1. Introduction
The E. coli expression system is the most commonly used bacterial expression system. However, unfortunately, it suffers from the formation of insoluble inclusion bodies (IBs), often requiring tedious and lengthy refolding procedures. In addition, the Gram-negative E. coli produces toxic endotoxins.
As a microbial expression system, the S. cerevisiae system also has been popularly used, so far. Some characteristics, like easy cultivation, simple genetic manipulation, stable expression, etc., make S. cerevisiae an attractive host. However, a major disadvantage of it is that the expression level is usually not high enough compared to bacterial hosts, like E. coli.
Those difficulties may be overcome, if a target protein can be secreted in the extracellular environment with high efficiency using an endotoxin-free, Gram-positive bacterium. Takagi
et al. conducted an extensive screening to find such a bacterial strain that satisfies the above conditions, i.e., secretory production and no endotoxin, which has led to the discovery of
Brevibacillus choshinensis HPD31 (
Bacillus brevis) [
1]. This bacterium was isolated from the soil at the shrine of Higeta Shoyu, a well-established soy sauce manufacturer in business for almost 400 years.
Brevibacillus choshinensis is a Gram-positive bacillus with the spore-forming ability that is characteristic of Gram-positive bacteria. Spore-forming bacterium is not ideal as a host bacterial strain for industrial production, because spores have a strong environmental tolerance and can cause cross-contamination. Namely, in the case of spore-forming bacteria, it is quite difficult to exterminate them completely after finishing a culture, because of the still surviving resistant spores. In addition, residual, although weak, protease activity was detected in the culture media. To circumvent these issues, multiple genes were disrupted, resulting in the elimination of spore formation and extracellular proteases.
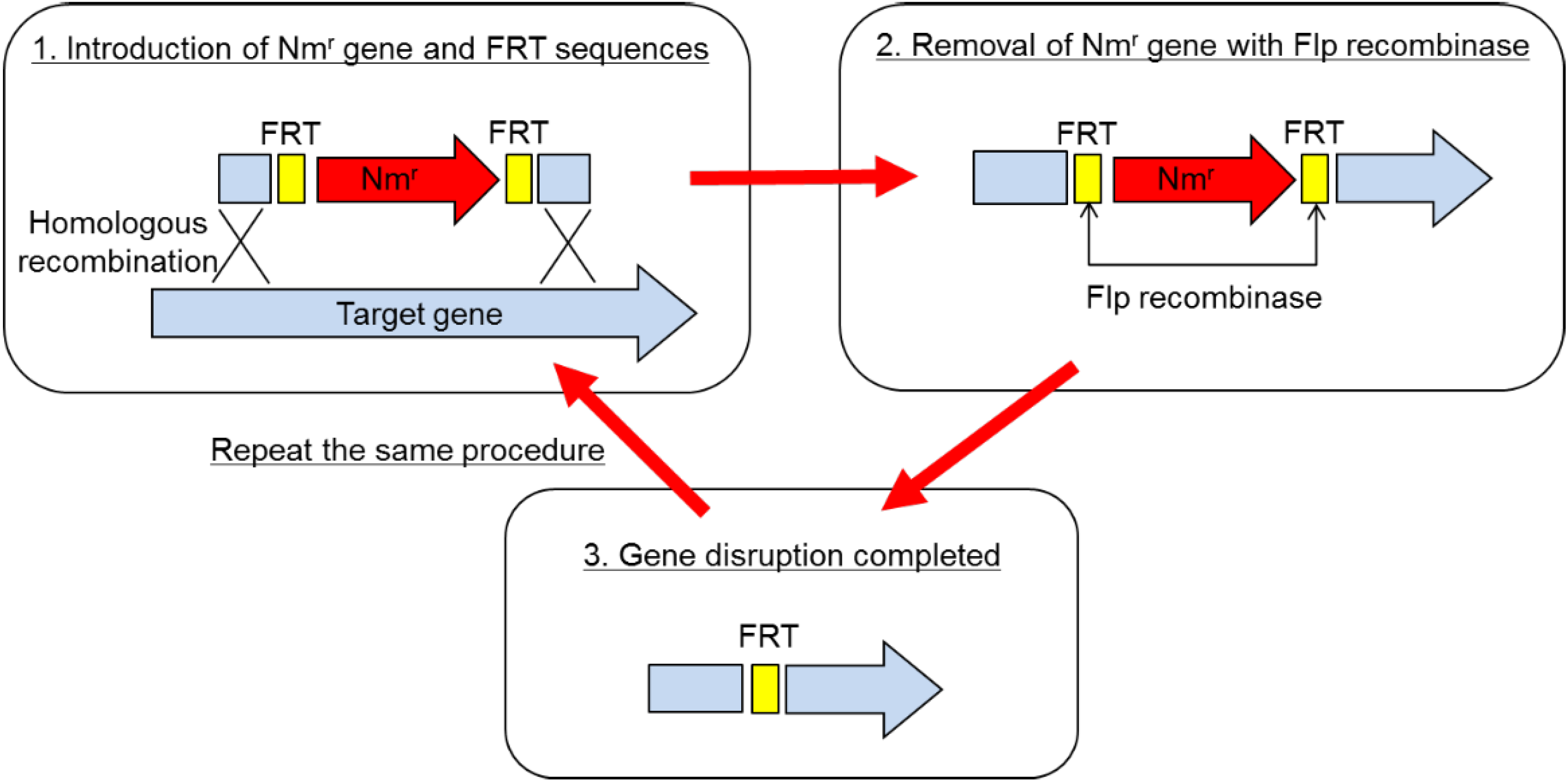
The method of gene disruption is schematically depicted in
Figure 1. Basically, it is a partial modification of the one employed in the gene replacement in
P. aeruginosa [
2]. In short, the disruption procedure consisted of two steps. (1) Gene disruption using the pBR vector on which a partial sequence of the target gene was cloned with an antibiotic resistance marker gene: The marker gene was sandwiched between two FRT sequences within the target gene, which was recognized by Flp recombinase. Disruptants were selected as drug-resistant clones on the plate containing the antibiotic. In the disruptant, the target gene was replaced by the partial sequence of the target gene interrupted by the drug resistance marker gene, which was sandwiched by FRT. As the next step, excision of the marker gene was necessary, so that multiple gene disruption was possible. (2) Removal of the marker gene using Flp recombinase: The second plasmid, which expressed Flp recombinase, was transformed into the disruptant. The marker gene was excised out from the genome due to the homologous recombination between 2 FRT sequences caused by Flp recombinase. As a final step, the Flp expressing plasmid was cured in the absence of drug. Thus, the whole procedure is completed.
Employing this method, two protease genes, imp and emp, and a spore formation gene, spoIIAC gene, were disrupted. The end result is
B. choshinensis HPD31-SP3, a versatile bacterial host for protein production [
3].
Figure 1.
Gene disruption procedure for Brevibacillus choshinensis.
Figure 1.
Gene disruption procedure for Brevibacillus choshinensis.
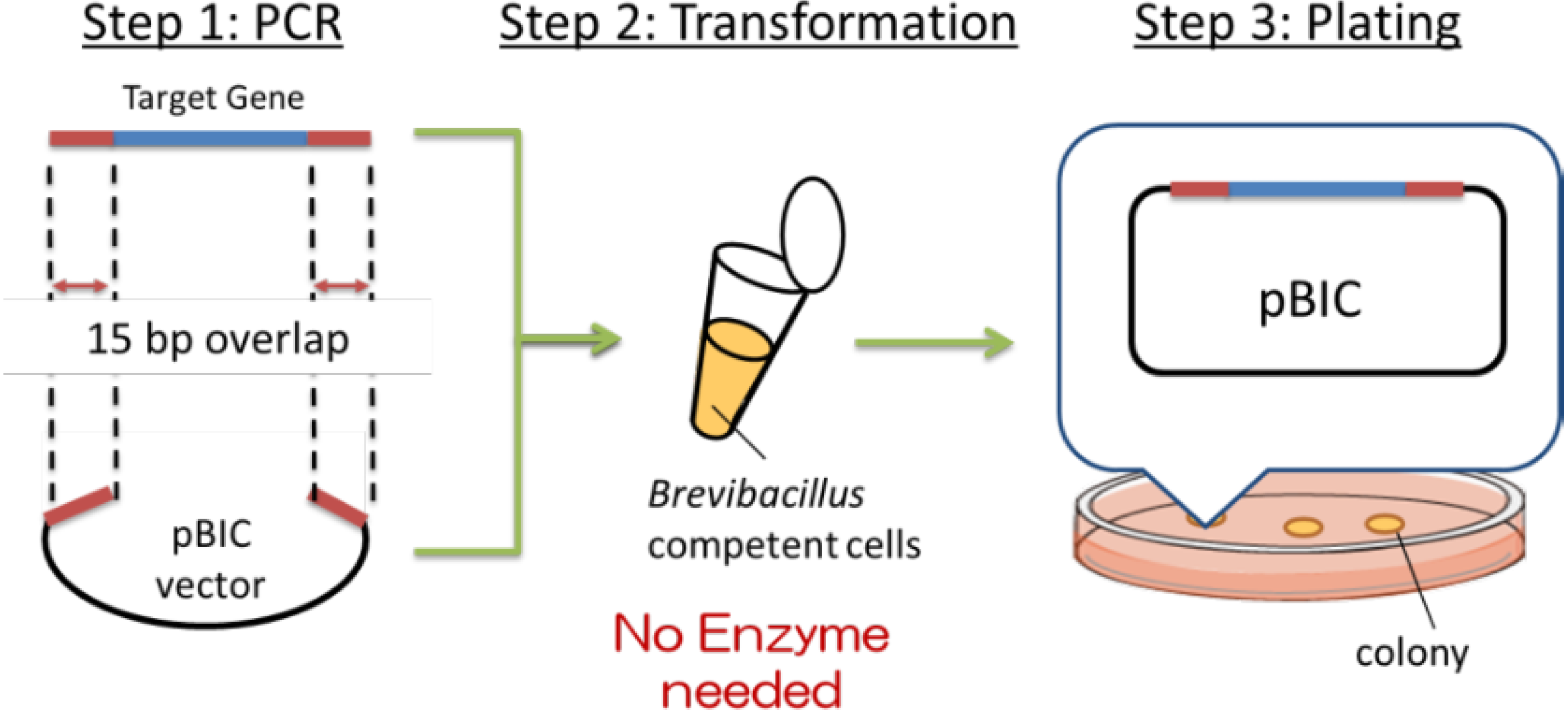
3. Robust Cloning with the BIC Method (Brevibacillus In Vivo Cloning Method)
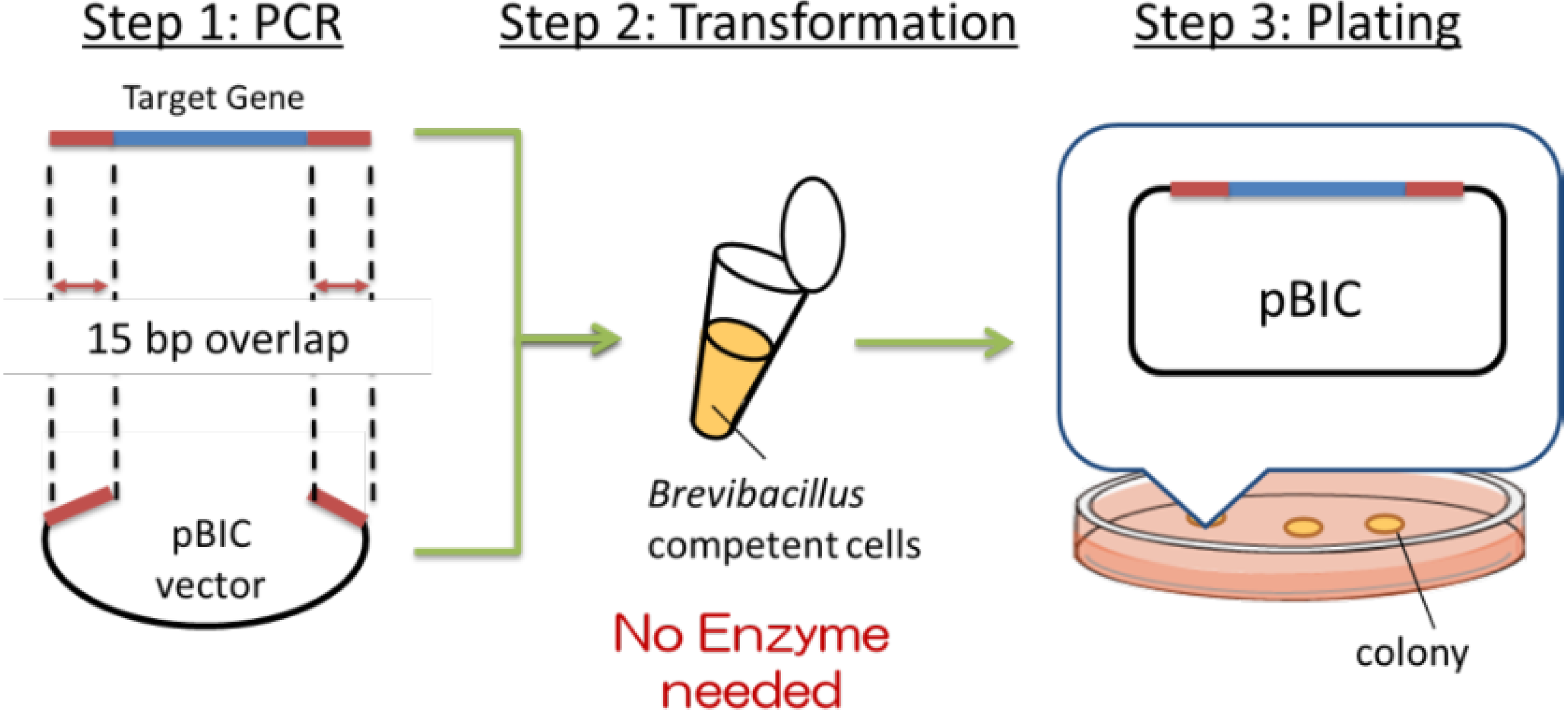
Recently, a simple method of gene cloning to construct high-expression plasmids has been developed at Higeta Shoyu Co. The whole process of gene cloning, termed the BIC (
Brevibacillus in vivo) method is schematically depicted in
Figure 2.
Figure 2.
Summary of BIC (Brevibacillus in vivo cloning) method.
Figure 2.
Summary of BIC (Brevibacillus in vivo cloning) method.
First (Step 1), the target gene (blue) was amplified by PCR. Primers were designed to contain 15 base pairs (red) that are homologous to the terminal sequences (red) of the linearized cloning vector (black),
i.e., at the desired site of insertion of the target gene. The expression plasmid was prepared by mixing the amplified target gene and the linearized vector and adding the mixture to the competent cells, as shown in Step 2 (
Figure 2). The transformation was induced by the addition of a concentrated polyethylene glycol (PEG) solution in Tris buffer, termed the NTP method (new Tris-PEG method). The target gene and the cloning vector introduced into the competent cells spontaneously formed the expression plasmid by homologous recombination of the corresponding 15 base pairs described above. This transformation efficiency is routinely ~10
6 CFU/µg DNA, comparable to the efficiency observed for the
E. coli expression system. The transformed cells were plated to select expression colonies in the presence of antibiotic neomycin (50 mg/L) (Step 3). Gene cloning efficiency by the BIC method (Steps 1, 2 and 3) has been confirmed to be very high, ranging from 80% to 100% (inserted gene size: 0.3–3.2 kbp). The electroporation method was unsuccessful in this case. The reason that electroporation did not work here remains to be clarified. However, a similar phenomenon has been reported in the case of
E. coli, too [
13]. Namely, plasmid construction exploiting the natural recombination machinery was possible only when the chemical transformation method was employed, not electroporation. There may be common background between the 2 cases. The BIC method has dramatically simplified the cloning experiments, thus precluding the need to select restriction sites. In addition to the vector termini, genes can be introduced to the internal sequence by manipulating the target gene primer sequences.
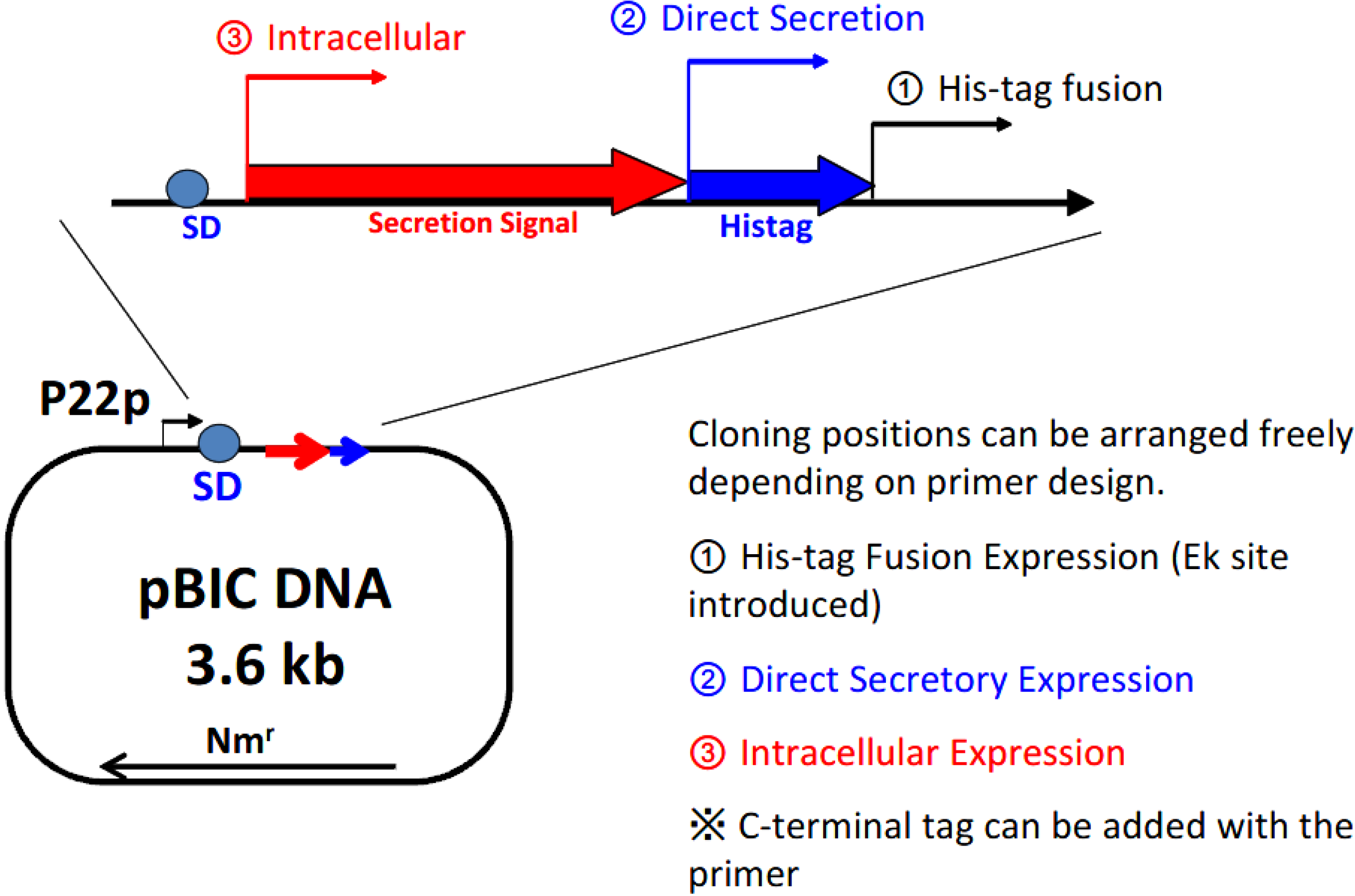
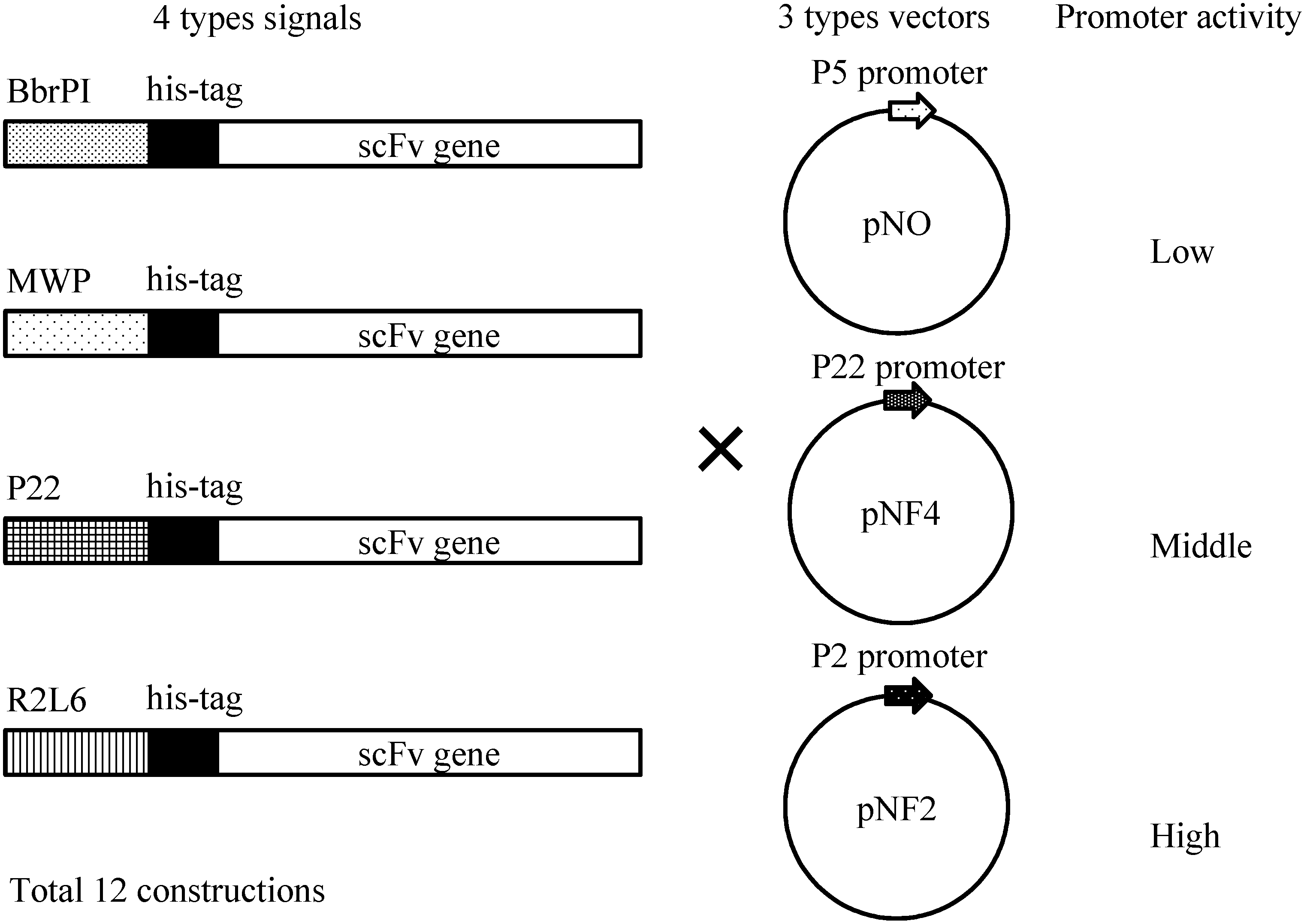
Figure 3 shows the structure of the expression plasmid, 3.6 kb pBIC DNA. A single vector can be applied in 3 different types of expression modes: (1) direct secretory production; (2) secretory production with N-terminal his-tag; and (3) intracellular production.
Figure 3.
Selection of the expression mode with the BIC system. P22p, P22 promoter; SD, Shine–Dalgarno sequence; Nmr, neomycin-resistant gene.
Figure 3.
Selection of the expression mode with the BIC system. P22p, P22 promoter; SD, Shine–Dalgarno sequence; Nmr, neomycin-resistant gene.
The prominent feature of the
Brevibacillus expression system is the highly efficient extracellular secretion of the target protein being produced, with the exception of membrane proteins. Concerning the challenge of the efficient production of membrane proteins, few successful results have been reported so far [
14].
The optimization of the promoter and secretion signal combination is essential for improving the secretory production efficiency. Protein productivity is markedly affected by the type of signal peptide used, as is evident in the
Bacillus subtilis system [
15,
16]. We have examined the secretion efficiency of different signal sequences with a variety of proteins and found it to vary from one signal sequence to another. However, there appears to be no universal sequence that guarantees uniformly high secretion efficiency for any proteins: i.e., the performance of the signal sequence varies according to the nature of the protein to be secreted. The BIC method facilitates the speedy testing of multiple secretion signals. In fact, four secretion signals, with well-proven high productivity, are currently employed in combination with promoters to select high-productivity strains. Three promoters were selected as described later.
4. Efficient Production of scFv Fragment [17]
The production in
E. coli of antibody fragments, e.g., scFv production, typically encounters the problem of necessitating the refolding of the protein upon expression in the cytoplasm as IBs [
18]. Furthermore, the secretion efficiency is typically low if expressed using secretion signals. In contrast, the
Brevibacillus expression system does not suffer from such shortcomings, because the produced scFv is efficiently transported to the extracellular milieu without making IB.
A production strain was constructed using the gene for the scFv antibody FLU [
19], derived from an antibody against the fluorescent dye, fluorescein.
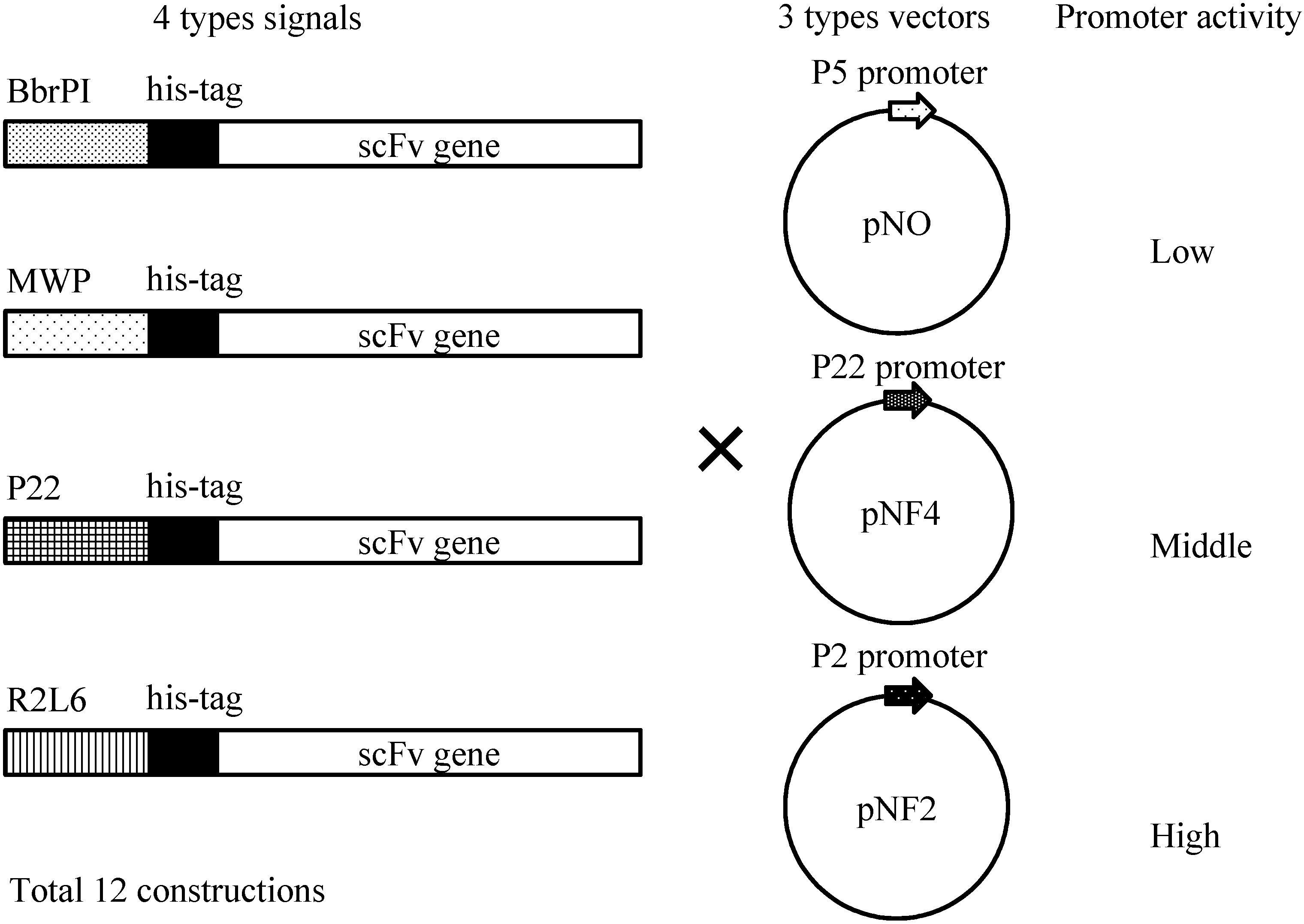
Figure 4 shows four different gene constructs comprising a
Bacillus brevis-derived middle wall protein (MWP) signal sequence, a R2L6 (modified MWP) signal sequence, a
B. choshinensis-derived BprPI signal sequence and a P22 signal sequence. These signals derive from the proteins abundantly secreted by
B. choshinensis. These four constructs were combined with three promoters, as shown in
Figure 4, i.e., P5 (pNO vector), P22 (pNF4 vector) and P2 (pNF2), resulting in 12 combinations, indicated in
Figure 4.
Figure 4.
Schematic illustration of His-scFv expression constructs. Secretion signals, His-tag and the scFv gene are schematically drawn. Three types of vectors, pNO, pNF4 and pNF2, are presented.
Figure 4.
Schematic illustration of His-scFv expression constructs. Secretion signals, His-tag and the scFv gene are schematically drawn. Three types of vectors, pNO, pNF4 and pNF2, are presented.
The N-terminal His-tag was introduced for detection and purification. The transformants harboring the correct plasmids were tested in culture production. A shake culture was performed using 3 mL 2SY medium (2% glucose, 4% Phytone Peptone (Difco, Detroit, MI), 0.5% Bacto yeast extract (Difco, Detroit, MI), 0.001% FeSO
4∙7H
2O, 0.001% MnSO
4∙4H
2O and 0.0001% ZnSO
4∙7H
2O, pH 7.2) on the test-tube scale for 48 h at 30 °C. Culture supernatants were prepared by centrifugation and subjected to SDS-PAGE to compare production levels.
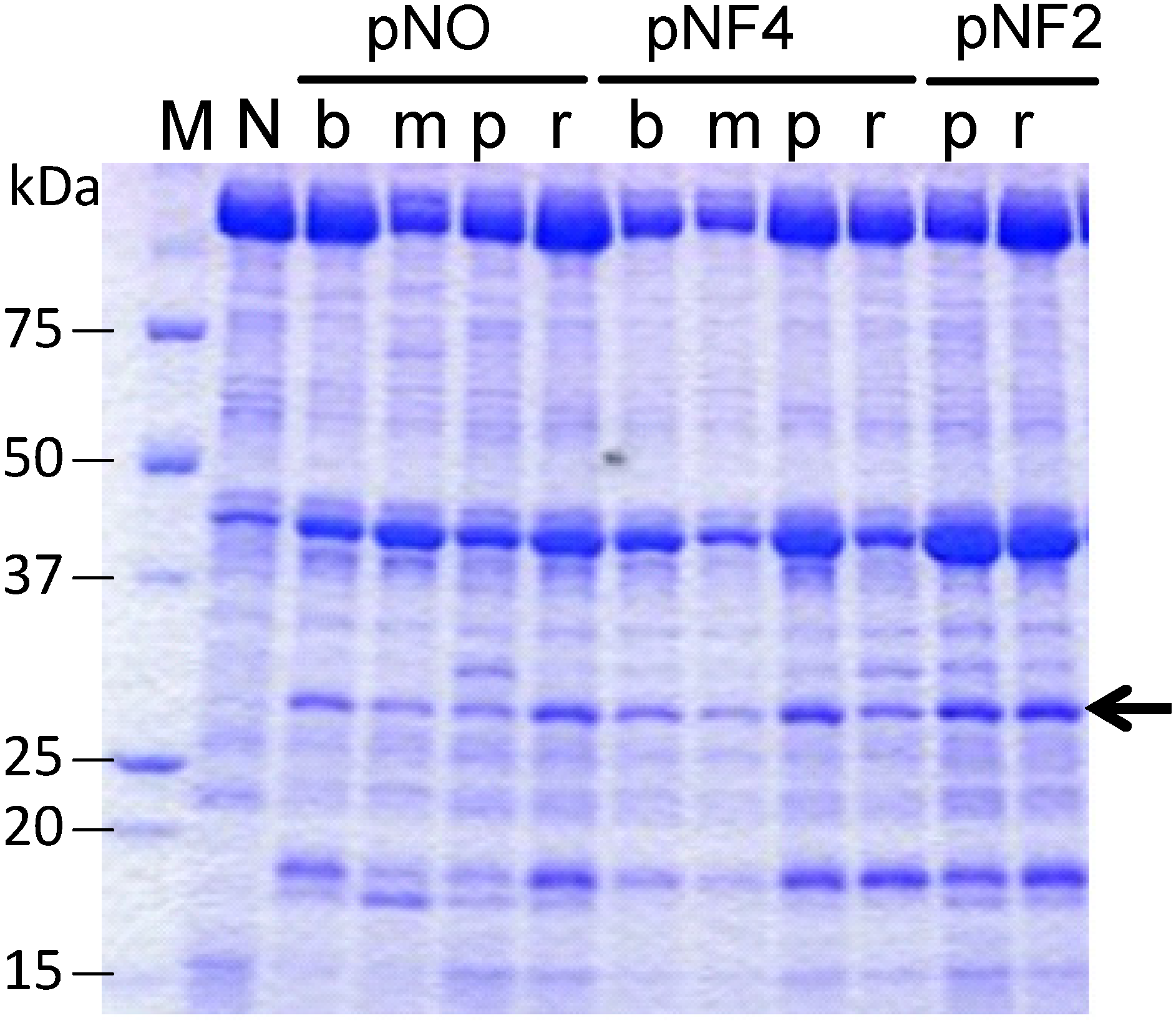
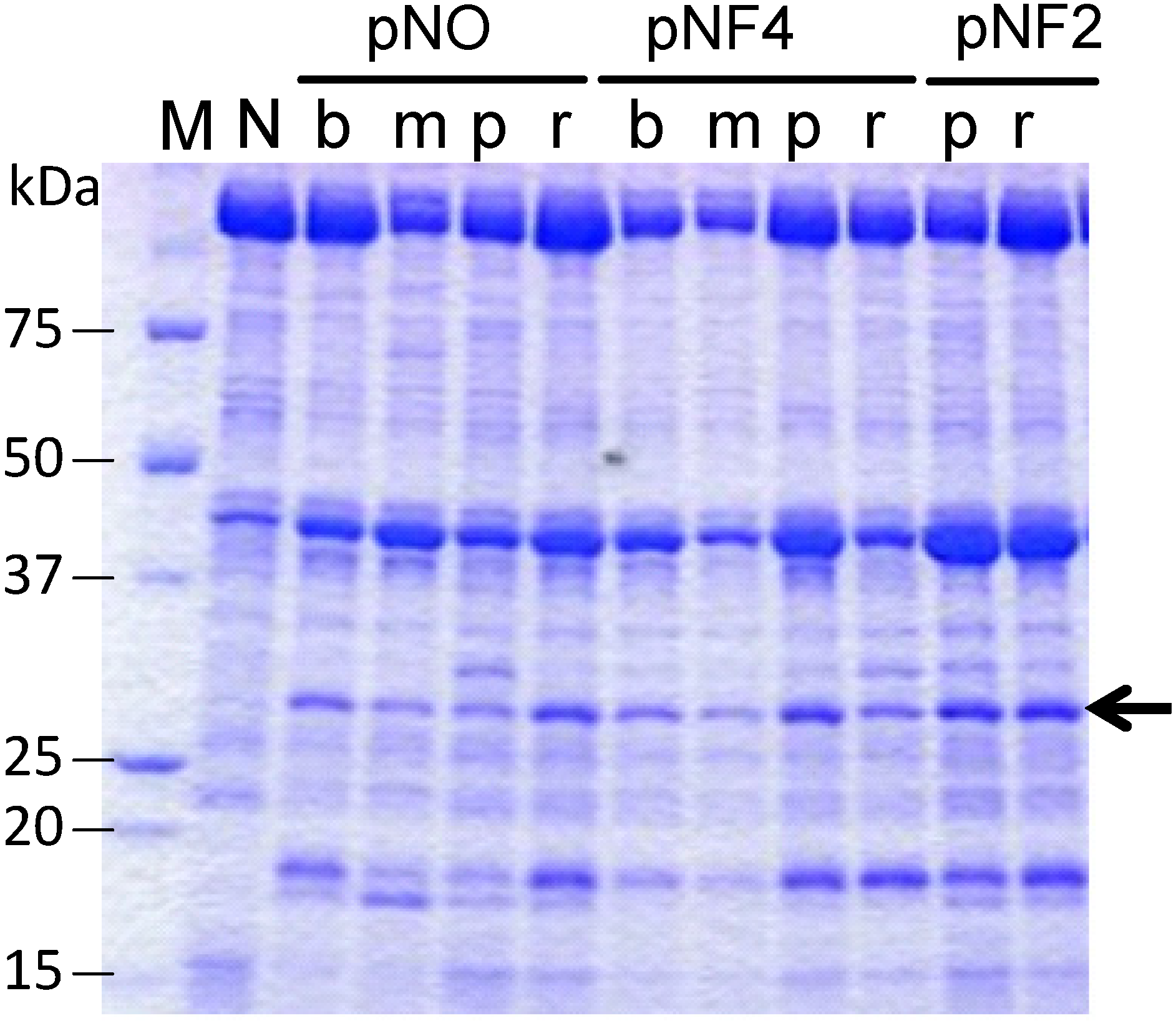
Figure 5 shows the expression of scFvFLU with different combinations of three promoters and four signal sequences (abbreviated as b, m, p and r; see
Figure 4). The production level of scFvFLU, indicated by the arrow, varied with the expression plasmid used, i.e., with the signal sequence and the promoter. The strain with the pNF2-R2L6 plasmid (far right lane) exhibited the highest productivity among those tested, at approximately 80 mg/L. Further improvement in productivity may be achieved by optimizing culture conditions. The scFvFLU was purified by Ni-sepharose Fast Flow (GE Healthcare) and gel filtration chromatography to the level of single-band purity and then assessed for fluorescein binding activity. The result indicated that approximately 95% of the inhibition of fluorescein fluorescence was observed by scFvFLU at a 1:1 ratio, demonstrating that the scFv produced in
Brevibacillus possesses binding activity.
Figure 5.
Expression of the scFvFLU protein. Cells were grown in 3 mL of culture medium at 30 °C for two days. N, negative control with blank vector; b, BbrPI signal; m, MWP signal; p, P22 signal; r, R2L6 signal. To compare the productivity of each clone with different signal or different promoter, the same amount, 4 µL/lane, of each culture supernatant was applied. pNO, pNF4, and pNF2 are expression vectors.
Figure 5.
Expression of the scFvFLU protein. Cells were grown in 3 mL of culture medium at 30 °C for two days. N, negative control with blank vector; b, BbrPI signal; m, MWP signal; p, P22 signal; r, R2L6 signal. To compare the productivity of each clone with different signal or different promoter, the same amount, 4 µL/lane, of each culture supernatant was applied. pNO, pNF4, and pNF2 are expression vectors.
In a heterologous protein expression system, fusion protein technology is sometimes effective [
20,
21]. For example, a target protein, which is sparingly soluble when expressed alone, can often be expressed as a soluble protein through its expression as a fusion protein. Additional advantages include improved detection sensitivity, that is, owing to the increased expression level, the fused protein band becomes more recognizable on SDS-PAGE gel. Furthermore, if the partner protein is applicable to affinity separation, the target protein also can be purified together with it. Although various partner proteins have been developed, “halophilic proteins” possess unique characteristics suitable as a fusion partner, including their high solubility, difficulty in salting out and ability to reversibly unfold and refold under denaturation. In the
Brevibacillus-based expression production of scFv molecules, halophilic β-lactamase (BLA) was tested as an expression partner [
22]. Anti-hen egg white lysozyme-scFv antibody (scFvHEL) was expressed and secreted as a BLA fusion protein (BLA-scFv-His-tag). The resultant scFvHELHis was successfully purified following proteolytic removal of BLA with thrombin. The purified product was shown to completely inhibit lysozyme activity at a 1:1 molar ratio. Gel-filtration-light scattering analysis revealed that the fusion scFv was present as a monomer. Similarly, scFvFLU-His was also successfully expressed as a fusion to BLA. Another fusion partner, the 45-kDa protein (P45, highly productive host derived protein), also afforded soluble expression of scFvs.
5. Expression of Fab Fragment
According to the report by Inoue
et al. [
23], Fab was successfully produced using
Brevibacillus. The expression of an antibody fragment to human urokinase-type plasminogen activator [
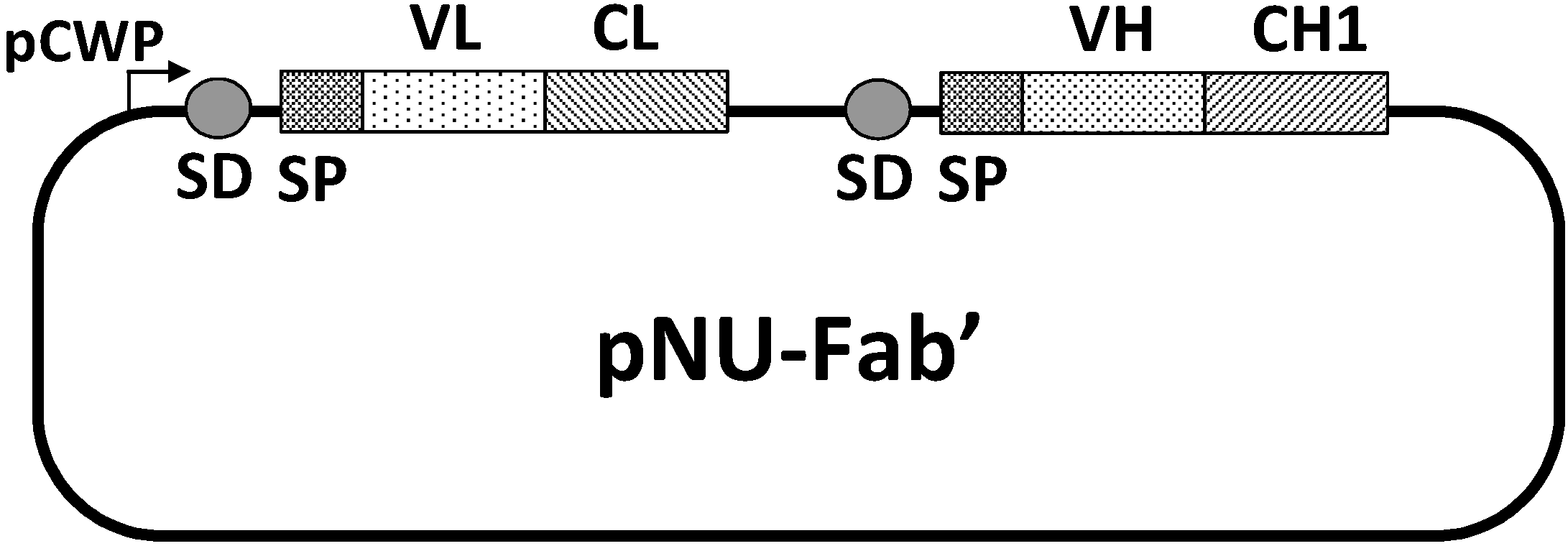
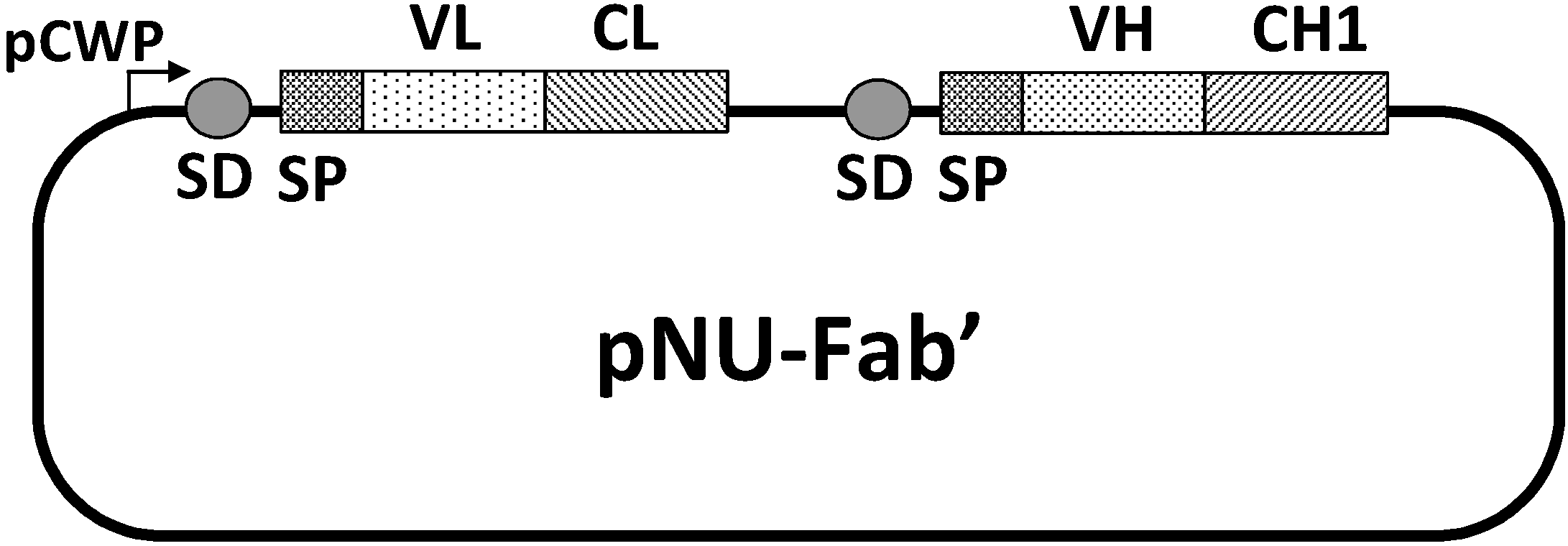
24] was similarly achieved. A pNU-Fab’ vector composed of variable regions of a murine light chain (VL) and heavy chain (VH) and constant regions of a human light chain (CL) and heavy chain (CH1) was constructed to co-express the light and heavy chains on the same plasmid in the order depicted in
Figure 6.
Figure 6.
Structure of the expression/secretion vector, pNU-Fab’. pCWP, the promoter of the B. brevis cell wall protein gene operon; SD, the ribosome binding site of the middle wall protein gene; SP, the signal peptide of the MWP; VL, the variable domain of the light chain; CL, the constant domain of the light chain; VH, the variable domain of the heavy chain; CH1, the first constant domain of the heavy chain. VL and VH are those from the mouse anti-(urokinase-type plasminogen activator) IgG gene, whereas CL and CH1 are from the human IgG gene.
Figure 6.
Structure of the expression/secretion vector, pNU-Fab’. pCWP, the promoter of the B. brevis cell wall protein gene operon; SD, the ribosome binding site of the middle wall protein gene; SP, the signal peptide of the MWP; VL, the variable domain of the light chain; CL, the constant domain of the light chain; VH, the variable domain of the heavy chain; CH1, the first constant domain of the heavy chain. VL and VH are those from the mouse anti-(urokinase-type plasminogen activator) IgG gene, whereas CL and CH1 are from the human IgG gene.
The active chimeric protein of Fab was successfully produced at a concentration of up to 100 mg/L in a simple shake flask culture setup. This value is substantially better than the results obtained with other production systems [
25], though a direct comparison of production is difficult, because productivity varies according to the protein species. For the construction of a plasmid, as described above, expressing a protein comprising plural subunits, genes of interest should be arranged in tandem. In such a case, too, the BIC method is very facilitatory for the whole construction work.
6. Prospects of Antibody Fragment Production Using the Brevibacillus Expression System
The efficient production of scFv in the active form using the BIC method to select an optimal combination of promoter and secretion signal was demonstrated. Although many proteins have been expressed using different combinations of secretion signals and promoters, there seems to be no universal rule to select one signal over others for a particular protein. Thus, there appears to be no choice, but to try multiple secretion signals and promoters in parallel. For proteins composed of plural subunits, such as Fab, a large number of combinations must be tested to find an optimal plasmid. The BIC method may be a powerful and critical tool for testing such a large number of combinations. In addition, there have been advancements in culture methods (unpublished), which dramatically increased production yield.
7. Conclusions
Currently, antibody fragments are being actively developed worldwide. In light of the multiple merits
Brevibacillus can offer, e.g., superb secretion capacity, ease of handling, negligible protease activity,
etc., this organism may become the first choice as the host to produce those proteins. The production of antibody fragments has also been tried many times using a wide variety of eukaryotic expression systems, like yeast, fungi, protozoa, insect cells, mammalian cells and plants. The results were neatly summarized and discussed in the review by Frenzel [
25]. Compared to the performance by
Brevibacillus, those results look less impressive, at least in terms of productivity, except for some cases in which a few hundred mg/L-level production was reached. However, even in those successful cases, considering the labor and the cost required for culture, extraction and purification, bacterial hosts seem apparently to get the upper hand for the production of antibody fragments. Recombinant whole antibody production may be a better target for those eukaryotic systems.
We expect the demand for Brevibacillus as an expression host may be on a steady increase from now on. The Brevibacillus expression system is commercially available for personal use. Researchers can try recombinant protein expression by themselves with much ease. This feature is another strong point of the system. It is worth being tried and may one day become the standard system for the production of secretory proteins in general.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}