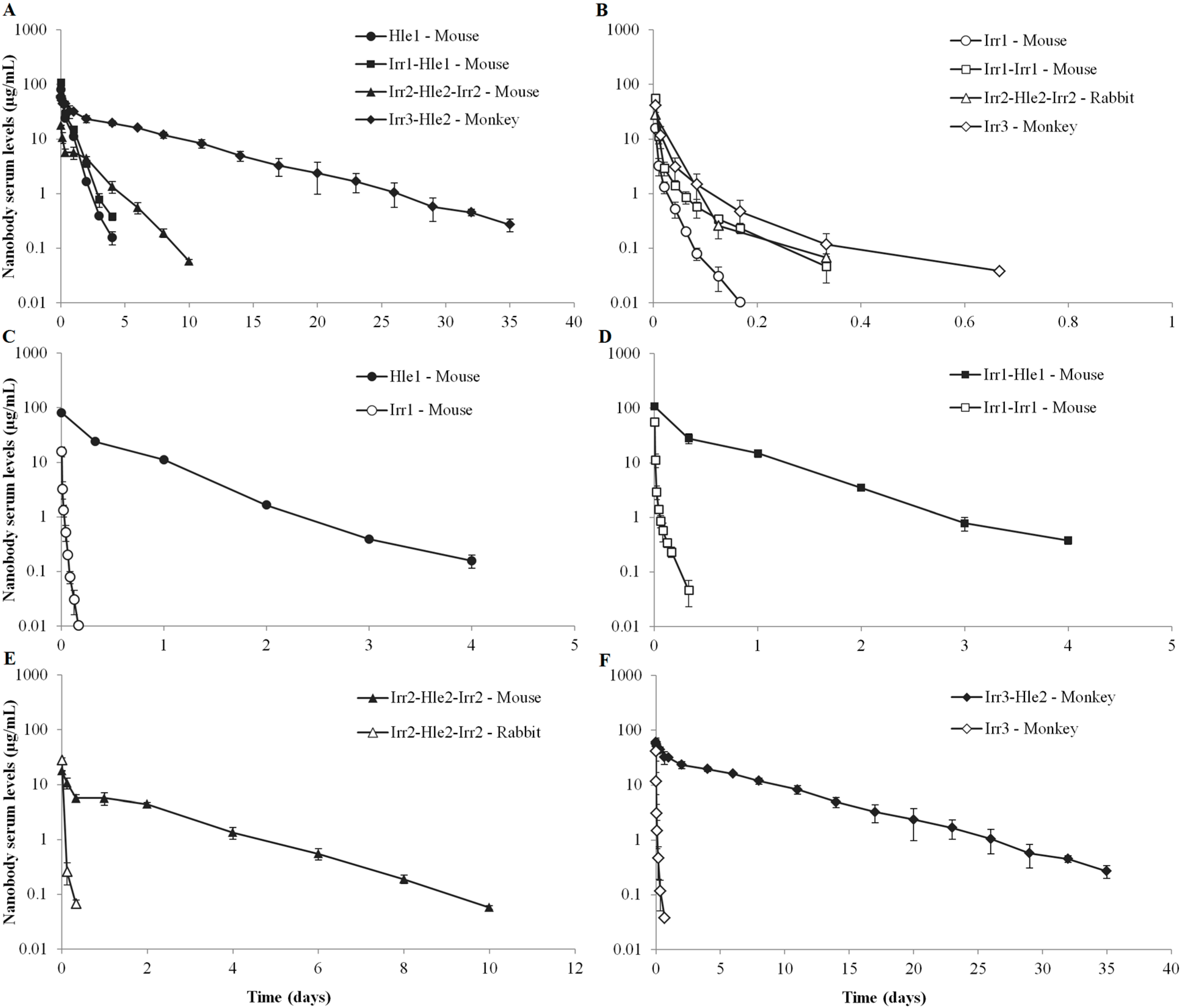
2. Results

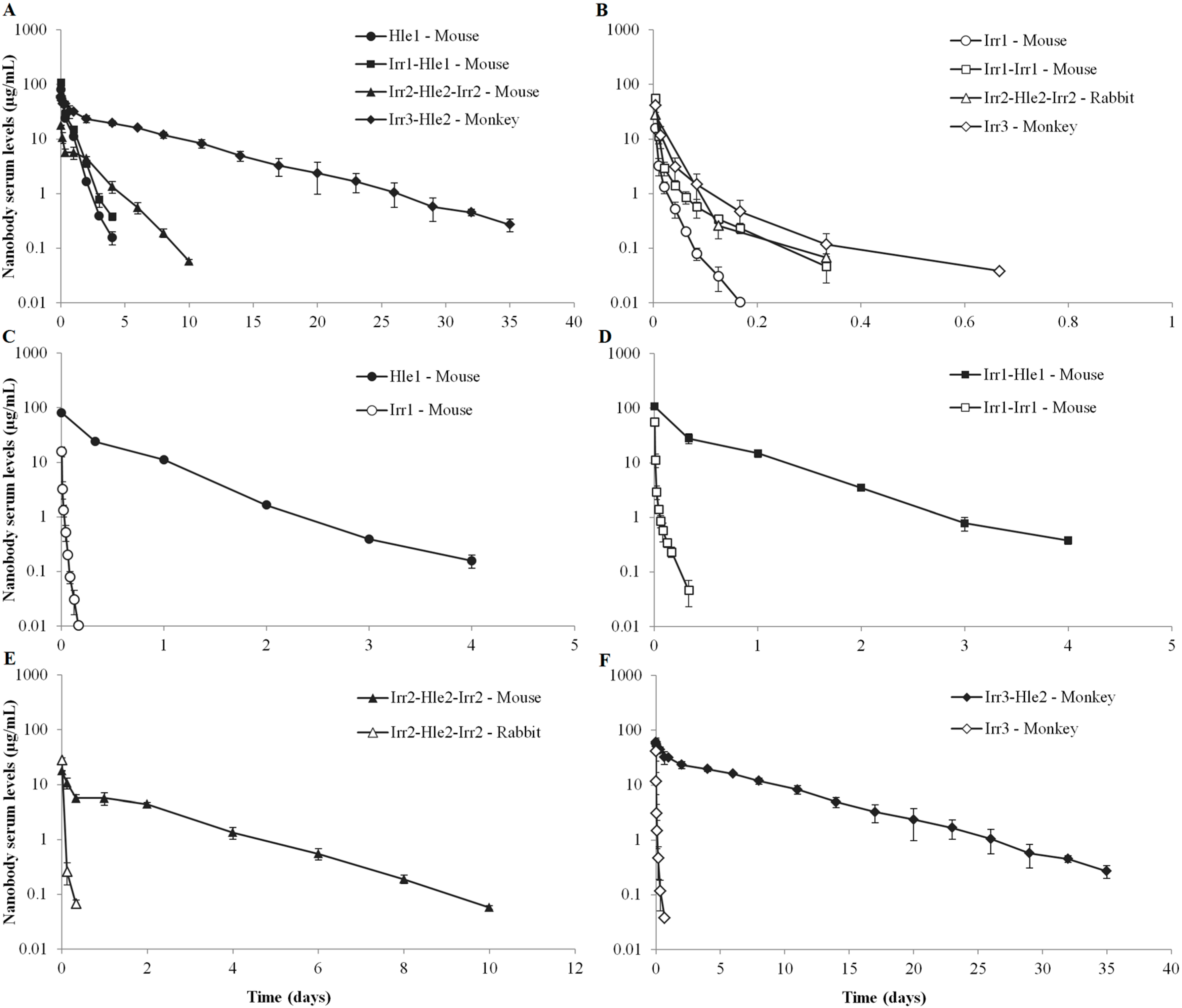{kind=link}
| Experi-ment | Nanobody | MW (kDa) | Mono-di-trimeric | pI | Dose (i.v.) | Species | Average body weight (kg) | KD for species albumin | Assay LLOQ (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Hle1 | 16 | Mono- | 6.4 | 100 µg | Mouse | 0.019 | 73 nM | 52 |
| 2 | Irr1 | 16 | Mono- | 5.5 | 100 µg | Mouse | 0.019 | - | 5 |
| 3 | Irr1-Hle1 | 31 | Di- | 6.1 | 100 µg | Mouse | 0.019 | 470 nM | 40 |
| 4 | Irr1-Irr1 | 31 | Di- | 5.4 | 100 µg | Mouse | 0.019 | - | 15 |
| 5 | Irr2-Hle2-Irr2 | 41 | Tri- | 9.0 | 1.2 mg/kg | Mouse | 0.019 | 180 nM | 40 |
| 6 | Irr2-Hle2-Irr2 | 41 | Tri- | 9.0 | 2.5 mg/kg | Rabbit | 1.99 | No binding | 40 |
| 7 | Irr3-Hle2 | 26 | Di- | 7.1 | 2 mg/kg | Monkey | 2.37 | NDa | 1.2 |
| 8 | Irr3 | 13 | Mono- | 5.8 | 2 mg/kg | Monkey | 2.40 | - | 8 |
| Hle1 Mouse | Irr1 Mouse | Irr1-Hle1 Mouse | Irr1-Irr1 Mouse | Irr2-Hle2-Irr2 Mouse | Irr2-Hle2-Irr2 Rabbit b | Irr3-Hle2 Monkey c | Irr3 Monkey c | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Units a | Estimate | RSE% | Estimate | RSE% | Estimate | RSE% | Estimate | RSE% | Estimate | RSE% | Estimate | RSE% | Mean | %CV | Mean | %CV |
| Vc | mL/kg | 64.0 | 36 | 171 | 40 | 47.0 | 59 | 49.1 | 27 | 64.7 | 21 | - | - | 34.4 | 11 | 41.4 | 38 |
| Vp | mL/kg | 48.3 | 172 | 192 | 32 | 54.9 | 413 | 152 | 27 | 54.5 | 32 | - | - | 17.9 | 43 | 92.0 | 19 |
| CL | mL/(day*kg) | 176 | 36 | 29425 | 18 | 135 | 103 | 8102 | 14 | 60.4 | 8 | 2907 | - | 7.61 | 8 | 2857 | 42 |
| mL/(min*kg) d | 0.122 | 36 | 20.4 | 18 | 0.094 | 103 | 5.63 | 14 | 0.042 | 8 | 2.02 | - | 0.005 | 8 | 1.98 | 42 | |
| Q | mL/(day*kg) | 312 | 553 | 8028 | 41 | 427 | 1561 | 2215 | 31 | 493 | 107 | - | - | 50 | 46 | 1331 | 37 |
| mL/(min*kg) d | 0.217 | 553 | 5.58 | 41 | 0.296 | 1561 | 1.54 | 31 | 0.342 | 107 | - | - | 0.034 | 46 | 0.924 | 37 | |
| AUC | day*µg/mL | 29.8 | 36 | 0.179 | 18 | 38.9 | 103 | 0.650 | 14 | 19.9 | 8 | 0.860 | - | 264 | 8 | 0.793 | 43 |
| t1/2,beta | day | 0.494 | 11 | 0.022 | 8 | 0.575 | 10 | 0.062 | 8 | 1.40 | 5 | 0.107 | - | 4.89 | 15 | 0.080 | 29 |
| Vss | mL/kg | 112 | 69 | 363 | 33 | 102 | 199 | 201 | 26 | 119 | 10 | 107 | - | 52.3 | 9 | 133 | 25 |
3. Discussion
3.1. Serum Pharmacokinetics of Non-Half-Life Extended Nanobodies
3.2. Serum Pharmacokinetics of Half-Life Extended Nanobodies
3.3. Conclusion
4. Experimental Section
4.1. Test Item Nanobodies
4.2. Animal Studies
4.3. Determination of Nanobody Serum Concentrations
4.4. Pharmacokinetic Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Ann. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Van Bockstaele, F.; Holz, J.-B.; Revets, H. The development of nanobodies for therapeutic applications. Curr. Opin. Investig. Drugs 2009, 10, 1212–1224. [Google Scholar] [PubMed]
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; De Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Rennke, H.G. The structural and molecular basis of glomerular filtration. Circul. Res. 1978, 43, 337–347. [Google Scholar] [CrossRef]
- Lin, J.H. Pharmacokinetics of biotech drugs: peptides, proteins and monoclonal antibodies. Curr. Drug Metab. 2009, 10, 661–691. [Google Scholar] [CrossRef] [PubMed]
- Rennke, H.G.; Patel, Y.; Venkatachalam, M.A. Glomerular filtration of proteins: Clearance of anionic, neutral, and cationic horseradish peroxidase in the rat. Kidney Int. 1978, 13, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Peters, T., Jr. Serum albumin. Adv. Protein Chem. 1985, 37, 161–245. [Google Scholar] [PubMed]
- Tojo, A.; Endou, H. Intrarenal handling of proteins in rats using fractional micropuncture technique. Am. J. Physiol. 1992, 263, F601–606. [Google Scholar] [PubMed]
- Tojo, A.; Kinugasa, S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int. J. Nephrol. 2012, 2012. Article ID 481520. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, M.; Sorensson, J.; Haraldsson, B. Glomerular size and charge selectivity in the rat as revealed by FITC-ficoll and albumin. Am. J. Physiol. 2000, 279, F84–F91. [Google Scholar]
- Kuwabara, T.; Ishikawa, Y.; Kobayashi, H.; Kobayashi, S.; Sugiyama, Y. Renal clearance of a recombinant granulocyte colony-stimulating factor, nartograstim, in rats. Pharm. Res. 1995, 12, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.B.; Lum, P.K.; Hayashi, M.M.; Roskos, L.K. Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J. Pharm. Sci. 2004, 93, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.T.; Baker, K.; Yoshida, M.; Qiao, S.W.; Aveson, V.G.; Lencer, W.I.; Blumberg, R.S. Neonatal Fc receptor: From immunity to therapeutics. J. Clin. Immunol. 2010, 30, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Sandlie, I. The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab. Pharmacok. 2009, 24, 318–332. [Google Scholar] [CrossRef]
- Sand, K.M.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front. Immunol. 2014, 5, 682. [Google Scholar] [CrossRef] [PubMed]
- Berson, S.A.; Yalow, R.S.; Schreiber, S.S.; Post, J. Tracer experiments with I131 labeled human serum albumin: distribution and degradation studies. J. Clin. Invest. 1953, 32, 746–768. [Google Scholar] [CrossRef] [PubMed]
- Morell, A.; Terry, W.D.; Waldmann, T.A. Metabolic properties of IgG subclasses in man. J. Clin. Invest. 1970, 49, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.; Kanakaraj, P.; Osborn, B.L.; Schneider, H.; Pickeral, O.K.; Chen, C.; Zhang, G.; Kaithamana, S.; Singh, M.; Schulingkamp, R.; et al. Development of a long-acting insulin analog using albumin fusion technology. Diabetes 2005, 54, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, J.A.; Carroll, G.J. Fc-based cytokines: Prospects for engineering superior therapeutics. BioDrugs 2008, 22, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bailon, P.; Won, C.Y. PEG-modified biopharmaceuticals. Exp. Opin. Drug Deliv. 2009, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Pehrson, R.; Tolmachev, V.; Daba, M.B.; Abrahmsen, L.; Ekblad, C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J. Biol. Chem. 2011, 286, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Saunders, K.; Grace, C.; Jin, M.; Piche-Nicholas, N.; Steven, J.; O'Dwyer, R.; Wu, L.; Khetemenee, L.; Vugmeyster, Y.; et al. Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. mAbs 2012, 4, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Tijink, B.M.; Laeremans, T.; Budde, M.; Stigter-van Walsum, M.; Dreier, T.; de Haard, H.J.; Leemans, C.R.; van Dongen, G.A. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: Taking advantage of modular Nanobody technology. Mol. Cancer Therap. 2008, 7, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar] [PubMed]
- Ulrichts, H.; Silence, K.; Schoolmeester, A.; de Jaegere, P.; Rossenu, S.; Roodt, J.; Priem, S.; Lauwereys, M.; Casteels, P.; Van Bockstaele, F.; et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011, 118, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Vosjan, M.J.; Perk, L.R.; Roovers, R.C.; Visser, G.W.; Stigter-van Walsum, M.; van Bergen En Henegouwen, P.M.; van Dongen, G.A. Facile labelling of an anti-epidermal growth factor receptor Nanobody with 68Ga via a novel bifunctional desferal chelate for immuno-PET. Eur. J. Nucl. Med. Mol. Imag. 2011, 38, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Gainkam, L.O.; Huang, L.; Caveliers, V.; Keyaerts, M.; Hernot, S.; Vaneycken, I.; Vanhove, C.; Revets, H.; De Baetselier, P.; Lahoutte, T. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J. Nucl. Med. 2008, 49, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Lote, C. Principles of Renal Physiology, 5th ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Lenting, P.J.; Westein, E.; Terraube, V.; Ribba, A.S.; Huizinga, E.G.; Meyer, D.; de Groot, P.G.; Denis, C.V. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J. Biol. Chem. 2004, 279, 12102–12109. [Google Scholar] [CrossRef] [PubMed]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn reclaims albumin but facilitates elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bronson, C.L.; Hayton, W.L.; Radmacher, M.D.; Roopenian, D.C.; Robinson, J.M.; Anderson, C.L. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am. J. Physiol. 2006, 290, G352–G360. [Google Scholar] [CrossRef] [PubMed]
- Borvak, J.; Richardson, J.; Medesan, C.; Antohe, F.; Radu, C.; Simionescu, M.; Ghetie, V.; Ward, E.S. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int. Immunol. 1998, 10, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Reyes, A.E., 2nd; Zhang, M.; McDonald, P.; Wong, W.L.; Damico, L.A.; Dennis, M.S. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng. Des. Sel. 2006, 19, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Iyer, S.; Theil, F.P.; Mortensen, D.L.; Fielder, P.J.; Prabhu, S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: What have we learned? mAbs 2011, 3, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Petkova, S.B.; Akilesh, S.; Sproule, T.J.; Christianson, G.J.; Al Khabbaz, H.; Brown, A.C.; Presta, L.G.; Meng, Y.G.; Roopenian, D.C. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int. Immunol. 2006, 18, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Vugmeyster, Y.; Szklut, P.; Tchistiakova, L.; Abraham, W.; Kasaian, M.; Xu, X. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of humanized monoclonal anti-IL-13 antibodies with different IL-13 neutralization mechanisms. Int. Immunopharm. 2008, 8, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Nguyen, C.; Mendoza, J.L.; Escandon, E.; Fei, D.; Meng, Y.G.; Modi, N.B. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J. Pharm. Exp. Therap. 1999, 288, 371–378. [Google Scholar]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Holz, J.B. The TITAN trial--assessing the efficacy and safety of an anti-von Willebrand factor Nanobody in patients with acquired thrombotic thrombocytopenic purpura. Transf. Apheresis Sci. 2012, 46, 343–346. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, S.; De Smedt, T.; Allosery, K.; Crabbe, P.; De Brabandere, V.; Detalle, L.; Mortier, K.; Schoolmeester, A.; Wouters, A.; Stöhr, T.; et al. ALX-0171: Safety and Therapeutic Potential of an Inhaled Anti-RSV Nanobody. Respirat. Drug Deliv. Eur. 2015, in press. [Google Scholar]
- Holz, J.B.; Sargentini-Maier, L.; De Bruyn, S.; Gachályi, B.; Udvaros, I.; Rojkovich, B.; Bruk, S.; Sramek, P.; Korkosz, M.; Krause, K.; et al. Twenty-four weeks of treatment with a novel anti-IL6 receptor nanobody (ALX-0061) resulted in 84% ACR20 improvement and 58% DAS28 remission in a phase I/II study in RA. Ann. Rheumat. Dis. 2013, 72, 64. [Google Scholar] [CrossRef]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoefman, S.; Ottevaere, I.; Baumeister, J.; Sargentini-Maier, M.L. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies 2015, 4, 141-156. https://doi.org/10.3390/antib4030141
Hoefman S, Ottevaere I, Baumeister J, Sargentini-Maier ML. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies. 2015; 4(3):141-156. https://doi.org/10.3390/antib4030141
Chicago/Turabian StyleHoefman, Sven, Ingrid Ottevaere, Judith Baumeister, and Maria Laura Sargentini-Maier. 2015. "Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®" Antibodies 4, no. 3: 141-156. https://doi.org/10.3390/antib4030141
APA StyleHoefman, S., Ottevaere, I., Baumeister, J., & Sargentini-Maier, M. L. (2015). Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies, 4(3), 141-156. https://doi.org/10.3390/antib4030141