1. Introduction
Human cytomegalovirus, a member of the
Herpesviridae family, infects a great majority of people worldwide; a recent meta-analysis estimated global IgG seroprevalence at ~83%, with prevalence varying markedly by region and socioeconomic status [
1,
2]. Although most immunocompetent individuals experience asymptomatic or mild infections, CMV can cause severe disease in fetuses, neonates, and immunocompromised individuals such as transplant recipients and patients with AIDS [
3,
4,
5]. In these high-risk groups, CMV activation often leads to life-threatening conditions, including pneumonia, retinitis, and systemic disease, underscoring the importance of enhanced diagnostic and therapeutic strategies.
The CMV genome encodes numerous proteins, among which pp71, encoded by the UL82 gene, has been recognized as a pivotal tegument protein that stimulates immediate-early (IE) gene expression and facilitates the initiation of lytic replication [
6,
7]. Furthermore, pp71 can disrupt host immune surveillance via the downregulation of MHC class I molecules [
8]. These functions make pp71 an attractive target for studying CMV pathogenesis and for developing potential diagnostic tools or antiviral strategies. However, despite its critical role in CMV infection, progress in diagnostics and basic research is hampered by the scarcity of commercially available monoclonal antibodies (mAbs) against pp71. Although leading academic groups in CMV research have provided pp71-specific mAbs as kind gifts for research purposes [
9,
10,
11], there remains a need to expand the repertoire of antibodies targeting this essential protein.
Monoclonal antibodies (mAbs) are integral to biomedical research and clinical diagnostics due to their high specificity for a single epitope [
12]. However, conventional hybridoma-based approaches for mAb generation are often time-consuming, labor-intensive, and dependent on animal immunization. In contrast, display technologies such as phage display, ribosome display, and yeast surface display enable the high-throughput screening and optimization of antibody fragments [
13]. Among these, yeast surface display offers notable advantages.
Saccharomyces cerevisiae provides a eukaryotic secretory apparatus that can promote oxidative protein folding and certain post-translational modifications (e.g., N-linked glycosylation), which may help in generating properly folded and functionally active antibody fragments [
14,
15]. Additionally, the isolation of high-affinity clones via fluorescence-activated cell sorting (FACS) is both quantitative and efficient.
Here, we report the isolation of a pp71-specific monoclonal antibody using a yeast-displayed single-chain variable fragment (scFv) library. We performed iterative selection rounds against recombinant pp71, followed by the characterization of the resulting clones using immunoassays (flow cytometry, immunofluorescence, ELISA, and BLI analysis). Our findings confirm the high specificity and affinity of the isolated mAb for pp71, thereby providing a novel tool for investigating CMV biology and facilitating the development of diagnostic and therapeutic approaches targeting this essential viral protein.
2. Materials and Methods
2.1. Construction of Random Human scFv Display Yeast
Peripheral blood mononuclear cells (PBMCs) from four healthy human donors were purchased from Lonza (#CC-2704). Total B cells were isolated from the PBMCs using part of an IgG+ Memory B Cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany, #130-094-350) and an AutoMACS Pro Separator (Miltenyi Biotec). The isolated total B cells were seeded at a density of 1.83 × 10
4–6.54 × 10
4 cells/mL in a 48-well plate and cultured for five days in 0.5 mL per well of ImmunoCult Human B Cell Expansion Kit medium (STEMCELL Technologies, Vancouver, BC, Canada, #ST-100-0645). The number of B cells used in this experiment is described in
Supplementary Table S1.
The expanded B cells were lysed in TRIzol™ Reagent (ThermoFisher Scientific, Waltham, MA, USA, #15596026), and RNA was extracted. Reverse transcription was performed using SuperScript™ IV Reverse Transcriptase (ThermoFisher Scientific, #18090010). The VH and VL regions were then amplified separately via PCR with cDNA, custom multiplex primers, and Platinum SuperFi II Green PCR Master Mix (ThermoFisher Scientific, #12369010). After gel extraction using NucleoSpin® Gel and PCR Clean-up (Takara, Shiga, Japan, #U0609C), random mutations were introduced into the amplicons using a Diversify PCR Random Mutagenesis Kit (Takara, #630703).
After gel extraction, to facilitate assembly, a complementary linker sequence for joining VH and VL was appended to each amplicon via a second PCR using custom primer sets and Q5 Hot Start High-Fidelity 2X Master Mix (NEB, Ipswich, MA, USA, #M0494L). These linker-tagged VH and VL fragments were gel-purified, mixed at equimolar ratios, and assembled into a full-length scFv using NEBuilder HiFi DNA Assembly Master Mix (NEB, #E2621L). After column purification was performed (Takara, #U0609C), adaptor sequences that overlapped with the pCTCON2 plasmid were added to the scFv amplicon via PCR, and then gel was extracted. The expression plasmid pCTCON2 was purchased from Addgene (Plasmid, Watertown, NY, USA, #41843) and cut using the restriction enzymes SalI (NEB, #R3138S), BamHI (NEB, # R3136S), and NheI (NEB, R3131S), and then gel was extracted. Finally, 6 μg of the cut pCTCON2 and 18 μg of the scFv amplicon were mixed and then electroporated into 1.6 × 10
9 AWY101 yeast cells, as previously reported [
16] (AWY101 was a kind gift from Eric V. Shusta, University of Wisconsin–Madison). Through homologous recombination in
S. cerevisiae, the scFv insert was integrated immediately downstream of the bidirectional GAL1/GAL10 promoter in the pCTCON2 vector, enabling the galactose-inducible expression of the Aga2–scFv fusion protein. The electroporated yeasts were cultured in SDCAA medium (20 g/L dextrose, 6.7 g/L Difco yeast nitrogen base w/o amino acids (BD, Franklin Lakes, NJ, USA, #291940), 5 g/L acid hydrolysate of casein (BIOKAR Diagnostics, Paris, France, #A1404HA), 5.4 g/L Na
2HPO
4, and 9.68 g/L NaH
2PO
4·2H
2O) for two days. After washing the yeasts once with SGDCAA medium (2 g/L dextrose, 20 g/L galactose, 6.7 g/L Difco yeast nitrogen base w/o amino acids, 5 g/L acid hydrolysate of casein, 5.4 g/L Na
2HPO
4, and 9.68 g/L NaH
2PO
4·2H
2O), they were cultured in SGDCAA at 20 °C for two days for the induction of surface antibodies. All SDCAA and SGDCAA media in this study contained penicillin–streptomycin (Wako, Osaka, Japan, #168-23191). The custom multiplex primer sequences used in this study are available from the corresponding author upon reasonable request.
2.2. Pp71 Expression and Purification
A DNA encoding Flag-pp71-His-SBP (where SBP denotes the streptavidin-binding peptide tag) was synthesized by Twist Bioscience (South San Francisco, CA, USA) and cloned into pcDNA3.1 (ThermoFisher Scientific, #V79020). The plasmid was transfected into Expi293F cells according to the manufacturer’s protocol (ThermoFisher Scientific, #A14635). After 7 days of culture, the cells were harvested and washed twice with PBS. The cell pellet was lysed in ice-cold lysis buffer (50 mM phosphate buffer, pH 7.5, 300 mM NaCl, 20 mM imidazole, 0.1% Igepal CO-630, 5 mM MgSO
4, and 1x Pierce Protease Inhibitor Tablet, EDTA-free (ThermoFisher Scientific, #A32965)). The lysate was incubated on ice for 30 min with occasional mixing, followed by centrifugation at 17,000×
g for 15 min at 4 °C. The supernatant was incubated overnight at 4 °C with Ni-NTA agarose resin (ThermoFisher Scientific, #88221) under gentle rotation. The resin was washed with washing buffer (50 mM phosphate buffer, pH 7.5, 300 mM NaCl, 50 mM imidazole) three times. The bound protein was eluted with elution buffer (50 mM phosphate buffer, pH 7.5, 300 mM NaCl, 250 mM imidazole). The eluate was concentrated and buffer-exchanged to PBS using Amicon Ultra-15 centrifugal filter units with a 10 kDa molecular weight cutoff (Merck, Darmstadt, Germany, #UFC901024). The purity of Flag-pp71-His-SBP was validated using SDS-PAGE, followed by CBB staining (
Supplementary Figure S1). The concentration of the purified Flag-pp71-His-SBP protein was determined using a DeNovix DS-11+ spectrophotometer (Wilmington, NC, USA).
2.3. MACS
The optical density (OD600) of the yeast culture was measured, and, assuming that an OD600 of 1 corresponds to ~1 × 107 cells/mL, cells equivalent to 2 × 109 yeast cells were washed once with rinsing buffer (Miltenyi Biotec, AutoMACS Rinsing Solution #130-091-222 supplemented with MACS® BSA Stock Solution #130-091-376). The yeasts were incubated with 4.6 μg of Flag-pp71-His-SBP at room temperature for one hour. After washing the yeasts twice with rinsing buffer, they were mixed with Anti-His-tag mAb-Alexa Fluor 647 (MBL, Tokyo, Japan, #D291-A64) and incubated for 30 min at 4 °C with rotation. Then, the yeasts were washed twice using the rinsing buffer and mixed with Anti-Cy5/Anti-Alexa Fluor 647 MicroBeads (Miltenyi Biotec, #130-091-395). After 15 min of rotation at 4 °C, the yeasts were washed once with rinsing buffer, and the magnetic separation Posseld program was performed using AutoMACS pro. The positive fraction was washed once with SDCAA medium and then cultured in SDCAA medium for one or two days at 30 °C, depending on the concentration of the yeasts. Following yeast expansion, the cells were washed once with SGDCAA medium and cultured in SGDCAA medium for two or three days at 20 °C. This screening process was repeated, with reagents alternated in each subsequent round. Specifically, the Anti-His-tag mAb-Alexa Fluor 647 and Anti-Cy5/Anti-Alexa Fluor 647 MicroBeads were replaced with FITC anti-His-Tag Antibody (BioLegend, San Diego, CA, USA, #362618) and Anti-FITC MicroBeads (Miltenyi Biotec, #130-048-701), respectively. MACS separation was performed up to round 5. From round 2 onward, 2.3 μg of Flag-pp71-His-SBP was used instead of 4.6 μg to enrich for higher-affinity clones.
2.4. Flow Cytometry Analysis of scFv-Expressing Yeasts
To evaluate pp71 binding to scFv-expressing yeasts, approximately 1 × 106 yeast cells (based on OD600 measurement) were washed once with rinsing buffer and then incubated with 0.1 μg of Flag-pp71-His-SBP for 30 min at 4 °C. After washing twice with rinsing buffer, the yeasts were incubated with FITC-streptavidin (BioLegend, #405202, 1:200) to detect the SBP tag on pp71 bound to the yeast surface and with Anti-Myc-tag mAb-Alexa Fluor 647 (MBL, #M047-A64, 1:100) to visualize the C-terminal Myc epitope of the surface-displayed scFv. After washing three times with rinsing buffer, the yeasts were analyzed using a FACSCantoII analyzer (BD Biosciences, San Jose, CA, USA) and FlowJo v10.
2.5. scFv-Fc Expression and Purification
Round 5 yeasts were spread on an SDCAA plate, and single colonies were picked up and cultured in SDCAA medium followed by the induction of the scFv in SGDCAA medium to assess Flag-pp71-His-SBP binding via flow cytometry. After the flow cytometry analysis, we isolated plasmid DNA using a Zymoprep Yeast Plasmid Miniprep Kit II.
In parallel, DNA coding a secretion signal peptide and the BALB/c mouse IgG1 Fc region sequence was synthesized by Twist Bioscience and cloned into pHEK293 Ultra Expression Vector I (Takara, #3390). This vector backbone was then PCR-linearized using PrimeSTAR Max DNA Polymerase Ver. 2 (Takara, #R047B) with primers designed to generate ends matching the signal peptide sequence at one terminus and the GS-linker–Fc junction at the other. The scFv region was PCR-amplified from the yeast plasmids with HA- and Myc-specific primers. The HA primer overlapped the signal peptide region, and the Myc primer overlapped the GS-linker–Fc region. Finally, the HA-scFv-Myc insert and PCR-linearized backbone were assembled using NEBuilder HiFi DNA Assembly Master Mix, yielding the final HA-scFv-Myc–Fc expression construct. Following assembly, the HA-scFv-Myc–Fc construct was transformed into E. coli (STBL3), and plasmid DNA was isolated using miniprep. Clone identity was confirmed via Sanger sequencing using vector-specific primers annealing to the pCTCON2 backbone and an internal primer annealing to the Fc region.
The plasmids were transfected into Expi293F cells according to the manufacturer’s protocol. One week after the transfection, the supernatant was filtered using Millex-HP 0.45 μm (Merck, #SLHPR33RS) and then incubated with Ab-Capcher beads (ProteNova, Kagawa, Japan, #P-002-10) at 4 °C overnight. The beads were washed with PBS three times, then eluted with Glycine-HCl pH 2.8 three times, and immediately neutralized with 1M Tris-HCl pH 8.0. Buffer was exchanged to PBS using Amicon Ultra-15 centrifugal filter units with a 10 kDa molecular weight cutoff (Merck, #UFC901024). The scFv-Fcs concentrations were quantified using a DeNovix DS-11+ spectrophotometer.
The scFv-Fc sequences used in this study are available from the corresponding author upon reasonable request.
2.6. Flow Cytometry Analysis Using scFv-Fcs
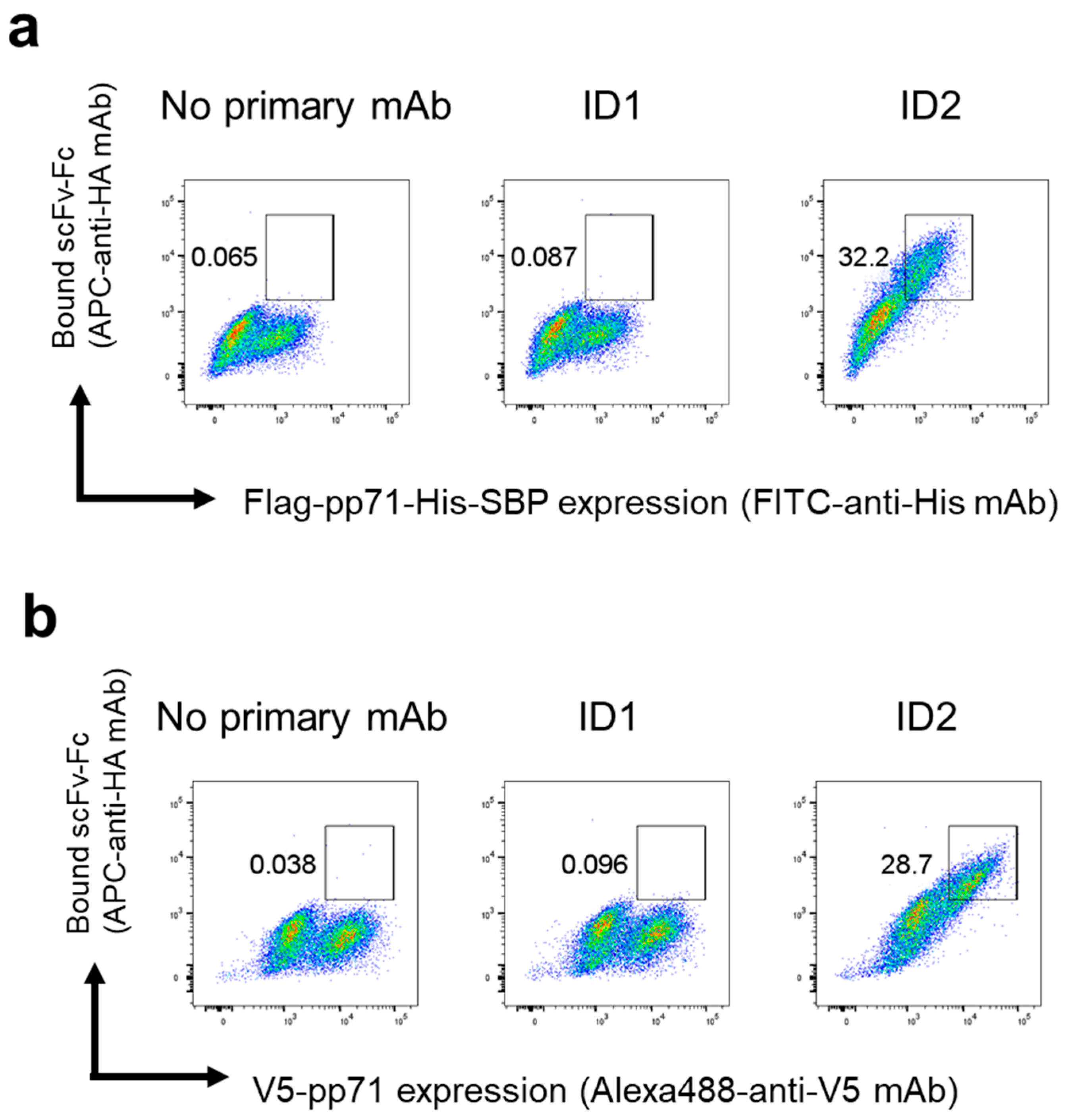
pp71-Flag-His or pp71-V5 plasmids were transfected into HEK293FT cells using PEI max. Two days after transfection, the cells were fixed and permeabilized with a FOXP3 Fix/Perm Buffer Set (BioLegend, #421403). The cells were incubated with the ID1 or ID2 scFv-Fc (1.3 μg/mL) and then stained with FITC anti-His tag antibody (BioLegend, #362618, 1:100) or Alexa Fluor 488 V5 Tag Monoclonal Antibody (Invitrogen, #37-7500-A488, 1:200) and APC anti-HA.11 tag antibody (BioLegend, #901524, 1:100). The cells were analyzed with a FACSCantoII analyzer (BD Biosciences).
2.7. Sandwich ELISA
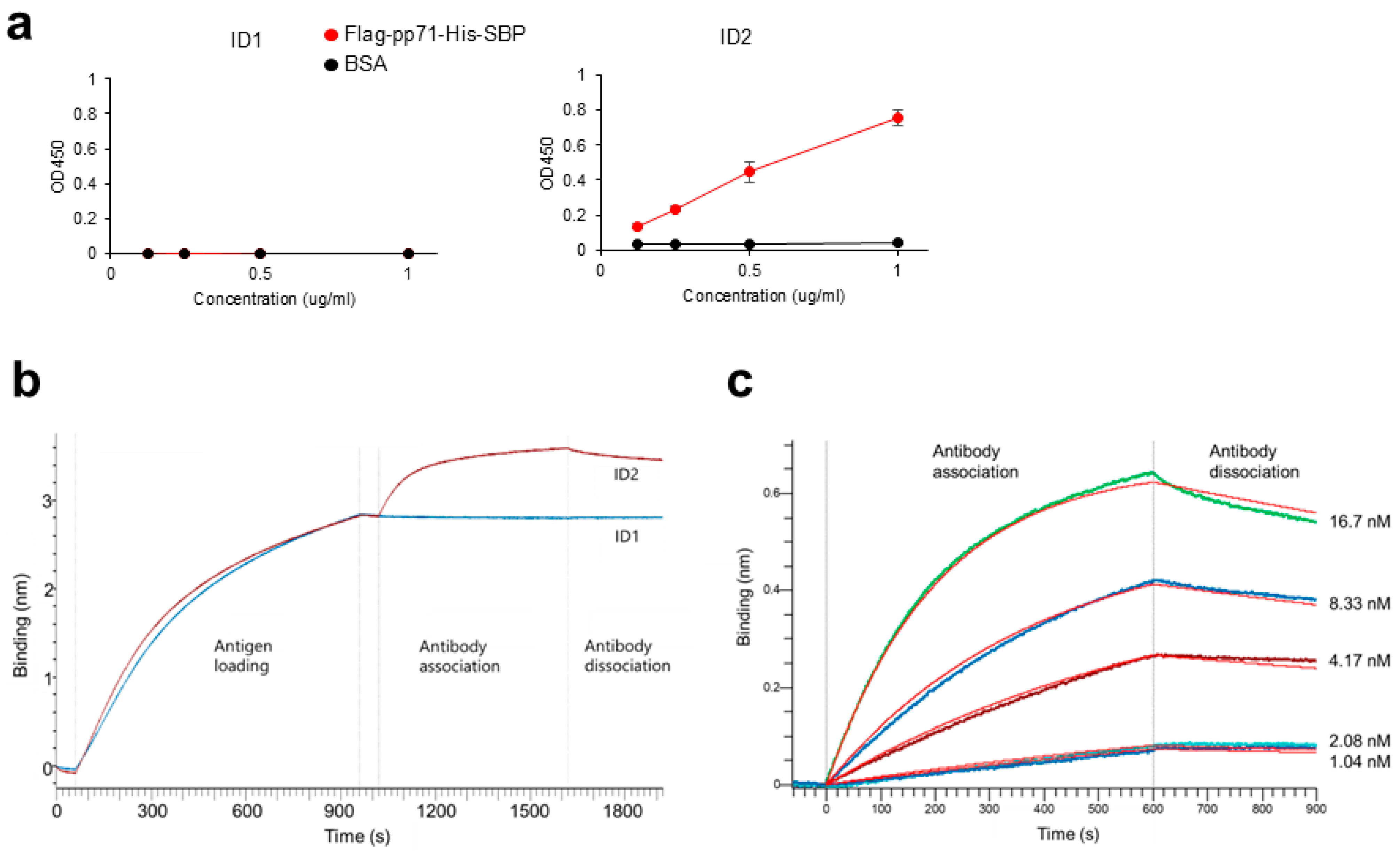
Anti-DYKDDDDK tag antibody (1 μg/mL; Clone L5; BioLegend, #637319) was precoated on MaxiSorp 96-well plates (ThermoFisher Scientific, #442404) at 4 °C overnight. The plates were washed with PBS/0.05% Tween20 and blocked with 10% FBS/PBS at room temperature for 1 h. After washing with PBS/0.05% Tween20, 0.125, 0.25, 0.5, and 1 μg/mL of Flag-pp71-His-SBP or BSA (Globulin-free; NACALAI TESQUE, Kyoto, Japan, #01281-26) in triplicate, the wells were incubated at room temperature for 2 h. After washing with PBS/0.05% Tween20, the wells were incubated with 5 μg/mL scFv-Fc (ID 1 or ID2) at room temperature for 1 h, followed by HRP-conjugated goat anti-mouse IgG (Clone Poly4053; 1:5000 dilution; BioLegend, #405306) at room temperature for 1 h. ELISA POD Substrate TMB solution (Easy) (NACALAI TESQUE, #05299-54) was added and stopped with 1 N HCl (NACALAI TESQUE, #18322-95). The absorbance at a dual wavelength of 450/595 nm was measured using a microplate reader, ARVO X3 (PerkinElmer, Shelton, CT, USA).
2.8. BLI Analysis
BLI binding assays were performed in 96-well black microplates (Sigma Aldrich, St. Louis, MO, USA, #781608) at 30 °C with shaking at 1000 rpm using an Octet R4 instrument (Sartorius, Göttingen, Germany) and anti-penta-His (HIS1K) biosensors (Sartorius, #18-5120). Flag-pp71-His-SBP was diluted to 5 µg/mL in assay buffer (PBS supplemented with 0.005% Tween20 and 0.02% BSA). ID1 and ID2 scFv-Fc were diluted to 66.7 nM (6.4 µg/mL) in assay buffer. For the ID2 antibody, 2-fold serial dilutions (1.04–16.7 nM) were prepared to determine the equilibrium dissociation constant.
Binding experiments consisted of the following steps: First Baseline, assay buffer for 60 s; Loading, 5 µg/mL His-tagged pp71 for 900 s; Second Baseline, assay buffer for 60 s; Association, analyte (antibodies) for 600 s; and Dissociation, assay buffer for 300 s. The resulting association curve (0–600 s) and disassociation curve (0–300 s) were analyzed using the Octet Analysis Studio 12.2.2.26 software (Sartorius). The data were fitted with a 1:1 binding model to obtain KD.
2.9. Immunofluorescence
Flag-pp71-His-SBP or V5-pp71 plasmids were transfected into HEK293FT cells using PEI max. The following day, the cells were seeded onto coverglasses in 6-well culture plates. Three days post-transfection, the cells attached to the coverglasses were fixed with 4% paraformaldehyde for 20 min at room temperature, followed by treatment with 1% Igepal CO-630 in PBS for 10 min at room temperature. The cells were then blocked with 3% BSA in PBS for 1 h at 37 °C. Subsequently, the cells were incubated with ID2 scFv-Fc (0.5 μg in 150 μL of 3% BSA in PBS) for 1 h at 37 °C. For the cells transfected with the Flag-pp71-His-SBP plasmid, staining was performed with Anti-His-tag mAb-Alexa Fluor 594 (MBL, #D291-A59, 1:250) and Alexa Fluor 488 anti-HA.11 Epitope Tag Antibody (BioLegend, #901509, 1:500). For the cells transfected with the V5-pp71 plasmid, staining was carried out with Alexa Fluor 594–anti-HA tag antibody (MBL, #M180-A59, 1:100) and Alexa Fluor 488 V5 Tag Monoclonal Antibody (ThermoFisher Scientific, #37-7500-A488, 1:1500) for 1 h at 37 °C. The coverglasses were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (VECTOR LABORATORIES, Newark, NJ, USA, #H-1200) and observed using a ZEISS LSM980 confocal microscope. Images were acquired and processed using the ZEN 3 software (Zeiss, Oberkochen, Germany).
4. Discussion
This study reaffirms that a combination of MACS and yeast display technology provides a rapid, low-cost route to obtain high-affinity antibodies [
17]. From a human scFv-expressing yeast library, we isolated ID2, which recognizes cytomegalovirus pp71 with sub-nanomolar affinity and performs reliably in immunofluorescence and ELISA assays. Because the relocalization of pp71 is a pivotal trigger of CMV replication [
6], ID2 offers a practical tool for dissecting the spatial regulation of the viral life-cycle.
Clinically, pp71 is readily detected by immunohistochemistry in normal prostate epithelium, prostate tumors, and glioblastoma specimen [
23,
24], highlighting the potential of ID2 for immunohistological analysis of biopsies.
In this study, only twelve individual scFv clones were tested in detail; deep sequencing of the post-selection yeast population would allow for a quantitative view of clonal convergence and might reveal additional anti-pp71 clones that were not recovered in single-colony screening.
Although ID2 itself is intended purely as a research reagent, its discovery highlights the broader translational capability of yeast display. A fully human anti-PD-1 antibody discovered using yeast display technology—sintilimab—received regulatory approval in China in 2018, demonstrating that binders isolated in yeast can meet clinical developability standards without the humanization step demanded by mouse-derived leads [
25].
Yeast-derived human scFvs are also advancing into cell therapy: the fully human BCMA-specific CAR CT103A, built with a yeast-selected scFv, achieved a 100% overall response rate and 72% complete/strict complete responses in a phase 1 trial of relapsed/refractory multiple-myeloma patients, with toxicity confined mainly to grade 1–2 cytokine-release syndrome [
26].
Together, these examples emphasize that the MACS-plus-yeast display workflow can deliver fully human, high-affinity antibodies that are readily adapted to modern therapeutic formats yet remain accessible to routine academic laboratories. At the same time, its performance is shaped by characteristic strengths and caveats.
Each
S. cerevisiae cell presents about 3 × 10
4 scFv molecules on its surface; during magnetic enrichment, this multivalency can capture clones whose intrinsic affinity is weak. A FACS step that simultaneously measures antigen binding and display level removes such avidity-driven binders [
13,
17,
27].
Another limitation of yeast display is library size: yeast display libraries are constrained by transformation efficiencies, commonly achieving practical library diversities of 10
8 to 10
9 transformants, considerably smaller than the theoretical diversities attainable with other display technologies such as phage display (10
10 to 10
12) and ribosome display (10
12 to 10
15) [
13,
28]
These limitations are balanced by notable advantages. As a eukaryotic host,
S. cerevisiae supports correct disulfide formation and secretion-quality control, allowing complex antibody fragments to fold properly on the cell surface—an edge over bacterial display systems [
14,
15]. The platform also enables quantitative, flow-cytometric screening: co-labelling antigen binding and antibody expression permits selection of high-affinity variants normalized for display level [
29]. When still greater diversity is required, the gap can be bridged by a hybrid workflow in which a very large phage library is first panned and the enriched pool is subsequently transferred into yeast for FACS-based enrichment [
30].
Recognizing both the constraints and unique benefits of the platform will guide its future application to increasingly demanding antiviral and anticancer targets.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}