
Immunotherapy in GI Cancers: Lessons from Key Trials and Future Clinical Applications

 , ,
, ,
Abstract
1. Introduction
2. Immunotherapy in Gastric and Gastroesophageal Junction Cancers
3. Immunotherapy in Hepatocellular Carcinoma (HCC)
4. Immunotherapy in Colorectal Cancer (CRC)
4.1. MSS CRC: Overcoming Immunotherapy Resistance
4.2. Clinical Decision-Making: When to Escalate Therapy in MSI-H CRC
4.3. Microbiome Modulation and Vitamin D: A Gut Feeling About Immunotherapy?
5. Immunotherapy in Pancreatic Cancer
5.1. Barriers to Immunotherapy in PDAC
5.1.1. Immunosuppressive Tumor Microenvironment
5.1.2. Low Tumor Mutational Burden
5.1.3. Poor Immune Cell Infiltration
5.2. Immunotherapeutic Strategies in Pancreatic Cancer
5.2.1. ICIs in Combination with Stroma-Modulating Agents
- Restoring tumor suppressor activity involving TP53, CDKN2A, and SMAD4 [77];
- Modulating the TGF-β–SMAD4 signaling axis to influence stromal and immune interactions [77].
5.2.2. CAR T-Cell Therapy
5.2.3. KRAS-Targeted Therapies
6. Limitations of Immunotherapy in GI Cancers
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AE | Adverse Event |
| AEG | Adenocarcinoma of the Esophagogastric Junction |
| CAFs | Cancer-Associated Fibroblasts |
| CAR | Chimeric Antigen Receptor |
| CEA | Carcinoembryonic Antigen |
| CPS | Combined Positive Score |
| CRC | Colorectal Cancer |
| CTLA-4 | Cytotoxic T-Lymphocyte Antigen 4 |
| CXCR4 | C-X-C Chemokine Receptor 4 |
| dMMR | Deficient DNA Mismatch Repair |
| DDR | DNA Damage Response |
| DOR | Duration of Response |
| EGFR | Epidermal Growth Factor Receptor |
| FDA | Food and Drug Administration |
| FGFR | Fibroblast Growth Factor Receptor |
| FMT | Fecal Microbiota Transplantation |
| FOLFOX | Leucovorin, 5-Fluorouracil, Oxaliplatin |
| GC | Gastric Cancer |
| GEJ | Gastroesophageal Junction |
| HCC | Hepatocellular Carcinoma |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HR | Hazard Ratio |
| ICI | Immune Checkpoint Inhibitor |
| ICB | Immune Checkpoint Blockade |
| IL-10 | Interleukin 10 |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene |
| LAG-3 | Lymphocyte Activation Gene 3 |
| MAPK | Mitogen-Activated Protein Kinase |
| MDSCs | Myeloid-Derived Suppressor Cells |
| MSS | Microsatellite Stable |
| MSI-H | Microsatellite Instability-High |
| mRNA | Messenger RNA |
| NCT | Neoadjuvant Chemotherapy |
| ORR | Objective Response Rate |
| OS | Overall Survival |
| PARP | Poly (ADP-Ribose) Polymerase |
| PD-1 | Programmed Death 1 |
| PD-L1 | Programmed Death Ligand 1 |
| PD-L2 | Programmed Death Ligand 2 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PFS | Progression-Free Survival |
| PSCA | Prostate Stem Cell Antigen |
| STRIDE | Single Tremelimumab Regular Interval Durvalumab |
| TGF-β | Transforming Growth Factor Beta |
| TIL | Tumor-Infiltrating Lymphocyte |
| TKI | Tyrosine Kinase Inhibitor |
| TMB | Tumor Mutational Burden |
| TME | Tumor Microenvironment |
| TRAEs | Treatment-Related Adverse Events |
| VEGF | Vascular Endothelial Growth Factor |
| XELOX | Capecitabine, Oxaliplatin |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Hasan, A.; Pozzoli, G.; Cenciarelli, C. Cancer Immunotherapy with Immune Checkpoint Inhibitors (ICIs): Potential, Mechanisms of Resistance, and Strategies for Reinvigorating T Cell Responsiveness When Resistance Is Acquired. Cancer Cell Int. 2023, 23, 64. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-Line Nivolumab plus Chemotherapy versus Chemotherapy Alone for Advanced Gastric, Gastro-Oesophageal Junction, and Oesophageal Adenocarcinoma (CheckMate 649): A Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Peshin, S.; Modi, S.; Singh, S. Advancements in Cancer Immunotherapy: A Comprehensive Review of Immune Checkpoint Inhibitors with a Focus on Pembrolizumab and Emerging Strategies. MCCRJ 2024, 2, 430–434. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for Patients with PD-L1-Positive Advanced Gastric Cancer (KEYNOTE-012): A Multicentre, Open-Label, Phase 1b Trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Kang, Y.-K.; Catenacci, D.V.; Muro, K.; Fuchs, C.S.; Geva, R.; Hara, H.; Golan, T.; Garrido, M.; Jalal, S.I.; et al. Pembrolizumab Alone or in Combination with Chemotherapy as First-Line Therapy for Patients with Advanced Gastric or Gastroesophageal Junction Adenocarcinoma: Results from the Phase II Nonrandomized KEYNOTE-059 Study. Gastric Cancer 2019, 22, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Boku, N.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; Yeh, K.-H.; et al. Nivolumab in Previously Treated Advanced Gastric Cancer (ATTRACTION-2): 3-Year Update and Outcome of Treatment beyond Progression with Nivolumab. Gastric Cancer 2021, 24, 946–958. [Google Scholar] [CrossRef]
- Kang, Y.-K.; Chen, L.-T.; Ryu, M.-H.; Oh, D.-Y.; Oh, S.C.; Chung, H.C.; Lee, K.-W.; Omori, T.; Shitara, K.; Sakuramoto, S.; et al. Nivolumab plus Chemotherapy versus Placebo plus Chemotherapy in Patients with HER2-Negative, Untreated, Unresectable Advanced or Recurrent Gastric or Gastro-Oesophageal Junction Cancer (ATTRACTION-4): A Randomised, Multicentre, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2022, 23, 234–247. [Google Scholar] [CrossRef]
- FDA Approves Nivolumab in Combination with Chemotherapy for Metastatic Gastric Cancer and Esophageal Adenocarcinoma; Food and Drug Administration: Silver Spring, MD, USA, 2024.
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients with Metastatic Esophagogastric Cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef]
- Zhang, W.; Guo, K.; Zheng, S. Immunotherapy Combined with Chemotherapy in the First-Line Treatment of Advanced Gastric Cancer: Systematic Review and Bayesian Network Meta-Analysis Based on Specific PD-L1 CPS. Curr. Oncol. 2025, 32, 112. [Google Scholar] [CrossRef]
- Pan, S.; Li, K.; Huang, B.; Huang, J.; Xu, H.; Zhu, Z. Efficacy and Safety of Immune Checkpoint Inhibitors in Gastric Cancer: A Network Meta-Analysis of Well-Designed Randomized Controlled Trials. Ann. Transl. Med. 2021, 9, 290. [Google Scholar] [CrossRef]
- Qian, M.; Fang, Y.; Xiang, Z.; Zhang, Y.; Zhan, H.; Chen, X.; Chen, Y.; Xu, T. The Efficacy of Neoadjuvant Immunotherapy in Gastric Cancer, Adenocarcinoma of the Esophagogastric Junction, and Esophageal Cancer: A Meta-Analysis. Front. Oncol. 2024, 14, 1502611. [Google Scholar] [CrossRef]
- Liu, S.; Wong, H.Y.; Xie, L.; Kim, Y.; Shu, D.; Zheng, B.; Liu, N.; Xing, C.; Chen, X.; Dong, Q. Comparative Efficacy and Tolerability of Targeted and Immunotherapy Combined with Chemotherapy as First-Line Treatment for Advanced Gastric Cancer: A Bayesian Network Meta-Analysis. Sci. Rep. 2022, 12, 22024. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A Global View of Hepatocellular Carcinoma: Trends, Risk, Prevention and Management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Mukund, A.; Vats, P.; Jindal, A.; Patidar, Y.; Sarin, S.K. Early Hepatocellular Carcinoma Treated by Radiofrequency Ablation-Mid- and Long-Term Outcomes. J. Clin. Exp. Hepatol. 2020, 10, 563–573. [Google Scholar] [CrossRef]
- Han, S.; Sung, P.S.; Park, S.Y.; Kim, J.W.; Hong, H.P.; Yoon, J.-H.; Chung, D.J.; Kwon, J.H.; Lim, S.; Kim, J.H.; et al. Local Ablation for Hepatocellular Carcinoma: 2024 Expert Consensus-Based Practical Recommendation of the Korean Liver Cancer Association. J. Liver Cancer 2024, 24, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Govalan, R.; Lauzon, M.; Luu, M.; Ahn, J.C.; Kosari, K.; Todo, T.; Kim, I.K.; Noureddin, M.; Kuo, A.; Walid, A.S.; et al. Comparison of Surgical Resection and Systemic Treatment for Hepatocellular Carcinoma with Vascular Invasion: National Cancer Database Analysis. Liver Cancer 2021, 10, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, Q.; Xu, S.; Ye, C.; Tian, T.; Jiang, Q.; Shan, J.; Ruan, J. Modulation of the Tumour Microenvironment in Hepatocellular Carcinoma by Tyrosine Kinase Inhibitors: From Modulation to Combination Therapy Targeting the Microenvironment. Cancer Cell Int. 2022, 22, 73. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Ren, B.; Cheng, L.; Wang, X.; Ma, Z.; Yong, W.P.; Chen, X.; Wang, L.; Goh, B.C. Unleashing the Efficacy of Immune Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma: Factors, Strategies, and Ongoing Trials. Front. Pharmacol. 2023, 14, 1261575. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A Clinical Trial of CTLA-4 Blockade with Tremelimumab in Patients with Hepatocellular Carcinoma and Chronic Hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Sangro, B.; Park, J.; Finn, R.; Cheng, A.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.; Harding, J.; Merle, P.; et al. LBA-3 CheckMate 459: Long-Term (Minimum Follow-up 33.6 Months) Survival Outcomes with Nivolumab versus Sorafenib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma. Ann. Oncol. 2020, 31, S241–S242. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Vogel, A.; Zhu, A.X.; Cheng, A.-L.; Yau, T.; Zhou, J.; Uppot, R.N.; Kim, E.; Malhotra, U.; Siegel, A.B.; et al. P024 KEYNOTE-937 Trial in Progress: Adjuvant Pembrolizumab for Hepatocellular Carcinoma and Complete Radiologic Response after Surgical Resection or Local Ablation. Gut 2021, 70, A22. [Google Scholar] [CrossRef]
- Lee, M.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.; Spahn, J.; Liu, B.; Abdullah, H.; et al. LBA39—Randomised Efficacy and Safety Results for Atezolizumab (Atezo) + Bevacizumab (Bev) in Patients (Pts) with Previously Untreated, Unresectable Hepatocellular Carcinoma (HCC). Ann. Oncol. 2019, 30, v875. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Qin, S.; Chen, M.; Cheng, A.-L.; Kaseb, A.O.; Kudo, M.; Lee, H.C.; Yopp, A.C.; Zhou, J.; Wang, L.; Wen, X.; et al. Atezolizumab plus Bevacizumab versus Active Surveillance in Patients with Resected or Ablated High-Risk Hepatocellular Carcinoma (IMbrave050): A Randomised, Open-Label, Multicentre, Phase 3 Trial. Lancet 2023, 402, 1835–1847. [Google Scholar] [CrossRef]
- AstraZeneca. A Phase III, Randomized, Double-Blind, Placebo-Controlled, Multi Center Study of Durvalumab Monotherapy or in Combination with Bevacizumab as Adjuvant Therapy in Patients with Hepatocellular Carcinoma Who Are at High Risk of Recurrence After Curative Hepatic Resection or Ablation. 2025. Available online: https://clinicaltrials.gov/study/NCT03847428 (accessed on 15 April 2025).
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Golan, T.; Lin, C.-C.; Dahan, L.; Fu, S.; Moreno, V.; Geva, R.; Reck, M.; Wasserstrom, H.A.; Mi, G.; et al. Ramucirumab (Ram) and Durvalumab (Durva) Treatment of Metastatic Non-Small Cell Lung Cancer (NSCLC), Gastric/Gastroesophageal Junction (G/GEJ) Adenocarcinoma, and Hepatocellular Carcinoma (HCC) Following Progression on Systemic Treatment(s). J. Clin. Oncol. 2019, 37, 2528. [Google Scholar] [CrossRef]
- Zai Lab (Shanghai) Co., Ltd. A Multicenter, Open-Label, Phase I/II Dose Escalation and Expansion Clinical Study to Assess the Safety and Efficacy of MGD013 Monotherapy and in Combination with Brivanib Alaninate (ZL-2301) in Patients with Advanced Liver Cancer. 2024. Available online: https://clinicaltrials.gov/study/NCT04212221 (accessed on 15 April 2025).
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Gujarathi, R.; Peshin, S.; Zhang, X.; Bachini, M.; Meeks, M.N.; Shroff, R.T.; Pillai, A. Intrahepatic Cholangiocarcinoma: Insights on Molecular Testing, Targeted Therapies, and Future Directions from a Multidisciplinary Panel. Hepatol. Commun. 2025, 9, e0743. [Google Scholar] [CrossRef]
- Randrian, V.; Evrard, C.; Tougeron, D. Microsatellite Instability in Colorectal Cancers: Carcinogenesis, Neo-Antigens, Immuno-Resistance and Emerging Therapies. Cancers 2021, 13, 3063. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Kuang, C.; Hao, S.; Yu, J.; Huang, Y.; Zhang, L. Immunotherapy Efficacy on Mismatch Repair-Deficient Colorectal Cancer: From Bench to Bedside. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188447. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Nivolumab plus Low-Dose Ipilimumab in Previously Treated Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: 4-Year Follow-up from CheckMate 142. Ann. Oncol. 2022, 33, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Yoshino, T.; Van Cutsem, E.; Eng, C.; Kim, T.W.; Wasan, H.S.; Desai, J.; Ciardiello, F.; Yaeger, R.; Maughan, T.S.; et al. Encorafenib, Cetuximab and Chemotherapy in BRAF-Mutant Colorectal Cancer: A Randomized Phase 3 Trial. Nat. Med. 2025, 31, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Verschoor, Y.L.; Tan, P.B.; Balduzzi, S.; Van Lent, A.U.; Grootscholten, C.; Dokter, S.; Büller, N.V.; Grotenhuis, B.A.; Kuhlmann, K.; et al. Neoadjuvant Immunotherapy in Locally Advanced Mismatch Repair-Deficient Colon Cancer. N. Engl. J. Med. 2024, 390, 1949–1958. [Google Scholar] [CrossRef]
- Chalabi, M.; van den Dungen, L.D.W.; Verschoor, Y.L.; Balduzzi, S.; de Gooyer, P.G.M.; Kok, N.; Kerver, E.; Grootscholten, C.; Voest, E.E.; Burger, J.W.A.; et al. LBA24 Neoadjuvant Immunotherapy in Locally Advanced MMR-Deficient Colon Cancer: 3-Year Disease-Free Survival from NICHE-2. Ann. Oncol. 2024, 35, S1217–S1218. [Google Scholar] [CrossRef]
- Verschoor, Y.L.; van den Berg, J.; Beets, G.; Sikorska, K.; Aalbers, A.; van Lent, A.; Grootscholten, C.; Huibregtse, I.; Marsman, H.; Oosterling, S.; et al. Neoadjuvant Nivolumab, Ipilimumab, and Celecoxib in MMR-Proficient and MMR-Deficient Colon Cancers: Final Clinical Analysis of the NICHE Study. J. Clin. Oncol. 2022, 40, 3511. [Google Scholar] [CrossRef]
- Dasari, A.; Lonardi, S.; Garcia-Carbonero, R.; Elez, E.; Yoshino, T.; Sobrero, A.; Yao, J.; García-Alfonso, P.; Kocsis, J.; Cubillo Gracian, A.; et al. Fruquintinib versus Placebo in Patients with Refractory Metastatic Colorectal Cancer (FRESCO-2): An International, Multicentre, Randomised, Double-Blind, Phase 3 Study. Lancet 2023, 402, 41–53. [Google Scholar] [CrossRef]
- Overman, M.J.; Yothers, G.; Jacobs, S.A.; Sanoff, H.K.; Cohen, D.J.; Guthrie, K.A.; Henry, N.L.; Ganz, P.A.; Kopetz, S.; Lucas, P.C.; et al. Colorectal Cancer Metastatic dMMR Immuno-Therapy (COMMIT) Study: A Randomized Phase III Study of Atezolizumab (Atezo) Monotherapy versus mFOLFOX6/Bevacizumab/Atezo in the First-Line Treatment of Patients (Pts) with Deficient DNA Mismatch Repair (dMMR) or Microsatellite Instability-High (MSI-H) Metastatic Colorectal Cancer (mCRC)—NRG-GI004/SWOG-S1610. J. Clin. Oncol. 2024, 42, TPS231. [Google Scholar] [CrossRef]
- Zhao, W.; Lei, J.; Ke, S.; Chen, Y.; Xiao, J.; Tang, Z.; Wang, L.; Ren, Y.; Alnaggar, M.; Qiu, H.; et al. Fecal Microbiota Transplantation plus Tislelizumab and Fruquintinib in Refractory Microsatellite Stable Metastatic Colorectal Cancer: An Open-Label, Single-Arm, Phase II Trial (RENMIN-215). eClinicalMedicine 2023, 66, 102315. [Google Scholar] [CrossRef]
- Saunders, M.P.; Graham, J.; Cunningham, D.; Plummer, R.; Church, D.; Kerr, R.; Cook, S.; Zheng, S.; La Thangue, N.; Kerr, D. CXD101 and Nivolumab in Patients with Metastatic Microsatellite-Stable Colorectal Cancer (CAROSELL): A Multicentre, Open-Label, Single-Arm, Phase II Trial. ESMO Open 2022, 7, 100594. [Google Scholar] [CrossRef]
- Heregger, R.; Huemer, F.; Steiner, M.; Gonzalez-Martinez, A.; Greil, R.; Weiss, L. Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer. Cancers 2023, 15, 5090. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Puglisi, S.; Pirrone, C.; Martelli, V.; Catalano, F.; Nardin, S.; Seeber, A.; Puccini, A.; Sciallero, S. The Role of Immunotherapy in Microsatellites Stable Metastatic Colorectal Cancer: State of the Art and Future Perspectives. Front. Oncol. 2023, 13, 1161048. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Mendes, F. CTLA-4 Blockade in the Treatment of Colorectal Cancer with Microsatellite Instability. Iran. J. Color. Res. 2021, 9, 1–6. [Google Scholar] [CrossRef]
- Haynes, J.; Manogaran, P. Mechanisms and Strategies to Overcome Drug Resistance in Colorectal Cancer. Int. J. Mol. Sci. 2025, 26, 1988. [Google Scholar] [CrossRef]
- Ibrahim, R.; Saleh, K.; Chahine, C.; Khoury, R.; Khalife, N.; Cesne, A.L. LAG-3 Inhibitors: Novel Immune Checkpoint Inhibitors Changing the Landscape of Immunotherapy. Biomedicines 2023, 11, 1878. [Google Scholar] [CrossRef]
- Yu, Y.; Jia, H.; Zhang, T.; Zhang, W. Advances in DNA Damage Response Inhibitors in Colorectal Cancer Therapy. Acta Biochim. Biophys. Sin. 2024, 56, 15–22. [Google Scholar] [CrossRef]
- Bai, Z.; Zhou, Y.; Ye, Z.; Xiong, J.; Lan, H.; Wang, F. Tumor-Infiltrating Lymphocytes in Colorectal Cancer: The Fundamental Indication and Application on Immunotherapy. Front. Immunol. 2021, 12, 808964. [Google Scholar] [CrossRef]
- Zalila-Kolsi, I.; Dhieb, D.; Osman, H.A.; Mekideche, H. The Gut Microbiota and Colorectal Cancer: Understanding the Link and Exploring Therapeutic Interventions. Biology 2025, 14, 251. [Google Scholar] [CrossRef]
- Vaughan-Shaw, P.G.; Buijs, L.F.; Blackmur, J.P.; Theodoratou, E.; Zgaga, L.; Din, F.V.N.; Farrington, S.M.; Dunlop, M.G. The Effect of Vitamin D Supplementation on Survival in Patients with Colorectal Cancer: Systematic Review and Meta-Analysis of Randomised Controlled Trials. Br. J. Cancer 2020, 123, 1705–1712. [Google Scholar] [CrossRef]
- Peters, C.; Klein, K.; Kabelitz, D. Vitamin C and Vitamin D-Friends or Foes in Modulating Γδ T-Cell Differentiation? Cell Mol. Immunol. 2022, 19, 1198–1200. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Algül, H.; Tuveson, D.A.; Gress, T.M. Stromal Biology and Therapy in Pancreatic Cancer: A Changing Paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef]
- Watt, J.; Kocher, H.M. The Desmoplastic Stroma of Pancreatic Cancer Is a Barrier to Immune Cell Infiltration. Oncoimmunology 2013, 2, e26788. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T Cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef]
- Beatty, G.L.; Eghbali, S.; Kim, R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 267–278. [Google Scholar] [CrossRef]
- Delitto, D.; Wallet, S.M.; Hughes, S.J. Targeting Tumor Tolerance: A New Hope for Pancreatic Cancer Therapy? Pharmacol. Ther. 2016, 166, 9–29. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells from Pancreatic Carcinomas in Mice Is Regulated by Ly6C(Low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef]
- Evans, R.A.; Diamond, M.S.; Rech, A.J.; Chao, T.; Richardson, M.W.; Lin, J.H.; Bajor, D.L.; Byrne, K.T.; Stanger, B.Z.; Riley, J.L.; et al. Lack of Immunoediting in Murine Pancreatic Cancer Reversed with Neoantigen. JCI Insight 2016, 1, e88328. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. CANCER IMMUNOLOGY. The “Cancer Immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial Contribution of Extrinsic Risk Factors to Cancer Development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic Cancer Biology and Genetics from an Evolutionary Perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti–PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef]
- Peng, H.; Shen, J.; Long, X.; Zhou, X.; Zhang, J.; Xu, X.; Huang, T.; Xu, H.; Sun, S.; Li, C.; et al. Local Release of TGF-β Inhibitor Modulates Tumor-Associated Neutrophils and Enhances Pancreatic Cancer Response to Combined Irreversible Electroporation and Immunotherapy. Adv. Sci. 2022, 9, e2105240. [Google Scholar] [CrossRef]
- Gueorguieva, I.; Tabernero, J.; Melisi, D.; Macarulla, T.; Merz, V.; Waterhouse, T.H.; Miles, C.; Lahn, M.M.; Cleverly, A.; Benhadji, K.A. Population Pharmacokinetics and Exposure-Overall Survival Analysis of the Transforming Growth Factor-β Inhibitor Galunisertib in Patients with Pancreatic Cancer. Cancer Chemother. Pharmacol. 2019, 84, 1003–1015. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ Receptor Inhibitor Galunisertib Is Linked to Inflammation- and Remodeling-Related Proteins in Patients with Pancreatic Cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Varghese, A.M. Chimeric Antigen Receptor (CAR) T and Other T Cell Strategies for Pancreas Adenocarcinoma. Chin. Clin. Oncol. 2017, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Baghel, K.; Mehrotra, S.; Prajapati, V.K. Revolutionizing Pancreatic Cancer Treatment with CAR-T Therapy. Adv. Protein Chem. Struct. Biol. 2025, 144, 331–353. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight Toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Baghdadi, T.A.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor Hypoxia as a Driving Force in Genetic Instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic Hypoxia Compromises Repair of DNA Double-Strand Breaks to Drive Genetic Instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Vyas, M.; Müller, R.; Pogge von Strandmann, E. Antigen Loss Variants: Catching Hold of Escaping Foes. Front. Immunol. 2017, 8, 175. [Google Scholar] [CrossRef]
- de Vries, T.J.; Fourkour, A.; Ruiter, D.J.; Van, M.G. Expression of Immunotherapy Candidate Proteins GP100, Mart-1/Melan-A and Tyrosinase in Human Melanocytic Lesions and Melanoma Cell Lines: 497. Melanoma Res. 1997, 7, S143. [Google Scholar] [CrossRef]
- Dobosz, P.; Stempor, P.A.; Roszik, J.; Herman, A.; Layani, A.; Berger, R.; Avni, D.; Sidi, Y.; Leibowitz-Amit, R. Checkpoint Genes at the Cancer Side of the Immunological Synapse in Bladder Cancer. Transl. Oncol. 2020, 13, 193–200. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Takeda, K.; Nakayama, M.; Hayakawa, Y.; Kojima, Y.; Ikeda, H.; Imai, N.; Ogasawara, K.; Okumura, K.; Thomas, D.M.; Smyth, M.J. IFN-γ Is Required for Cytotoxic T Cell-Dependent Cancer Genome Immunoediting. Nat. Commun. 2017, 8, 14607. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 Antibody Therapy Inhibits Breast Tumor Growth and Metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef]
- Beavis, P.A.; Slaney, C.Y.; Milenkovski, N.; Henderson, M.A.; Loi, S.; Stagg, J.; Kershaw, M.H.; Darcy, P.K. CD73: A Potential Biomarker for Anti-PD-1 Therapy. Oncoimmunology 2015, 4, e1046675. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Zhou, Y.L.; Guo, X.; Ge, J.; Fu, J. Immune-Related Adverse Events from Combination Immunotherapy in Cancer Patients: A Comprehensive Meta-Analysis of Randomized Controlled Trials. Int. Immunopharmacol. 2018, 63, 292–298. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.-E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Vanterpool, K.B.; Gacki-Smith, J.; Downey, M.C.; Nordstrom, M.; Luken, M.; Riggleman, T.; Fichter, S.; Altema, W.; Jensen, S.E.; Dumanian, G.A.; et al. Patient Preferences of Patient Selection Criteria for Upper Extremity Vascularized Composite Allotransplantation: A Qualitative Study. SAGE Open Med. 2023, 11, 20503121231181236. [Google Scholar] [CrossRef]
- Simmet, V.; Eberst, L.; Marabelle, A.; Cassier, P.A. Immune Checkpoint Inhibitor-Based Combinations: Is Dose Escalation Mandatory for Phase I Trials? Ann. Oncol. 2019, 30, 1751–1759. [Google Scholar] [CrossRef]
- Zhou, X.; Yao, Z.; Bai, H.; Duan, J.; Wang, Z.; Wang, X.; Zhang, X.; Xu, J.; Fei, K.; Zhang, Z.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitor-Based Combination Therapies in Clinical Trials: A Systematic Review and Meta-Analysis. Lancet Oncol. 2021, 22, 1265–1274. [Google Scholar] [CrossRef]
- Iwama, S.; De Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary Expression of CTLA-4 Mediates Hypophysitis Secondary to Administration of CTLA-4 Blocking Antibody. Sci. Transl. Med. 2014, 6, 230ra45. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Kim, C.; Baer, L.; Zhu, X. Bevacizumab Increases Risk for Severe Proteinuria in Cancer Patients. J. Am. Soc. Nephrol. 2010, 21, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- An, M.M.; Zou, Z.; Shen, H.; Liu, P.; Chen, M.L.; Cao, Y.B.; Jiang, Y.Y. Incidence and Risk of Significantly Raised Blood Pressure in Cancer Patients Treated with Bevacizumab: An Updated Meta-Analysis. Eur. J. Clin. Pharmacol. 2010, 66, 813–821. [Google Scholar] [CrossRef] [PubMed]

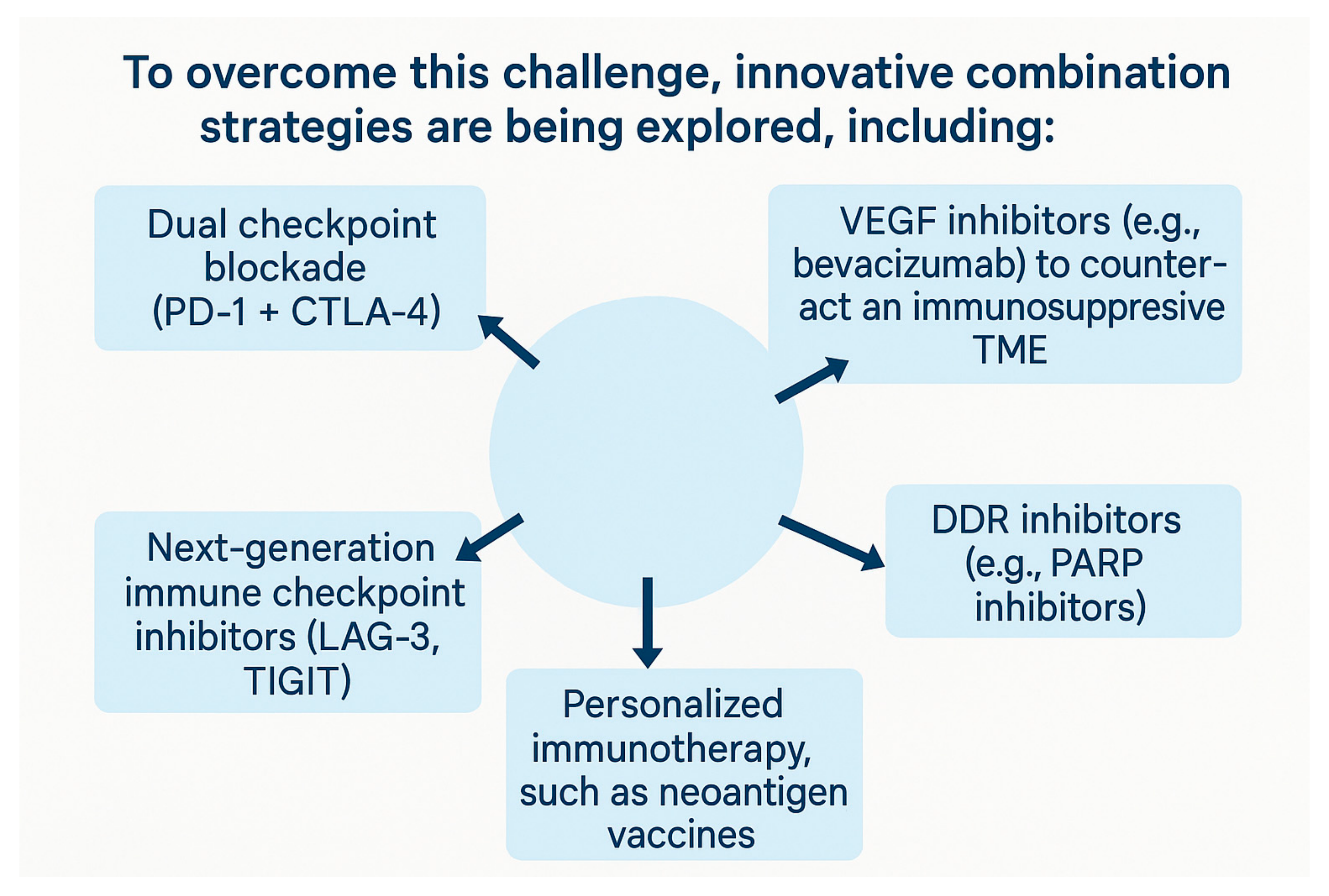
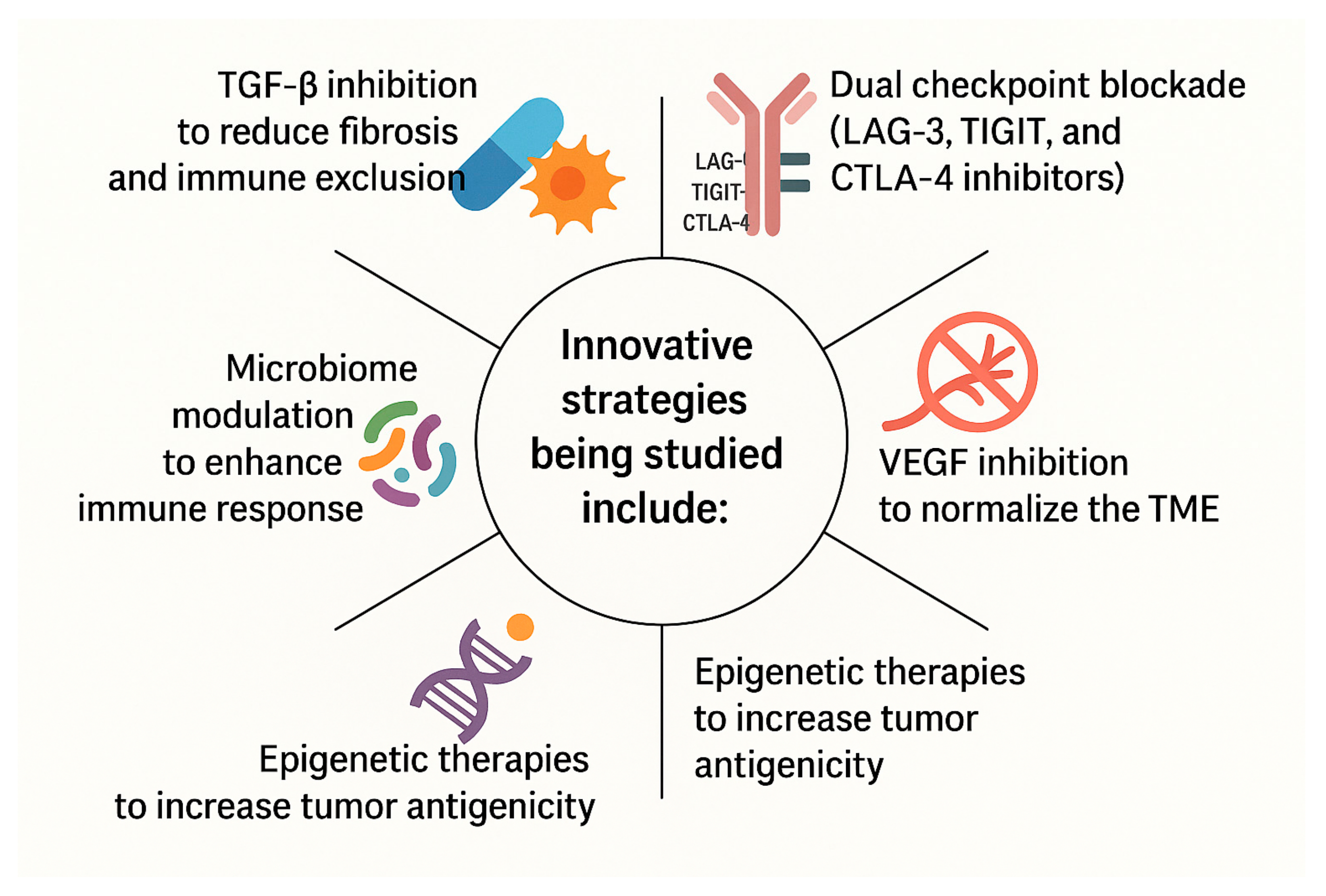

{kind=link}
{kind=link}
{kind=link}
| Trial Name | Phase | Therapy Evaluated | Patient Population/Key Findings |
|---|---|---|---|
| KEYNOTE-012 | 1b | Pembrolizumab monotherapy | PD-L1+ advanced/metastatic GC after chemotherapy; demonstrated clinical benefit with manageable toxicity. |
| KEYNOTE-059 | 2 | Pembrolizumab ± chemotherapy | Chemo-refractory or treatment-naïve advanced GC/GEJ cancer; ORR ~11.6%, higher in PD-L1+; durable responses in subsets. |
| ATTRACTION-2 | 3 | Nivolumab vs. placebo | Advanced GC post ≥ 2 chemotherapy lines; significant OS improvement; first ICI showing survival benefit in this setting. |
| ATTRACTION-4 | 2–3 | Nivolumab + oxaliplatin-based chemotherapy | 1st-line, HER2–unresectable/recurrent GC; improved PFS; OS benefit not significant; favorable long-term outcomes. |
| CheckMate-649 | 3 | Nivolumab + chemotherapy vs. chemo alone | 1st-line advanced GC/GEJ/esophageal adenocarcinoma; improved OS and PFS, especially in PD-L1 CPS ≥ 5; established new standard of care. |
| CheckMate-032 | 2 | Nivolumab ± ipilimumab | Heavily pretreated advanced GC/GEJ cancer; ORR up to 24% (NIVO1+IPI3); durable responses; comparable OS across arms. |
| Drug class | Name of Study | Phase | Article | Drug |
|---|---|---|---|---|
| ICI | GO30140 study | Phase Ib | Lee et al. [34] | Atezolizumab + bevacizumab (vs. sorafenib) |
| ICI | -- | Phase II | Sangro B, Gomez et al. [28] | Tremelimumab |
| ICI | CheckMate 040 | Phase II | El-Khoueiry et al.[29] | Nivolumab |
| ICI | Keynote 224 | Phase II | Zhu et al. [32] | Pembrolizumab |
| ICI | Himalaya trial | Phase III | Abou-Alfa et al. [38] | Tremelimumab + durvalumab |
| ICI | CheckMate 459 | Phase III | Yau et al. [30] | Nivolumab |
| ICI | Keynote 240 | Phase III | Finn, Ryoo et al. [41] | Pembrolizumab |
| ICI | IMbrave050 | Phase III | Qin et al. [36] | Atezolizumab + bevacizumab (vs active surveillance) |
| ICI | IMbrave150 | Phase III | Finn et al. [35] | Atezolizumab + bevacizumab (vs sorafenib) |
| ICI | Keynote-937 | Phase III | NCT03867084 | Pembrolizumab |
| ICI | EMERALD-2 | Phase III | NCT03847428 | Durvalumab +/- bevacizumab |
| ICI + TKI | Phase Ib | Bang et al. [39] | Durvalumab + remucirumab | |
| ICI + TKI | Phase I/II | NCT04212221 | MGD013 + brivanib |
| Trial Name | Combination Therapy | Target Mechanism |
|---|---|---|
| BREAKWATER | Encorafenib + Cetuximab ± Chemotherapy | BRAF V600E-targeted therapy |
| FRESCO-2 | Fruquintinib + Standard Care | Anti-angiogenesis |
| COMMIT [51] | Atezolizumab + Bevacizumab ± Chemotherapy | PD-L1 + VEGF blockade |
| RENMIN-215 [52] | Fecal Microbiota Transplantation + ICIs | Gut microbiome modulation |
| Zabadinostat + Nivolumab [53] | HDAC Inhibitor + PD-1 Blockade | Epigenetic modulation |
| Trial Name | Phase | Therapy Evaluated | Patient Population/Key Findings |
|---|---|---|---|
| KEYNOTE-158 | II | Pembrolizumab monotherapy | MSI-H/dMMR advanced PDAC; ORR 18.2%, median PFS 2.0 months, and median OS 4.0 months |
| Chemo4METPANC | II | Cemiplimab + Motixafortide + Gemcitabine/Nab-Paclitaxel | Metastatic PDAC; in a pilot study with 11 patients: 7 partial responses, 3 stable disease, 1 progression; promising early efficacy |
| Autogene Cevumeran | I | Personalized mRNA vaccine + Atezolizumab | Resected PDAC; among 16 patients, 8 responders showed no recurrence at 18 months; non-responders had a median recurrence-free survival of 13.4 months |
| IMM-101 Study | II | IMM-101 + Gemcitabine vs. Gemcitabine alone | Advanced PDAC; combination therapy was well-tolerated; suggested potential survival benefit over gemcitabine alone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peshin, S.; Bashir, F.; Kodali, N.A.; Dharia, A.; Zaiter, S.; Singal, S.; Moka, N. Immunotherapy in GI Cancers: Lessons from Key Trials and Future Clinical Applications. Antibodies 2025, 14, 58. https://doi.org/10.3390/antib14030058
Peshin S, Bashir F, Kodali NA, Dharia A, Zaiter S, Singal S, Moka N. Immunotherapy in GI Cancers: Lessons from Key Trials and Future Clinical Applications. Antibodies. 2025; 14(3):58. https://doi.org/10.3390/antib14030058
Chicago/Turabian StylePeshin, Supriya, Faizan Bashir, Naga Anvesh Kodali, Adit Dharia, Sajida Zaiter, Sakshi Singal, and Nagaishwarya Moka. 2025. "Immunotherapy in GI Cancers: Lessons from Key Trials and Future Clinical Applications" Antibodies 14, no. 3: 58. https://doi.org/10.3390/antib14030058
APA StylePeshin, S., Bashir, F., Kodali, N. A., Dharia, A., Zaiter, S., Singal, S., & Moka, N. (2025). Immunotherapy in GI Cancers: Lessons from Key Trials and Future Clinical Applications. Antibodies, 14(3), 58. https://doi.org/10.3390/antib14030058