
Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy

Abstract
:1. Introduction
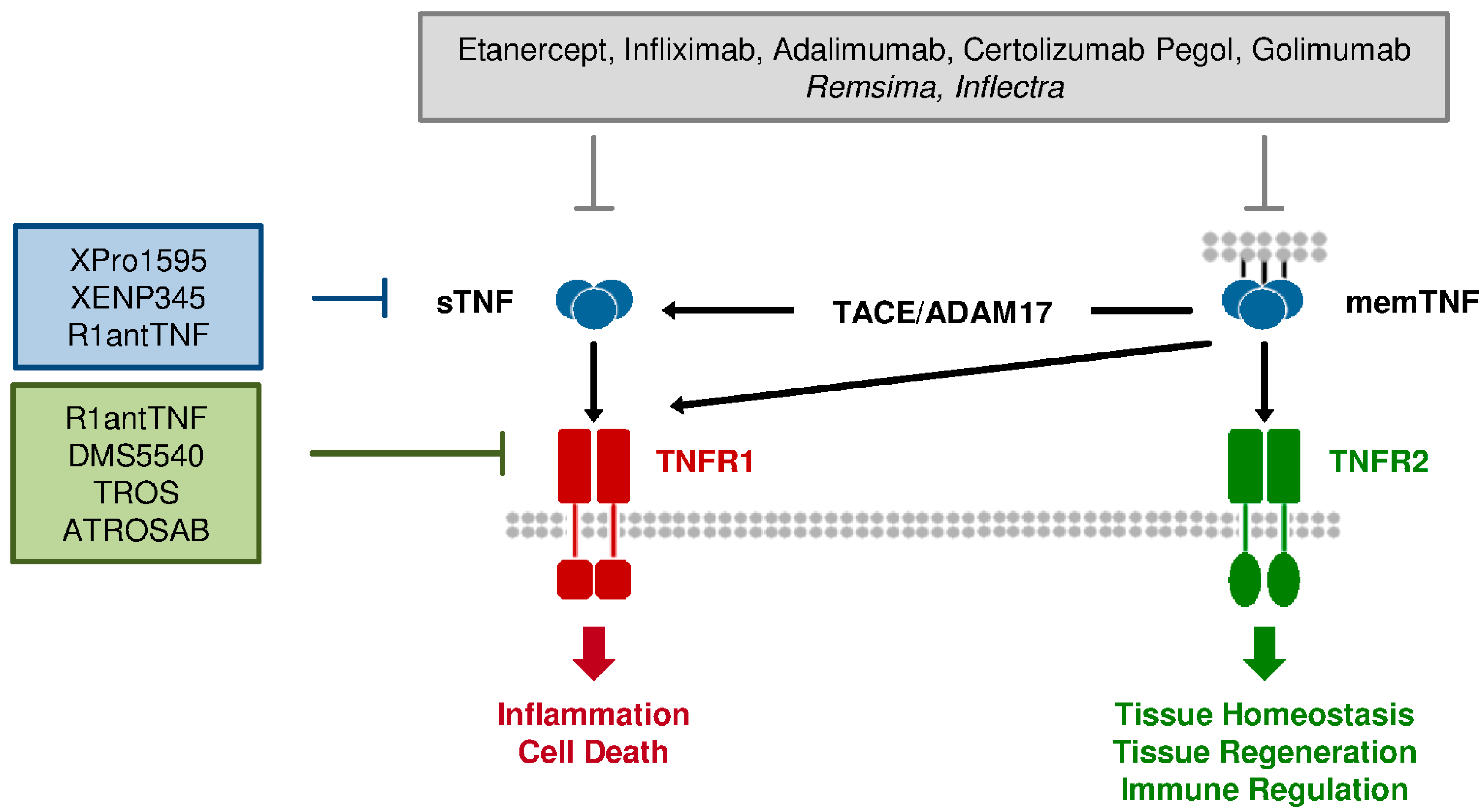
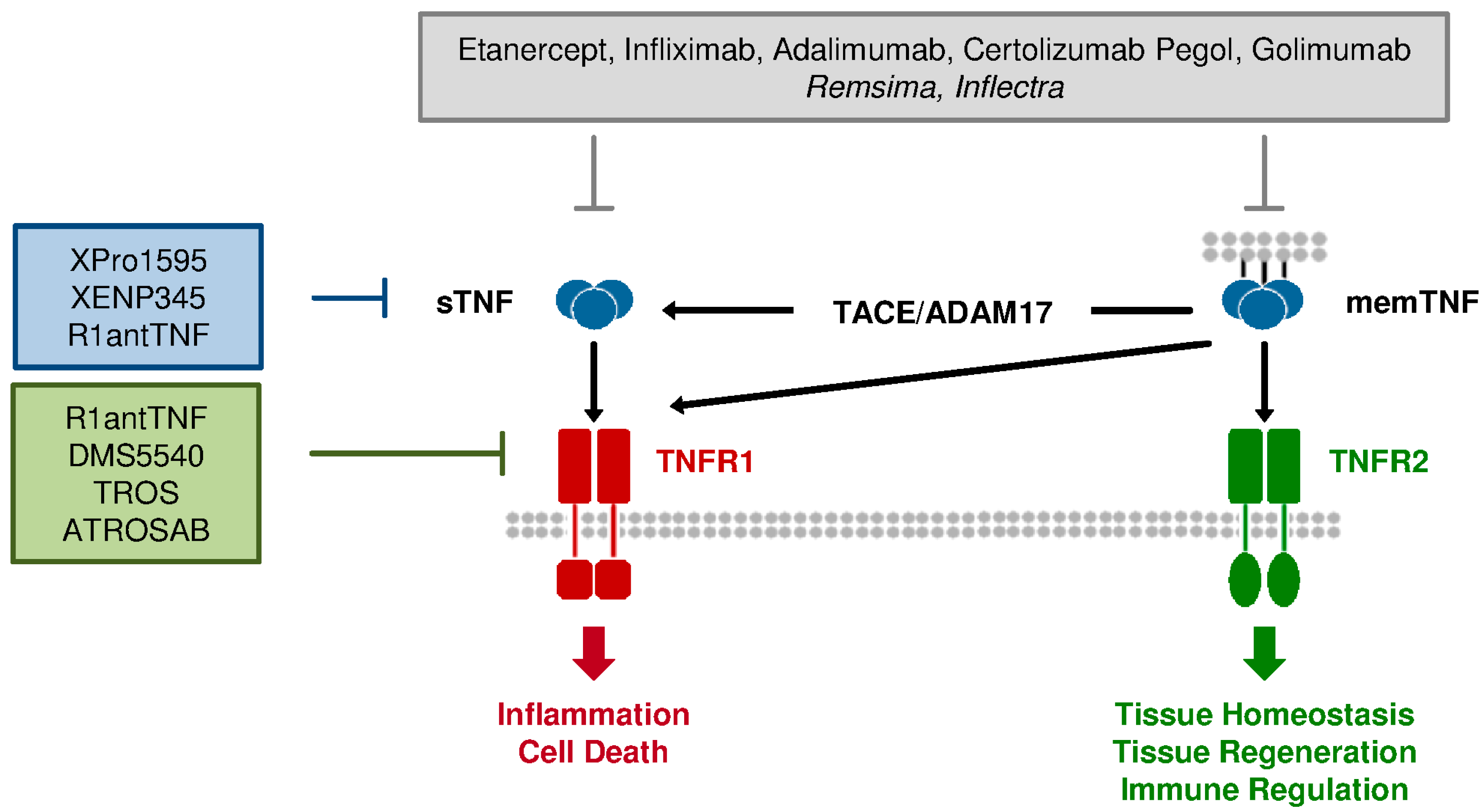
2. TNF Signaling
2.1. General Aspects
2.2. TNF Receptors
2.3. TNF Signaling in Immune Regulation
2.4. Pathophysiology of TNF Deregulation
3. TNF Inhibition in Disease
3.1. Current Anti-TNF Therapeutics: Application and Limitation

{kind=link}
| Approved anti-TNF therapeutics | ||
|---|---|---|
| Drug name (brand name) | Structure | Approved disease indications |
| Etanercept (Enbrel) | Human TNFR2exc:IgG1-Fc | RA, JIA, PA, PP, AS |
| Infliximab (Remicade) | humanized chimeric IgG/k mAb | RA, PA, PP, AS, CD, UC |
| Adalimumab (Humira) | fully human IgG1/k mAb | RA, PA, PP, AS, CD, JIA |
| Certolizumab Pegol (Cimzia) | PEGylated Fab’ fragment | CD, RA, AS, PA |
| Golimumab (Simponi) | fully human IgG1/k mAb | RA, PA, AS, UC |
| Approved anti-TNF biosimilars | ||
| Drug name | Biosimilar of | Disease indication |
| Remsima | Infliximab | RA, PA, PP, AS, CD, UC |
| Inflectra | Infliximab | RA, PA, PP, AS, CD, UC |
| Current sTNF/TNFR1 targeting agents under development | ||
| Name | Structure | Mechanism |
| XPro1595 | Dominant-negative TNF mutein | sTNF inhibitor |
| XENP345 | Dominant-negative TNF mutein | sTNF inhibitor |
| R1antTNF | TNFR1-selective antagonistic mutant TNF | TNFR1 antagonist (sTNF inhibitor?) |
| DMS5540 | TNFR1-selective monovalent domain antibody fused to albumin specific domain antibody | TNFR1 antagonist |
| TROS | Two TNFR1-selective single domain nanobodies fused to anti-albumin nanobody | TNFR1 antagonist |
| ATROSAB | Humanized TNFR1-selective IgG1 | TNFR1 antagonist |
3.2. Differences between TNFR1 & TNFR2: Requirement for Selective Therapeutics
4. Novel sTNF and TNFR1-Specific Antagonists
4.1. sTNF-Selective Dominant-Negative TNF Derivatives
4.2. TNFR1-Selective Antagonistic TNF
4.3. TNFR1-Specific Antibodies
4.4. Comparison of sTNF Inhibitors and TNFR1 Antagonists
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Apostolaki, M.; Armaka, M.; Victoratos, P.; Kollias, G. Cellular mechanisms of TNF function in models of inflammation and autoimmunity. Curr. Dir. Autoimmun. 2010, 11, 1–26. [Google Scholar] [PubMed]
- Kollias, G. TNF pathophysiology in murine models of chronic inflammation and autoimmunity. Semin. Arthritis Rheum. 2005, 34, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Diff. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Wong, M.; Ziring, D.; Korin, Y.; Desai, S.; Kim, S.; Lin, J.; Gjertson, D.; Braun, J.; Reed, E.; Singh, R.R. TNFα blockade in human diseases: Mechanisms and future directions. Clin. Immunol. 2008, 126, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Scheurich, P.; Pfizenmaier, K. Antagonists of TNF action: Clinical experience and new developments. Exp. Opin. Drug Discov. 2009, 4, 279–292. [Google Scholar] [CrossRef]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 177, 244–279. [Google Scholar] [CrossRef]
- Cessak, G.; Kuzawińska, O.; Burda, A.; Lis, K.; Wojnar, M.; Mirowska-Guzel, D.; Bałkowiec-Iskra, E. TNF inhibitors—Mechanisms of action, approved and off-label indications. Pharmacol. Rep. 2014, 66, 836–844. [Google Scholar] [CrossRef] [PubMed]
- van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.; Woody, J.N.; Hartung, H.P.; Polman, C.H. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- The Lenercept Study Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999, 53, 457–465. [Google Scholar]
- Slifman, N.R.; Gershon, S.K.; Lee, J.H.; Edwards, E.T.; Braun, M.M. Listeria monocytogenes infection as a complication of treatment with tumor necrosis actor alpha-neutralizing agents. Arthritis Rheum. 2003, 48, 319–324. [Google Scholar]
- Sicotte, N.L.; Voskuhl, R.R. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology 2001, 57, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Shakoor, N.; Michalska, M.; Harris, C.A.; Block, J.A. Drug-induced systemic lupus erythematosus associated with etanercept therapy. Lancet 2002, 359, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.L.; Greene, M.H.; Gershon, S.K.; Edwards, E.T.; Braun, M.M. Tumor necrosis factor antagonist therapy and lymphoma development: Twenty-six cases reported to the Food and Drug Administration. Arthritis Rheum. 2002, 46, 3151–3158. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Engelmann, H.; Nophar, Y.; Aderka, D.; Kemper, O.; Hornik, V.; Holtmann, H.; Brakebusch, C. Soluble and cell surface receptors for tumor necrosis factor. Agents Actions Suppl. 1991, 35, 51–57. [Google Scholar] [PubMed]
- Solomon, K.A.; Pesti, N.; Wu, G.; Newton, R.C. Cutting edge: A dominant negative form of TNF-alpha converting enzyme inhibits proTNF and TNFRII secretion. J. Immunol. 1999, 163, 4105–4108. [Google Scholar] [PubMed]
- Aderka, D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996, 7, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Szymkowski, D.E. The TNF superfamily in 2009: New pathways, new indications, and new drugs. Drug Discov. Today 2009, 14, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Pfizenmaier, K.; Szymkowski, D.E. Workshop Summary: Introduction to rational design of new means for therapeutic modulation of function of the TNF family. Adv. Exp. Med. Biol. 2011, 691, 487–491. [Google Scholar] [PubMed]
- van Hauwermeiren, F.; Vandenbroucke, R.E.; Libert, C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011, 22, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Scheurich, P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011, 278, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Baeuerle, P.A.; Kaltschmidt, C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol. Aspects Med. 1993, 14, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Firestein, G.S. c-Jun N-Terminal Kinase in inflammation and rheumatic diseases. Open Rheumatol. J. 2012, 6, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.S.; Rhee, M.H.; Sung, G.H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. 2014, 352371. [Google Scholar]
- Naudé, P.J.; den Boer, J.A.; Luiten, P.G.; Eisel, U.L. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Naumer, M.; Krippner-Heidenreich, A.; Scheurich, P.; Pfizenmaier, K. Ligand-induced internalization of TNF receptor 2 mediated by a di-leucin motif is dispensable for activation of the NFκB pathway. Cell Signal. 2011, 23, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.; Eisel, U.L. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alz. Dis. 2008, 13, 111–122. [Google Scholar]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF Receptor 2 selective agonist rescues neurons from oxidative stress-induced cell death. PLoS One 2011, 6, e27621. [Google Scholar] [CrossRef]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, P.; Ritter, U.; Labbow, S.; Donhauser, N.; Rollinghoff, M.; Bogdan, C.; Korner, H. Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 2001, 166, 4012–4019. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNFα-deficient mice: A critical requirement for TNFα in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.E.; Peters, C.; Hahn, H. Cytokines in the induction and expression of T-cell-mediated granuloma formation and protection in the murine model of listeriosis. Immunol. Rev. 1997, 158, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Lesslauer, W.; Lötscher, H.; Lang, Y.; Koebel, P.; Köntgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Mackay, F.; Bluethmann, H.; Zinkernagel, R.; Lesslauer, W. Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Circ. Shock 1994, 44, 51–56. [Google Scholar] [PubMed]
- Xu, J.; Chakrabarti, A.K.; Tan, J.L.; Ge, L.; Gambotto, A.; Vujanovic, N.L. Essential role of the TNF–TNFR2 cognate interaction in mouse dendritic cell-natural killer cell crosstalk. Blood 2007, 109, 333–3341. [Google Scholar]
- Musicki, K.; Briscoe, H.; Tran, S.M.; Britton, W.J.; Saunders, B.M. Differential requirements for soluble and transmembrane tumor necrosis factor in the immunological control of primary and secondary Listeria monocytogenes infection. Infect. Immun. 2006, 74, 3180–3189. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Janot, L.; Quesniaux, V.F.; Grivennikov, S.I.; Maillet, I.; Sedgwick, J.D.; Ryffel, B.; Erard, F. Membrane tumor necrosis factor confers partial protection to Listeria infection. Am. J. Pathol. 2005, 167, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Baumel, M.; Männel, D.N.; Howard, O.M.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.; Männel, D.N.; Oppenheim, J.J. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+ CD25+ FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. TNF-alpha: An activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr. Dir. Autoimmun. 2010, 11, 119–134. [Google Scholar] [PubMed]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.; Netea, M.G.; Oppenheim, J.J. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T cell phenotype in the inflammatory environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kimberley, F.C.; Lobito, A.A.; Siegel, R.M.; Screaton, G.R. Falling into TRAPS—Receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res. Ther. 2007, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. Acute pancreatitis as a model of SIRS. Front. Biosci. 2009, 14, 2042–2050. [Google Scholar] [CrossRef]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Path. 2004, 202, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Schulz, R.; Heusch, G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail. Rev. 2011, 16, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Piguet, P.F.; Grau, G.E.; Vesin, C.; Loetscher, H.; Gentz, R.; Lesslauer, W. Evolution of collagen arthritis in mice is arrested by treatment with anti-tumour necrosis factor (TNF) antibody or a recombinant soluble TNF receptor. Immunology 1992, 77, 510–514. [Google Scholar] [PubMed]
- Mori, L.; Iselin, S.; De Libero, G.; Lesslauer, W. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1-treated and TNFR1-deficient mice. J. Immunol. 1996, 157, 3178–3182. [Google Scholar] [PubMed]
- Dickson, D.W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 1997, 56, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; Merrill, J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989, 170, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Bachmann, R.; Bluethmann, H.; Lassmann, H.; Fontana, A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 1999, 29, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Kollias, G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: Implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 2001, 193, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One 2014, 9, e90117. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Kranidioti, K.; Xanthoulea, S.; Denis, M.; Kotanidou, A.; Douni, E.; Blackshear, P.J.; Kontoyiannis, D.L.; Kollias, G. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol. 2006, 36, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Reichert, J.M. Approval of the first biosimilar antibodies in Europe: A major landmark for the biopharmaceutical industry. MAbs 2013, 5, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Thuerkauf, R.; Stevens, R.; Lesslauer, W. Immune response to a recombinant human TNFR55-IgG1 fusion protein: auto-antibodies in rheumatoid arthritis (RA) and multiple sclerosis (MS) patients have neither neutralizing nor agonist activities. Hum. Immunol. 1999, 60, 774–790. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Laterre, P.F.; Garbino, J.; Pingleton, S.; Butler, T.; Dugernier, T.; Margolis, B.; Kudsk, K.; Zimmerli, W.; Anderson, P.; et al. Lenercept (p55 tumor necrosis factor fusion protein) in severe sepsis and early septic shock: A randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit. Care Med. 2001, 29, 503–570. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Faustman, D. NOD mice are defective in proteasome production and activation of NF-kappaB. Mol. Cell. Biol. 1999, 19, 8646–8659. [Google Scholar] [PubMed]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell. Mol. Life Sci. 2005, 62, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N.; Kaminitz, A.; Yarkoni, S. Mechanisms of T regulatory cell function. Autoimmun. Rev. 2008, 7, 370–375. [Google Scholar] [CrossRef]
- Nagar, M.; Jacob-Hirsch, J.; Vernitsky, H.; Berkun, Y.; Ben-Horin, S.; Amariglio, N.; Bank, I.; Kloog, Y.; Rechavi, G.; Goldstein, I. TNF activates a NF-kappaB-regulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J. Immunol. 2010, 184, 3570–3581. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Lazzeri, E.; Manetti, R.; Vanini, V.; Romagnani, P.; Maggi, E.; Romagnani, S. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J. Exp. Med. 2002, 196, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Fischer, R.; Agresti, C.; Pfizenmaier, K. TNFR2 activation protects oligodendrocyte progenitor cells against oxidative stress, Bioch. Biophys. Res. Comm. 2013, 440, 336–341. [Google Scholar] [CrossRef]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Ruuls, S.R.; Hoek, R.M.; Ngo, V.N.; McNeil, T.; Lucian, L.A.; Janatpour, M.J.; Körner, H.; Scheerens, H.; Hessel, E.M.; Cyster, J.G.; et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity 2001, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Saunders, B.M.; Tran, S.; Ruuls, S.; Sedgwick, J.D.; Briscoe, H.; Britton, W.J. Transmembrane TNF is sufficient to initiate cell migration and granuloma formation and provide acute, but not long-term, control of Mycobacterium tuberculosis infection. J. Immunol. 2005, 174, 4852–4859. [Google Scholar] [CrossRef] [PubMed]
- Fremond, C.; Allie, N.; Dambuza, I.; Grivennikov, S.I.; Yeremeev, V.; Quesniaux, V.F.; Jacobs, M.; Ryffel, B. Membrane TNF confers protection to acute mycobacterial infection. Respir. Res. 2005, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Münkel, S.; Neumeyer, J.; Müller, D.; Branschädel, M.; Scheurich, P.; Pfizenmaier, K. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J. Immunother. 2008, 31, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Secher, T.; Ezhevsky, S.A.; Janot, L.; Steed, P.M.; O'Brien, C.; Eivazi, A.; Kung, J.; Nguyen, D.H.; Doberstein, S.K.; et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J. Immunol. 2007, 179, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Vesin, D.; Fotio, A.L.; Santiago-Raber, M.L.; Tauzin, S.; Szymkowski, D.E.; Garcia, I. Soluble TNF, but not membrane TNF, is critical in LPS-induced hepatitis. J. Hepatol. 2010, 53, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Martinez, T.N.; McAlpine, F.E.; Blesch, A.; Tansey, M.G. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in Hemiparkinsonian rats. Mol. Ther. 2008, 16, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-κB. Brain 2011, 134, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.J.; Popovich, P.G. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp. Neurol. 2008, 209, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, in press. [Google Scholar]
- Novrup, H.G.; Bracchi-Ricard, V.; Ellman, D.G.; Ricard, J.; Jain, A.; Runko, E.; Lyck, L.; Yli-Karjanmaa, M.; Szymkowski, D.E.; Pearse, D.D.; et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J. Neuroinflamm. 2014, 11, 159. [Google Scholar] [CrossRef]
- Clausen, B.; Degn, M.; Martin, N.; Couch, Y.; Karimi, L.; Ormhøj, M.; Mortensen, M.L.; Gredal, H.; Gardiner, C.; Sargent, I.I.; et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J. Neuroinflamm. 2014, 11, 203. [Google Scholar] [CrossRef]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Abe, Y.; Nomura, T.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; Mayumi, T.; et al. The therapeutic effect of TNFR1-selective antagonistic mutant TNF-alpha in murine hepatitis models. Cytokine 2008, 44, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Abe, Y.; Ohkawa, A.; Nomura, T.; Minowa, K.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 2009, 30, 6638–6647. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; Kawara, T.; Arita, S.; Furuya, T.; Yamashita, T.; et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J. Control. Release 2011, 149, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, M.; Isoda, K.; Kamada, H.; Kobayashi, T.; Tsunoda, S.; Tsutsumi, Y.; Niida, T.; Kujiraoka, T.; Ishigami, N.; Ishihara, M.; et al. Novel TNFα receptor 1 antagonist treatment attenuates arterial inflammation and intimal hyperplasia in mice. J. Atheroscler. Thromb. 2012, 19, 36–46. [Google Scholar] [CrossRef] [PubMed]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.; Stoop, A.A.; Williams, R.O. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Puimège, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and characterization of small single domain antibodies inhibiting human TNF receptor 1. J. Biol. Chem. 2014, 290, 4022–4037. [Google Scholar] [CrossRef] [PubMed]
- Thoma, B.; Grell, M.; Pfizenmaier, K.; Scheurich, P. Identification of a 60-kD tumor necrosis factor (TNF) receptor as the major signal transducing component in TNF responses. J. Exp. Med. 1990, 172, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Zettlitz, K.A.; Lorenz, V.; Landauer, K.H.; Münkel, S.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs 2010, 2, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF receptor one-specific antibody (ATROSAB): Receptor binding and in vitro bioactivity. PLoS One 2013, 8, e72156. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, K.C.; Pinckard, J.K.; Arthur, C.D.; Dehner, L.P.; Goeddel, D.V.; Schreiber, R.D. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: Identification of a novel in vivo role for p75. J. Exp. Med. 1995, 181, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [PubMed]
- Ottoboni, L.; Frohlich, I.Y.; Lee, M.; Healy, B.C.; Keenan, B.T.; Xia, Z.; Chitnis, T.; Guttmann, C.R.; Khoury, S.J.; Weiner, H.L.; et al. Clinical relevance and functional consequences of the TNFRSF1A multiple sclerosis locus. Neurology 2013, 81, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, N.H.; Steinman, L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int. Immunol. 1990, 2, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Suen, W.E.; Bergman, C.M.; Hjelmström, P.; Ruddle, N.H. A critical role of lymphotoxin in experimental allergic encephalomyelitis. J. Exp. Med. 1997, 186, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.Y.; Kolumam, G.A.; Yu, X.; Francesco, M.; Ivelja, S.; Peng, I.; Gribling, P.; Shu, J.; Lee, W.P.; Refino, C.J.; et al. Targeted depletion of lymphotoxin-a-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat. Med. 2009, 15, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Conaghan, P.G.; Quinn, M.A.; Bingham, S.J.; Veale, D.; Emery, P. True infliximab resistance in rheumatoid arthritis: A role for lymphotoxin a? Ann. Rheum. Dis. 2004, 63, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Calmon-Hamaty, F.; Combe, B.; Hahne, M.; Morel, J. Lymphotoxin a revisted: General features and implications in rheumatoid arthritis. Arthritis Res. 2011, 13, 232. [Google Scholar] [CrossRef]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, R.; Kontermann, R.E.; Maier, O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies 2015, 4, 48-70. https://doi.org/10.3390/antib4010048
Fischer R, Kontermann RE, Maier O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies. 2015; 4(1):48-70. https://doi.org/10.3390/antib4010048
Chicago/Turabian StyleFischer, Roman, Roland E. Kontermann, and Olaf Maier. 2015. "Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy" Antibodies 4, no. 1: 48-70. https://doi.org/10.3390/antib4010048
APA StyleFischer, R., Kontermann, R. E., & Maier, O. (2015). Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies, 4(1), 48-70. https://doi.org/10.3390/antib4010048