1. Introduction
The market for biopharmaceuticals has grown exponentially over the past 2–3 decades, leading to the treatment and prevention of a vast range of diseases and disorders. In parallel, more sophisticated biopharmaceuticals are emerging on the market, including antibody drug conjugates and cell and gene therapies. This growth in biologics is, in part, due to their continued demonstration of high specificity, thereby enabling more targeted actions with fewer and/or less severe adverse events. Moreover, the efficiency of manufacturing biopharmaceuticals has improved greatly over the years, which has ensured more streamlined development, consistent products, and decreased cost.
Nonetheless, manufacturing and control challenges persist, even for simple biopharmaceuticals. For example, protein-based biotherapeutics are prone to numerous chemical and biochemical post-translational modifications (PTMs) during manufacturing, processing, and storage. These PTMs—including glycosylation, glycation, oxidation, and deamidation—can impact the stability, potency, pharmacokinetics, and/or immunogenicity of the product, thereby compromising clinical efficacy and safety [
1].
Asparagine deamidation is one PTM that converts asparagine residues into iso-aspartate and/or aspartate [
2,
3]. The converted aspartate and iso-aspartate residues are largely of the L-isomer; however, D-isomers have been detected at low levels following deamidation [
4]. Deamidation can occur both enzymatically [
5] and non-enzymatically [
6,
7,
8]. The susceptibility and rate of non-enzymatic deamidation is dependent on many variables, including pH, temperature, solvent/buffer and excipients, and physical state (i.e., liquid vs. lyophilized) [
2,
9]. Protein primary structure, as well as secondary and tertiary structure, also influence the susceptibility and rate of protein deamidation [
10,
11,
12,
13].
Deamidation has a high potential to impact protein structure and function, as it induces a change in residue charge, hydrophobicity, and mass. In nature, deamidation is ubiquitously and frequently observed, leading some to hypothesize that endogenous deamidation is a mechanism for regulating physiological processes. For instance, some claim that non-enzymatic deamidation regulates protein turnover, thereby acting as a biomolecular clock, in accordance with deamidation rates, to regulate organism development and aging [
14,
15]. Others report deamidation as a switch for apoptosis following DNA damage [
16], or even a tool for pathogens to evade the innate immune system [
17].
As in nature, deamidation is observed frequently in protein-based therapeutics. Depending on the location of the deamidated site, this PTM has the potential to significantly impact the stability, potency, pharmacokinetics, and/or clinical safety of biotherapeutics. For example, Lu et al. report that deamidation of the antibody variable fragment (Fv) of Moxetumomab pasudotox results in impaired target binding, internalization, and delivery of cytotoxic payload to cancer cells [
18]. As another example, deamidation in the crystallizable fragment (Fc) region of an anonymized therapeutic antibody was reported to decrease Fc effector functions through decreased FcyRIIIa binding [
19]. Hence, deamidation is routinely monitored as a potential critical quality attribute (pCQA) in the biopharmaceutical industry.
CQA evaluations for deamidation usually begin with the identification of relevant amino acid sequence liabilities, as, again, the primary structure is known to influence deamidation susceptibility. The amino acid in the n + 1 position is of particular interest when identifying liabilities for asparagine deamidation. Amino acid motifs NG, NS, NN, NG, and NH are commonly found within the complementarity-determining region (CDR) of antibodies and are considered canonical for deamidation, in which NG is more labile than NS, and NS more labile than NT or NN and NH. On the other hand, the common CDR motifs NY, NW, NQ, and NF are considered non-canonical, with a low risk for deamidation [
20].
ADC-A is an anonymized antibody drug conjugate (ADC) that was under recent development for cancer indication. ADC-A is composed of a monoclonal antibody (mAb) intermediate conjugated to a cytotoxic small molecule warhead via a chemical linker. The cytotoxic warhead is specifically conjugated at an inserted cysteine site in the hinge region of each mAb intermediate heavy chain (HC) [
21,
22], which allows tight control of a targeted drug-to-antibody ratio (DAR) of two. The primary mechanism of action of ADC-A is warhead-induced cancer cell death, which is initiated via the ADC-A binding to its target antigen, expressed on the surface of cancer cells. Once bound, the ADC-A-antigen complex is internalized within the cancer cell and degraded through lysosomal activity, thereby freeing the cytotoxic warhead to induce cell death. The specific warhead of ADC-A elicits cytotoxicity by binding and crosslinking DNA, thereby inhibiting transcription and replication processes.
The initial analysis of sequence liabilities for this mAb intermediate identified an N102–W103 motif in the CDR. Observing the NW motif itself was not surprising nor a concern, as again NW is found frequently within the CDR of antibodies, but demonstrates low risk for deamidation [
12,
13,
20]. In fact, in one study that evaluated over 100 clinical-stage therapeutic antibodies, NW was identified as the second most commonly occurring asparagine motif within CDRs but demonstrated the lowest rate of deamidation among all canonical and non-canonical motifs [
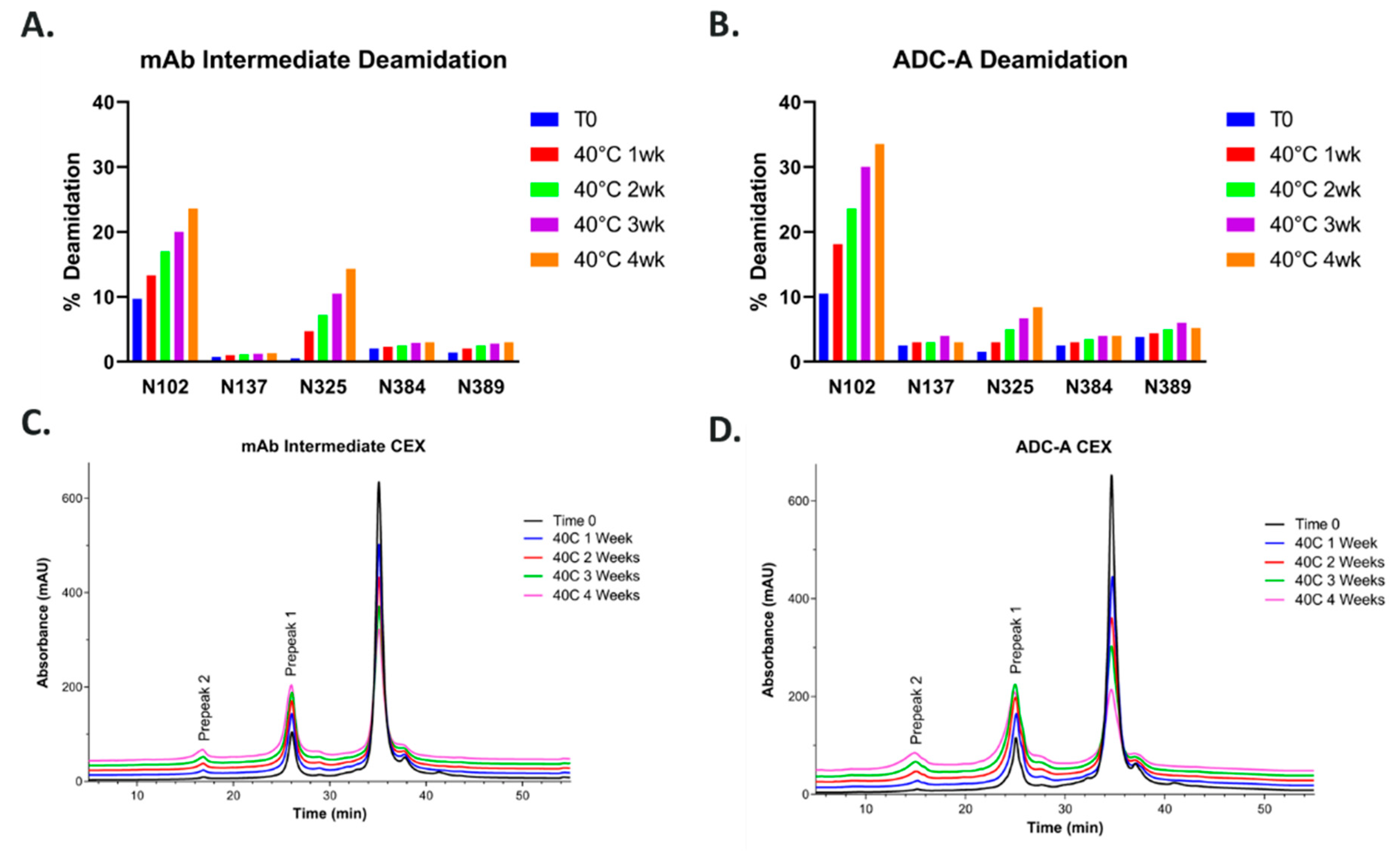
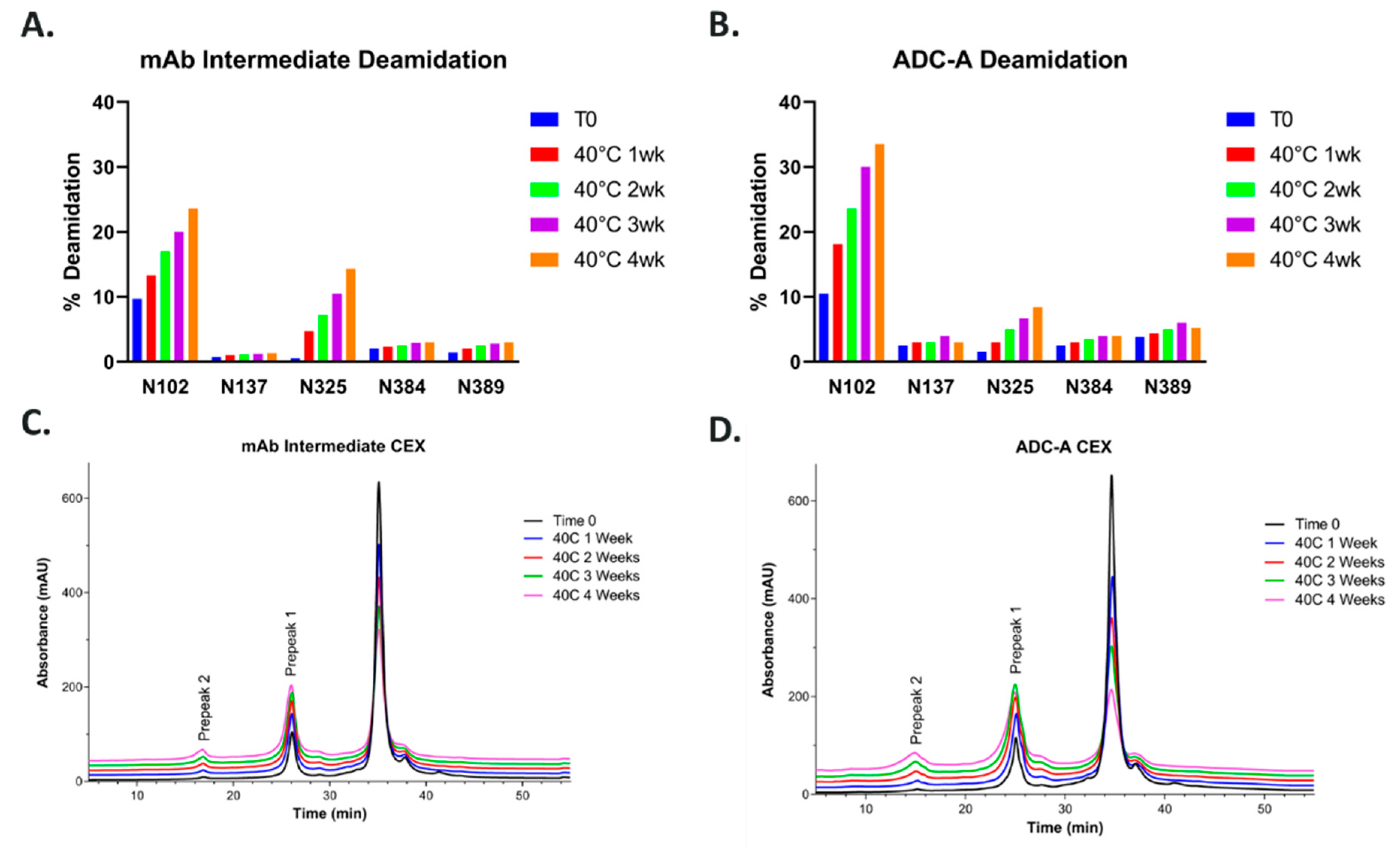
20]. Therefore, N102–W103 was not deemed a sequence liability. However, during later forced-degradation/CQA evaluation studies, ADC-A and its mAb intermediate were found to readily deamidate at N102. Moreover, N102 deamidation was observed not only under forced degradation conditions, but also during normal manufacturing process conditions, resulting in up to approximately 15–20% of mAb intermediate product being deamidated at this residue.
Further investigations of mAb intermediate/ADC-A deamidation revealed that N102 deamidation converts the asparagine residue into nearly 100% iso-aspartate. This finding is strikingly different from previous reports, where a mixture of aspartate and iso-aspartate is typically observed following deamidation through a succinimide intermediate [
2,
3]. Regarding biological activity, N102 deamidation significantly attenuates the binding affinity of the mAb intermediate to the target antigen. This impact on target binding persists after payload conjugation, which consequentially impairs ADC-A’s ability to internalize into tumor cells and induce a cytotoxic effect in vitro. Interestingly, the biological activity of ADC-A is more severely reduced when N102 is deamidated into isoD102, as compared to when N102 is mutated to N102D. Here, we report the discovery of this atypical NW deamidation, characterize its impact on biological activity, and use structural data and modeling to explain the differential impact of isoD102 versus N102D on activity.
2. Materials and Methods
2.1. Reagents and Materials
Engineered antibody intermediate and antibody drug conjugates were produced by AstraZeneca (Gaithersburg, MD, USA). Synthetic peptides were obtained from Tufts University (Boston, MA, USA). HepG2 and T47D cells were obtained from ATCC (Gaithersburg, MD, USA). Dulbecco’s Modified Eagle Medium (DMEM), Advanced DMEM, MEM non-essential amino acids (NEAA), and GlutaMax™ were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Ammonium sulfate, sodium acetate N-ethylmaleimide, sodium phosphate dibasic, sodium phosphate monobasic monohydrate, sodium chloride, formic acid, trifluoroacetic acid (TFA), Fetal Bovine Serum (FBS), and 10× Phosphate Buffered Saline (PBS) pH 7.4 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Urea (OmniPur), water (OmniSolv, HPLC and spectrophotometry grade), acetonitrile (OmniSolv, HPLC and spectrophotometry grade), and isopropyl alcohol (OmniSolv, HPLC and spectrophotometry grade) were obtained from EMD Serono (Rockland, MA, USA). LysC and CellTiter-Glo® were obtained from Promega (Madison, WI, USA). Dithiothreitol (no-weight format) was obtained from Pierce Protein Biology (Waltham, MA, USA). All 96-well microplates were obtained from VWR (Radnor, PA, USA). DELFIA® Eu-N1 anti-human IgG, DELFIA® assay buffer, DELFIA® wash buffer, and DELFIA® enhancement solution were obtained from PerkinElmer (Waltham, MA, USA). Incucyte® Human Fabfluor-pH Red antibody labeling dye was obtained from Sartorious (Göttingen, Germany).
2.2. Purification of Non-Deamidated and Single-Deamidated Species
ADC-A contained a mixture of double deamidated, single deamidated, and non-deamidated species. Separation of these materials was performed using Source 15S cation exchange resin (Cytiva, Marlborough, MA, USA). The resin was packed into a 2.2 cm × 20 cm Vantage Column (MilliporeSigma, Burlington, MA, USA) which was operated at 150 cm/h on an Akta Avant (Cytiva, Marlborough, MA, USA). Prior to separation, the resin was sanitized with 1 N sodium hydroxide and then equilibrated with 25 mM acetate pH 5.0. ADC-A was applied to the equilibrated resin at 20 g/L. The loaded resin was re-equilibrated prior to a 20 column-volume gradient elution to 25 mM acetate and 500 mM sodium chloride, at pH 5.0. Fractions were collected during elution and analyzed for protein concentration, purity, and deamidation. As expected, the material eluted from the column according to charge, with double deamidated species eluting prior to the single and non-deamidated materials. There were 60 mg of single deamidated product and 300 mg of non-deamidated product obtained, both with monomeric purities greater than 99.5%. All chemicals were JTBaker USP grade (VWR, Radnor, PA, USA).
2.3. Non-Deamidated, Deamidated, and N102D Mutant mAb Intermediate Conformations Were Compared by Hydrogen–Deuterium Exchange (HDX)
Deamidated (isoD-form) mAb intermediate, collected by fractionation, and N102D mutant (D-form) were compared with the non-deamidated protein (N-form), using HDX, to investigate the protein conformational changes caused by deamidation or mutation around the PTM/mutation site or globally. The HDX experimental procedure was fully automated and conducted by Waters HDX Manager (Waters, Milford, MA, USA) with the temperature-controlled LEAP liquid handling system (Trajan, Morrisville, NC, USA). Two HDX experiments were performed: non-deamidated (N form) vs. deamidated (isoD-form), and non-deamidated (N form) vs. N102D mutant (D form). To initiate HDX, 3 µL of the sample, at approximately 10 mg/mL, was diluted tenfold with 50 mM sodium phosphate buffer at pH 7.0 in D2O. The exchange reaction was temperature controlled at 24 °C for 0.5, 1.5, 5, 15, and 60 min, in triplicates. The exchange reaction was quenched by adding the same volume of pre-chilled acidic quenching buffer, composed of 200 mM glycine, 8 M guanidine HCl, and 500 mM TCEP at pH 2.5 under 0 °C. The quenched samples were then diluted fourfold with 0.1% formic acid (pH 2.5, 0 °C) and injected onto the digestion column inside the Waters HDX manager with the cooling chamber at 0 °C. The samples were on-column digested by passing through a column with immobilized pepsin resin (Waters, 2.1 × 30 mm). The digested peptides were desalted on a VanGuard precolumn (Waters, 2.1 × 5 mm) for 2.5 min at a flow rate of 100 μL/min, and subsequently separated in an analytical column (Waters Acquity BEH300 column, 1.0 × 100 mm) at a flow rate of 40 μL/min. The automation method was created and controlled by Chronos software version 2.1 (Trajan Automation, Morrisville, NC, USA). Experiments were undertaken in triplicates for each time point.
2.4. Liquid Chromatography–Mass Spectrometry (LC–MS) Analysis of HDX Samples
The digested peptides were separated with a 6.8 min gradient of 15% to 28% Solvent B (Solvent B: acetonitrile with 0.1% formic acid; Solvent A, H2O with 0.1% formic acid). For N-form vs. isoD-form comparison, data were acquired on a QExactive HF-X mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA); and for the N-form vs. D-form experiment, the data were collected on Eclipse (Thermo Fisher Scientific, Waltham, MA, USA). Peptide digestion without D2O was undertaken on QE-HF and Eclipse, with the precursor ions mass range at 300–2000 m/z for all samples, with a 120,000 resolving power. From each MS scan, the top ten most abundant precursors were selected for fragmentation by collision-induced dissociation (CID) in a data-dependent acquisition (DDA) mode, with a fixed collisional energy of 27%. Deuterium labeling data were collected similarly without MS/MS scans. Xcalibur 2.2 (Waltham, MA, USA) was employed for data acquisition.
2.5. HDX Data Analysis
MS data in the digestion runs were analyzed by Byos to identified all the peptides. (Protein Metrics, San Carlos, CA, USA). The deconvoluted peptide masses were searched against the non-specific digestion of amino acid sequences of the mAb intermediate or the mAb intermediate with N102D. The search tolerance window was set to 10 ppm mass error for precursor ions and 50 ppm mass error for product ions. The identified peptide list was imported into HD Examiner software version 2.1 (Sierra Analytics, Modesto, CA, USA) to calculate the numbers of deuterium uptake (D-uptake) of each peptide at each time point and later compared between the N-form vs. isoD-form and N-form vs. D-form, to make the HDX differential plots.
Homology models for the N-form, isoD-form, and D-form were generated using an antibody modeler by Molecular Operating Environment (MOE). The MOE modeler in this study used Protein Data Bank (PDB) to build the homology models. In Fab homology modeling, mAb intermediate N-form and D-form sequences were searched against the database and templates with the highest similarity for the framework region sequences and CDR loop sequences were identified. The modeler then grafted CDR loop templates onto the light chain or heavy chain frameworks. The transition areas between CDRs and frameworks were fine-tuned by energy minimization using an AMBER99 force field. HDX data were visualized by the HDX module of MOE. The color code scheme “Jet” was applied to illustrate the D-uptake differences between the N-, isoD- and the D-forms.
2.6. Analytical CEX
The identity of each fraction was verified by analytical scale CEX analysis. Briefly, 75 μL of each sample and Reference Standard diluted to 1.0 mg/mL in mobile phase A were injected onto a Dionex Propac™ WCX-10 ion exchange column (4 × 250 mm). The mobile phases used were 20 mM sodium phosphate at pH 7.0 for mobile phase A and 20 mM sodium acetate containing 250 mM sodium chloride at pH 7.0 for mobile phase B. A gradient of 35.0 to 95.0% mobile phase B was run from 5 to 55 min at a flow rate of 1 mL/min. The eluted protein was detected using UV absorbance at 220 nm.
2.7. DAR Measurement by RP-HPLC
The ADC-A or mAb intermediate samples were made to 2 mg/mL in HPLC water. To each sample with 50 μL (100 μg), 50 μL denaturing buffer (8 M guanidine HCl, 160 mM Tris, 1 mM EDTA, pH 7.6) and 2 μL of 500 mM DTT (dithiothreitol) were added and the mixture was incubated at 37 °C for 30 min. From each sample, 10 μL was loaded onto a Waters BioResolve RP mAb polyphenyl column (2.1 × 150 mm, 2.7 µm, 450Å). Mobile phase A and B consisted of 0.1% TFA in water and 0.1% TFA in acetonitrile, respectively. A chromatography method with a linear gradient of 32.5% to 46.5% mobile phase B from 2 to 30 min at a flow rate of 0.5 mL/min was adopted, with the eluted protein detected at a UV wavelength of 280 nm. DAR and drug-load distribution were calculated based on peak areas.
2.8. Reduced Peptide Mapping
Ten µL of protein samples at 10 mg/mL were denatured, reduced, and alkylated. The denaturation and reduction were performed using 20 µL denaturing buffer (7.9 M Guanidine-HCl, 100 mM Tris, 0.1 mM EDTA, pH 7.3–7.4) and 2 µL DTT at 500 mM, incubated at 37 °C for 30 min. The alkylation was conducted by adding 5 µL 500 mM IAM (iodoacetamide) and incubated at room temperature for 30 min in the dark. Samples (volume ~37 µL) were then transferred to a Pierce™ 96-well Microdialysis Plate, 10 K MWCO (Thermo, Part # 88260), which were pre-conditioned with 1400 µL dialysis buffer (6 M urea, 150 mM Tris, pH 7.5). Dialysis was performed using a mixer at 500 rpm for 2 h, at room temperature in dark. After dialysis, the protein solution was recovered to Eppendorf tubes and 70 µL of digestion buffer (100 mM Tris, pH 7.5) was added. Half of the mixed solution (volume ~50 µL) was aliquoted for trypsin digestion, with the protein enzyme ratio being kept at 12.5:1. This was digested for 3.5 to 4 h at 37 °C and quenched by using v/v 2% TFA. The resulting solution contained approximately 1 mg/mL protein (tryptic digests), 2 M urea, and 2% TFA. Finally, the peptide fragments, generated by the enzymatic cleavage of ADC-A by trypsin, were separated using UPLC and analyzed by UV detection followed by tandem mass spectrometry. The peptide mapping analysis with LC–MS was performed using a Waters Acquity UPLC system and a Waters Acquity C18 BEH300, (1.7 µm, 2.1 × 150 mm) with mobile phase A and B, which consisted of 0.02% TFA in HPLC water and 0.02% TFA in acetonitrile, respectively, and a Thermo LTQ Orbitrap mass spectrometer.
2.9. Cell-Based Binding Assay
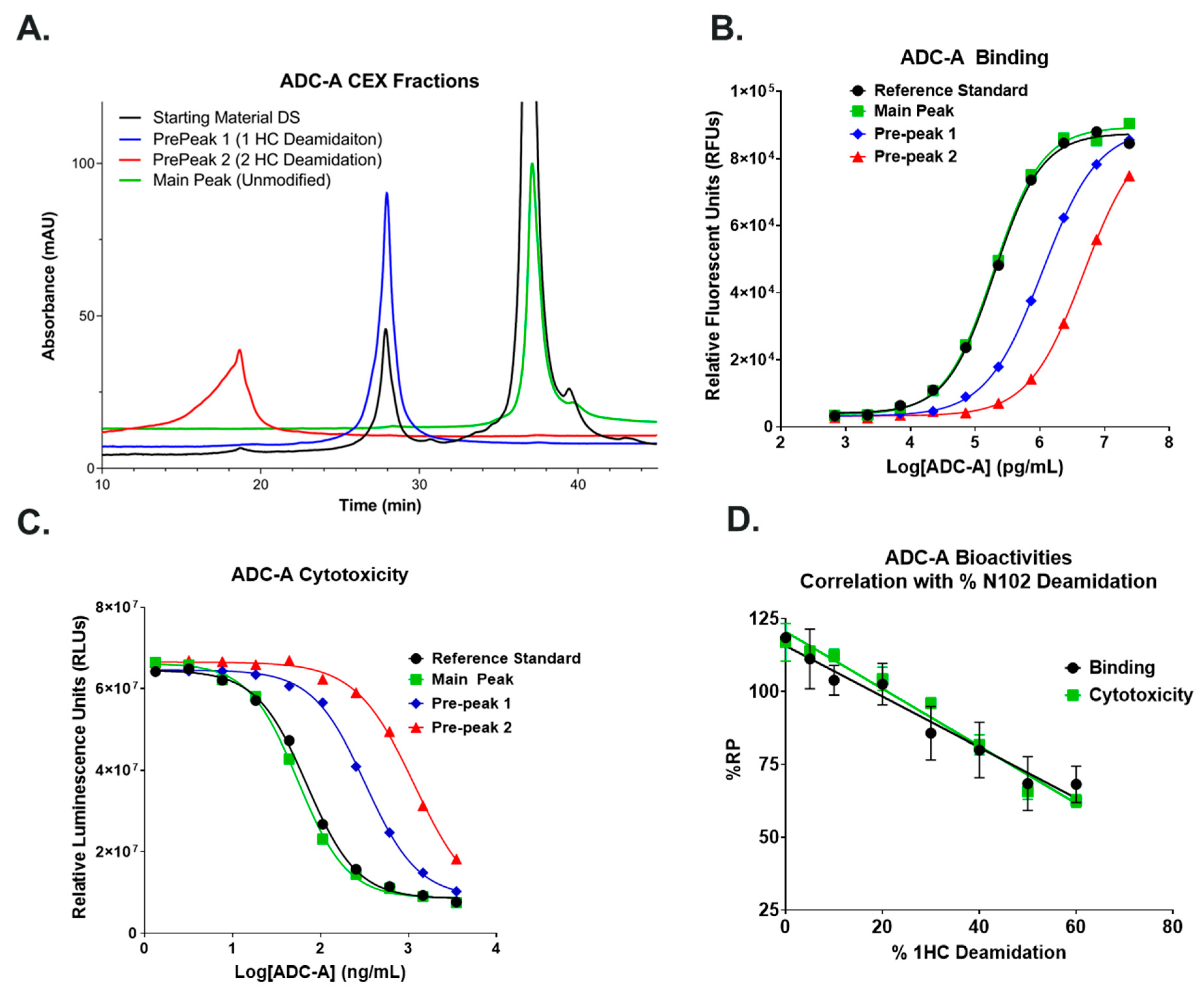
HepG2 cells, which endogenously express ADC-A’s target antigen at the cell surface, were prepared in cell culture media (DMEM, 10% FBS, and 1× MEM NEAA) and seeded in Corning® 96-well PureCoat™ amine plates at 1.5 × 105 cells/well. Plates were then incubated at 37 °C under 5% CO2 in a humified incubator overnight. The following day, plates were washed with 1× PBS pH 7.4 using a BioTek 405 LS plate washer. Increasing concentrations of ADC-A Reference Standard and test samples (24,000–0.68 ng/mL) were then added to the plate wells, and the plates were incubated at an ambient temperature for 40–60 min. Following incubation, the plates were washed with 1× PBS pH 7.4. A 65 ng/mL solution of DELFIA® Eu-N1 anti-human IgG prepared in DELFIA® assay buffer was then added to the plate wells, and the plates incubated at ambient temperature for 50–70 min. Following incubation, the plates were washed with 1× DELFIA® wash buffer. DELFIA® Enhancement solution was then added to the plate wells, and the plates were incubated for 15–60 min while being gently shaken at an ambient temperature. Bound DELFIA Eu-N1 anti-human IgG was measured via time-resolved fluorescence using a PerkinElmer Envision microplate reader (PerkinElmer; Waltham, MA, USA). The resulting relative fluorescence units (RFUs) were plotted against the ADC-A concentration using Prism version 9 (GraphPad Software; San Diego, CA, USA), and the data were fit using a 4PL curve fit. EC50 values were obtained from the 4PL curve fits. To calculate the %RP, the EC50 value of the Reference Standard was divided by the EC50 value of the test sample and multiplied by 100. Statistical analyses were performed in Prism 9 (Boston, MA, USA).
2.10. Cytotoxicity Assay
T47D cells, which endogenously expresses ADC-A’s target antigen, were prepared in cell culture media (Advanced DMEM, 5% FBS, and 1× GlutaMAX™) and seeded in Corning® 96-well white tissue culture-treated microplates. ADC-A Reference Standard and test samples were also prepared in cell culture media and added to the plate wells at final concentrations of 14,000–5.2 ng/mL. The plates were then incubated at 37 °C under 5% CO2 in a humidified incubator for 4 days. Following incubation, an equivalent volume of CellTiter-Glo® luminescent cell viability substrate was added to the plate wells, and the plates were incubated at an ambient temperature while being gently shaken for 20–40 min. Luminescence within the plate wells was then measured with a PerkinElmer Envision microplate reader. The resulting relative luminesce units (RLUs) were plotted against ADC-A concentration using Prism 9, and the data were fit using a 4PL curve fit. IC50 values were obtained from the 4PL curve fits. To calculate %RP, the IC50 value of the Reference Standard was divided by the IC50 value of the test sample and multiplied by 100%. Statistical analyses were performed in Prism 9.
2.11. Cell-Based ADC-A Internalization Assay
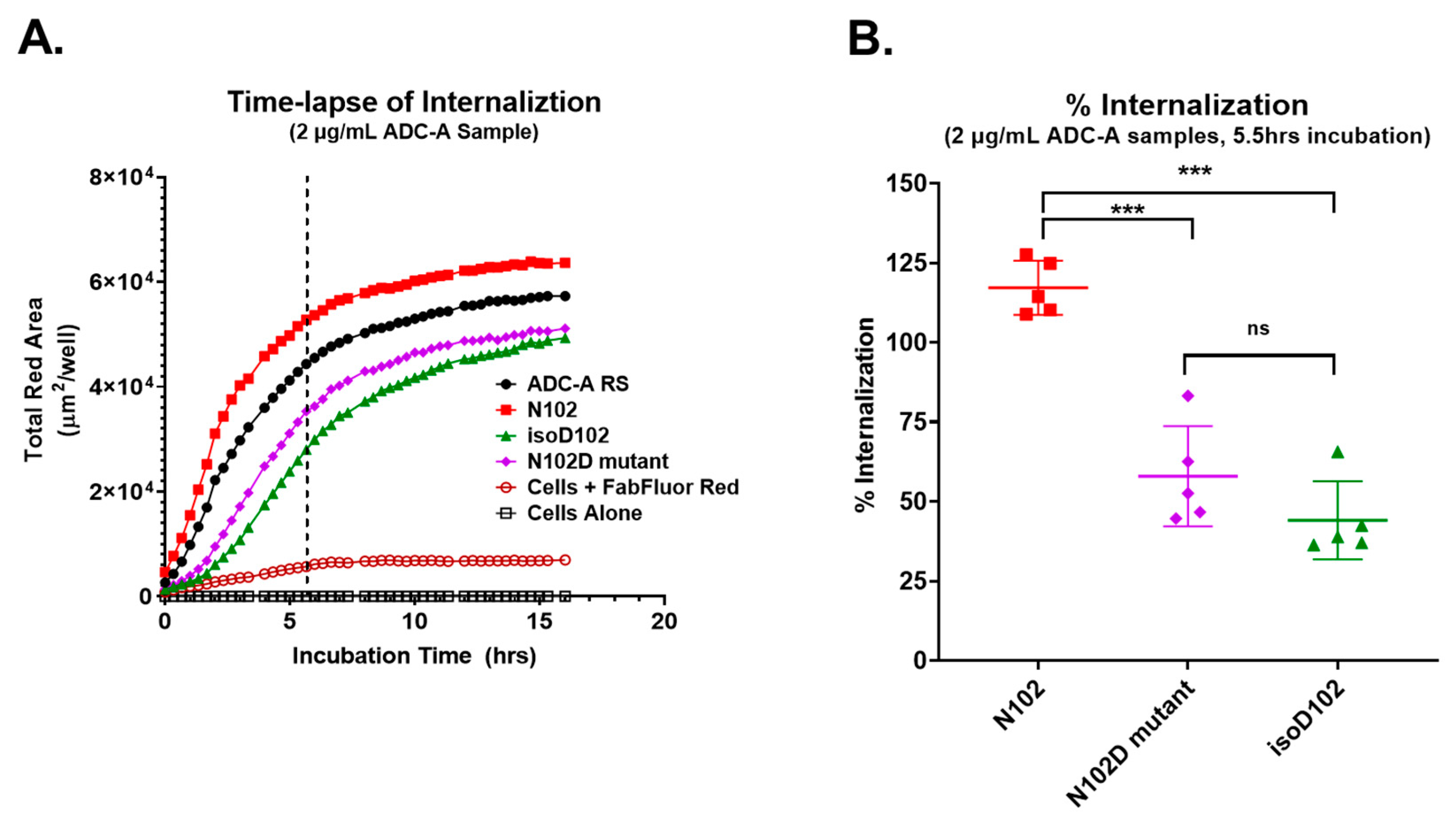
T47D cells were prepared in cell culture media (Advanced DMEM, 5% FBS, and 1× GlutaMAX™) and seeded in 96-well clear tissue culture-treated microplates at 2 × 104 cells/well. The plates were then incubated at 37 °C under 5% CO2 in a humified incubator overnight. The following day, serial dilutions of ADC-A were prepared in Advanced DMEM and incubated with 5 µg/mL Incucyte® Human Fabfluor-pH Red antibody labeling dye for 15–20 min at an ambient temperature. This mixture was then added to plate wells at a final ADC-A concentration of 2.0 µg/mL. Plates were immediately placed into an Incucyte S3 high-content fluorescent imager (Sartorious; Göttingen, Germany) and imaged for phase-contrast and red fluorescence under 20× objective every 20 min. Images were analyzed using Incucyte S3 software version 2020c rev1 (Sartorius, Göttingen, Germany). The confluency of T47D cells was detected within phase contrast images with a segmentation adjustment value of 0.5 and a minimum area filter set at 200 µm2. Red fluorescence was detected via top hat segmentation with the radius set at 10 µm and the threshold set at 0.3 RCU. The total red fluorescence area (µm2/well) was plotted against incubation time in GraphPad Prism 9. Statistical analyses were performed in Prism 9.
4. Discussion
Previous studies report that the NW amino acid motif is commonly found within the CDRs of biotherapeutic antibodies, but that this motif possesses a low propensity to deamidate [
12,
13,
20]. In contrast to these reports, we discovered a labile N102–W103 motif within the CDR-3 of ADC-A and its mAb intermediate, which is readily deamidated. The observed NW deamidation is increased under normal manufacturing and process conditions, as well as under thermal stress. Consistent with the deamidation of other motifs [
10,
11,
12,
13], the observed NW deamidation within the ADC-A/mAb intermediate is influenced by primary and higher-order structures, as evident from studies that have used synthetic peptides.
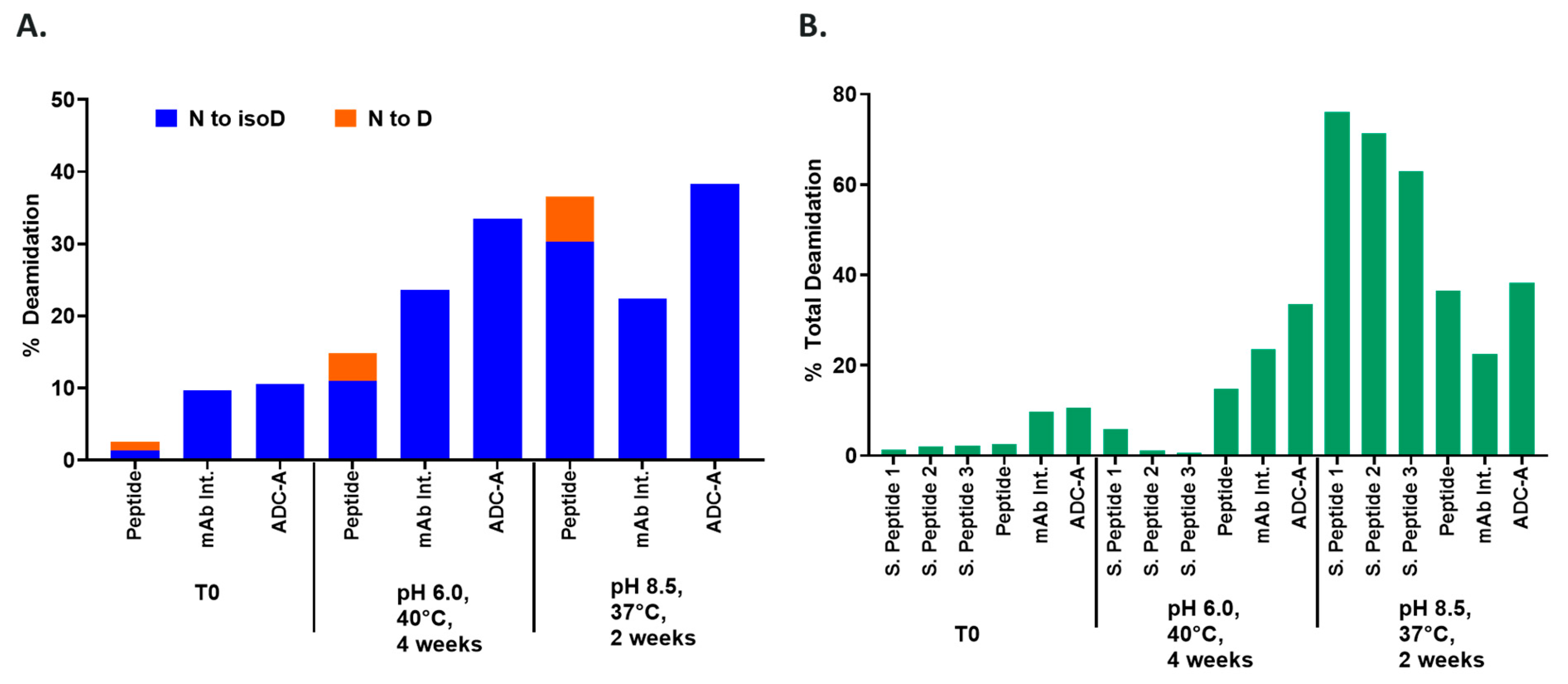
To elaborate, thermal stress increases the deamidation of both ADC-A and its mAb intermediate, which is not influenced by the pH of the solvent (
Figure 5A). In contrast, a synthetic peptide composed of the NW motif and with an identical sequence to the native ADC-A/mAb intermediate deamidates to significantly higher levels when in a basic solvent, compared to an acidic solvent (
Figure 5A). The deamidation’s dependency on the solvent pH is magnified when the peptide is shortened, in which deamidation levels reach < 10% at pH 6.0 versus > 60% at pH 8.5 (
Figure 5B; S. Peptide 1). Altogether, these results imply that NW deamidation becomes less dependent on solvent exposure as the peptide chain lengthens and becomes less flexible, and as the complexity of the surrounding structure increases. Thus, the deamidation of N102–W103 within ADC-A and its mAb intermediate appears predominantly influenced by higher-order protein structure.
Experiments with synthetic peptides also demonstrate that residues contiguous to the NW motif influence its deamidation, at least when the motif is solvent exposed. Specifically, S. Peptide 2 (H to V substitution at n + 2) deamidated to a lower level than S. Peptide 1 under both acidic and basic environments (
Figure 5B). S. Peptide 3 (K to V substituted at n − 1) deamidated to even lower levels under both environments (
Figure 5B). Future studies are needed to determine whether deamidation of NW is equally influenced by these contiguous residues, when contained within the higher-order structure of ADC-A and its mAb intermediate.
Another contributing factor to the liability of this NW motif, when contained within a full antibody, could be the presence of the inserted cysteines in the HCs, which again act as the site of payload conjugation near the hinge region to make the ADC-A [
21,
22]. These inserted mutations may perturb the otherwise stable NW micro-environment, rendering it more susceptible to deamidation. Furthermore, the addition of a bulky and hydrophobic linker-warhead to these sites might further perturb the NW micro-environment, thereby explaining why the N102 deamidation rate is more rapid in ADC-A than in its mAb intermediate, under all pH- and thermal-stress conditions (
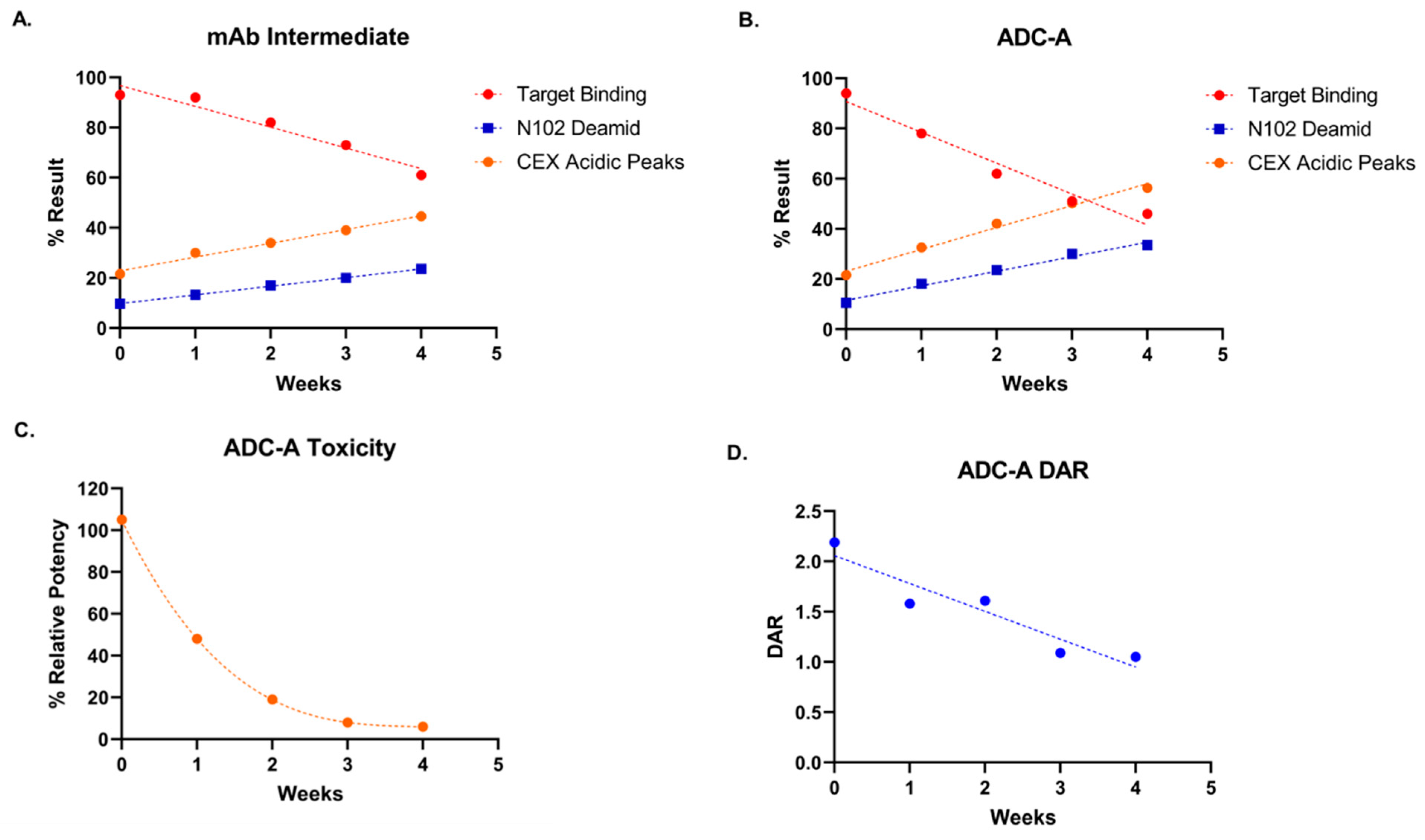
Figure 1,
Figure 2 and
Figure 5).
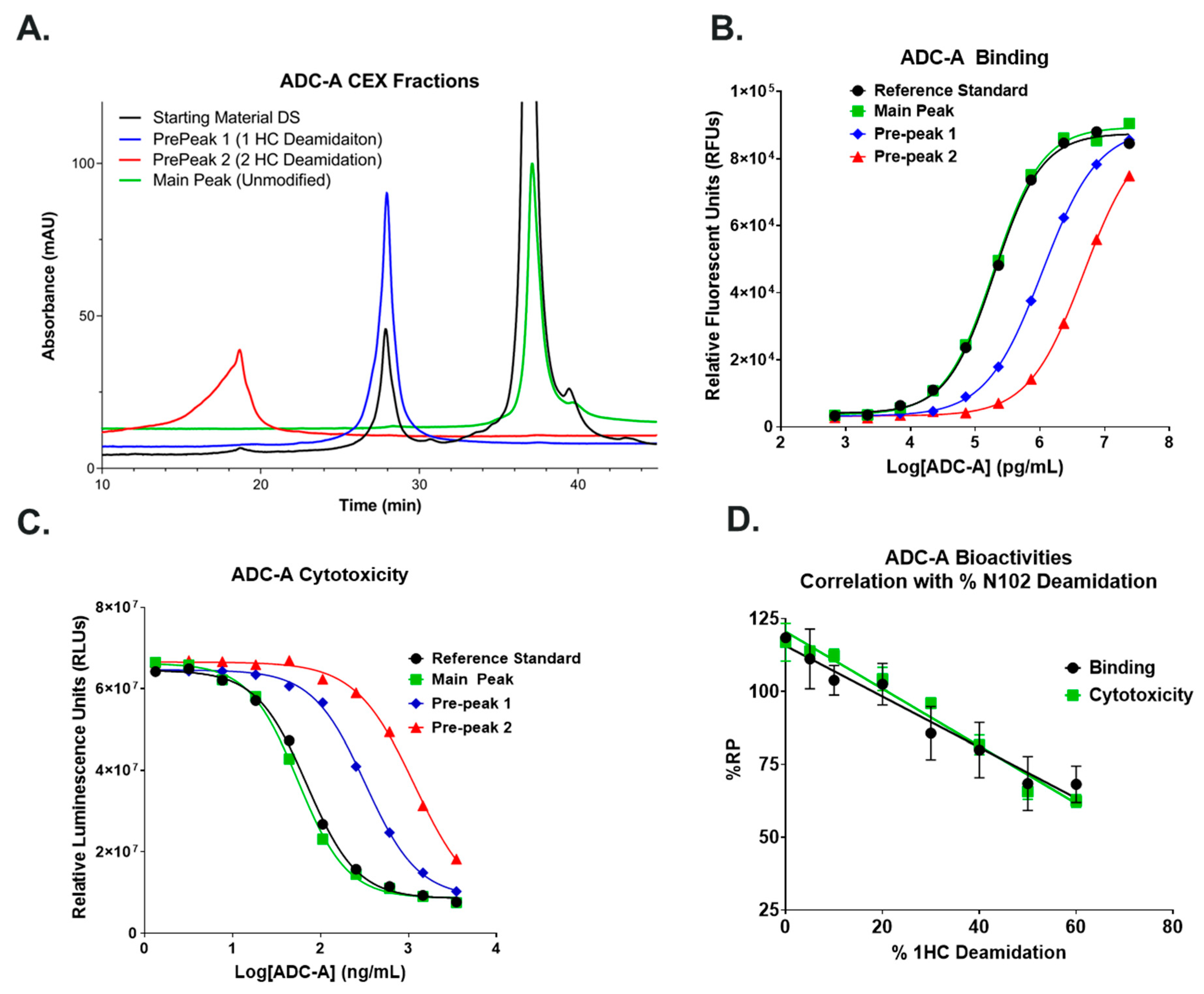
Further studies are needed to understand how the inserted cysteines and/or payload conjugation within the hinge region of ADC-A/mAb intermediate influence the CDR-3 3D micro-environment to enable NW deamidation. So far, preliminary data continue to support the hypothesis that the NW deamidation, when contained within the full antibody, is reliant on hinge engineering/conjugation. Specifically, we produced a recombinant Fab that has an identical sequence to the Fab in ADC-A or its mAb intermediate (verified by peptide mapping). The produced Fab and the CEX pre-peak 1 (fraction from ADC-A with 49% N102 deamidation) were incubated at pH 9 under 40 °C for 1 day and then 25 °C for a week. We then performed peptide mapping on the stressed recombinant Fab and pre-peak 1. As expected, the N102 deamidation in the pH-/thermal-stressed pre-peak 1 continued to increase from 49% to 87%. Surprisingly, the N102 in the pH-/thermal-stressed recombinant Fab was nearly unchanged, resulting in only a 1.1% increase in deamidation (
Supplementary Figures S4 and S5). UV chromatograms matched with the mass spectrometry data, in that only native N102 peak was observed in the pH-/thermal-stressed recombinant Fab—the deamidated N102 peak was not found. In contrast, the majority of the native N102 peak was deamidated into two isoD102 peaks (one from complete digestion, and one from incomplete digestion) in the stressed pre-peak 1.
So far, these data confirm that, without the hinge region and thus the inserted cysteines and payload conjugation, the NW motif within CDR-3 Fab is resistant to deamidation. Likewise, the payload conjugation of ADC-A also impacted N325-K326 motif in the Fc region. As shown in
Figure 1A,B, N325 deamidation was 14% in the 40 °C one-month-stressed mAb intermediate, as is typically observed in monoclonal antibodies [
19,
28]. However, N325 deamidation was significantly lower (8%) in ADC-A under the same stress condition, implying conjugation had a protective effect against deamidation in this case. Additional studies are ongoing to further understand the influence of the hinge region’s cysteine insertion and payload conjugation on these deamidation sites, which will be addressed in a future publication.
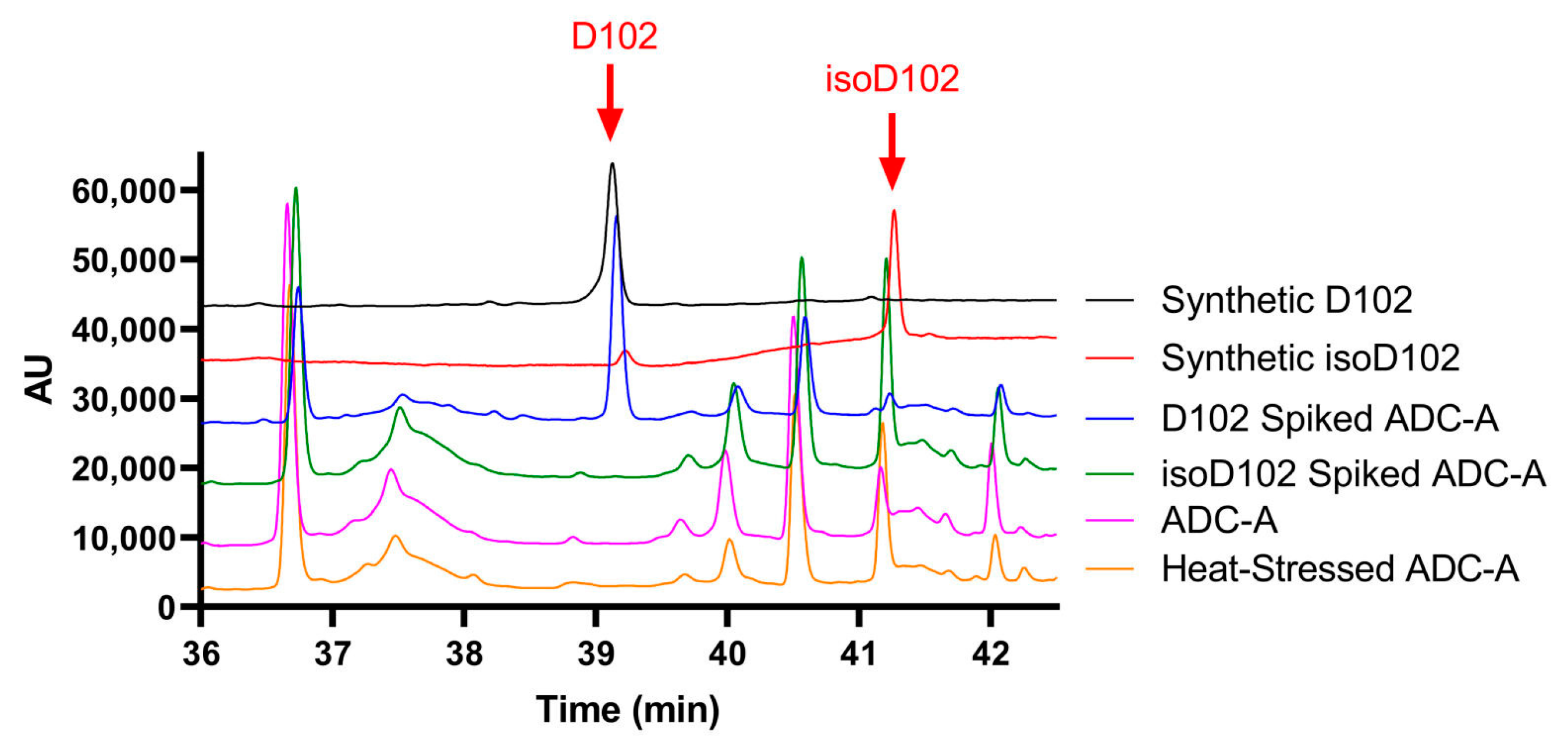
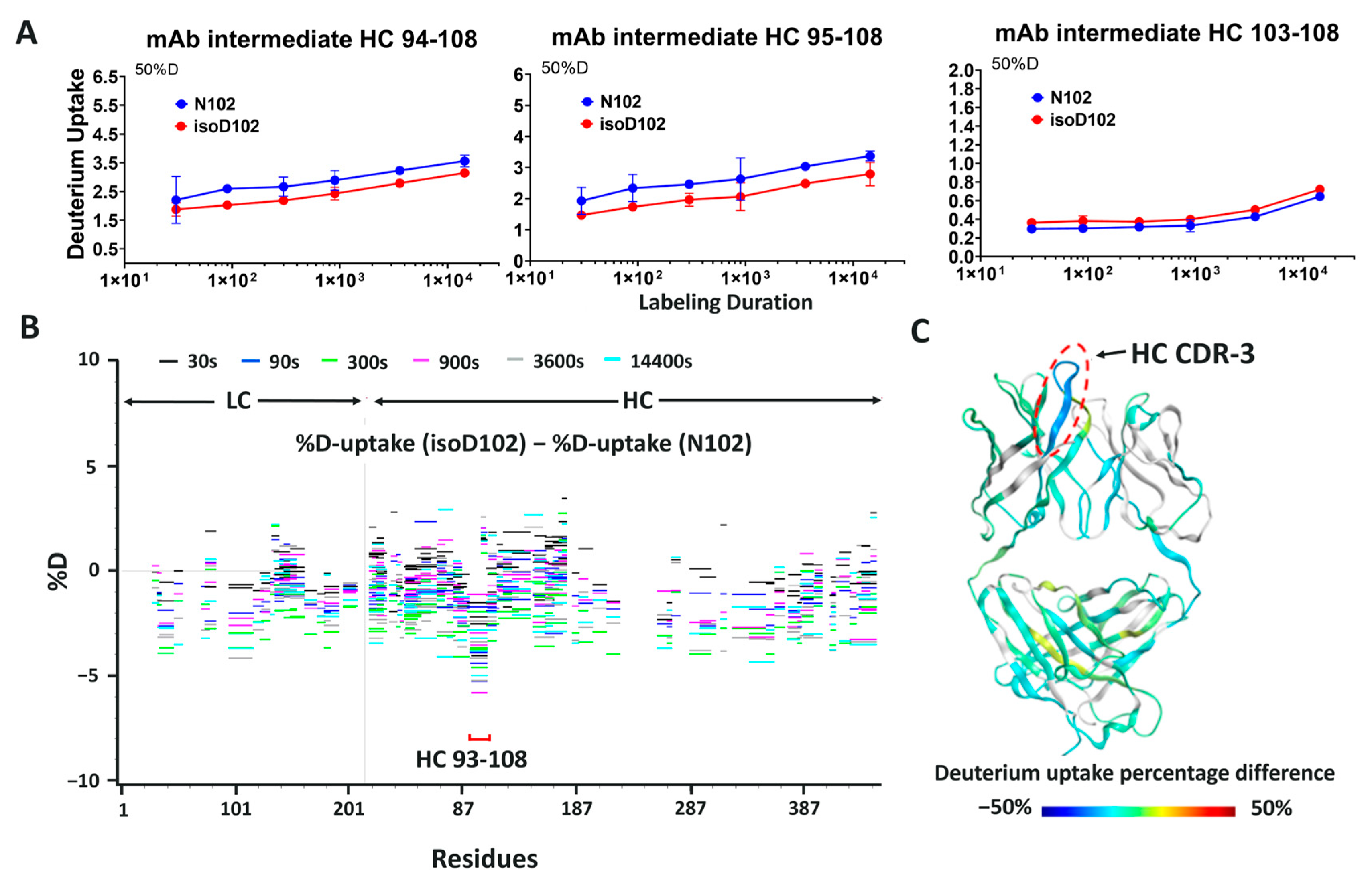
N102 within the ADC-A/mAb intermediate was shown to deamidate solely into iso-aspartate (
Figure 4), which is atypical [
2,
3,
4]. As previously mentioned, deamidation of asparagine could happen via the three degradation pathways: (1) nucleophilic attack of the alpha-NH group on the carbonyl carbon of the side chain, leading to a five-membered succinimide (cyclic imide) intermediate that hydrolyzes into a mixture of aspartate and iso-aspartate; (2) nucleophilic attack by the backbone carbonyl oxygen on the carbonyl carbon of the side chain, resulting in a cyclic isoimide intermediate, yielding only aspartate after hydrolysis; or (3) asparagine residues deamidating to aspartic acid only by direct water-assisted hydrolysis [
2,
12,
20]. As degradation pathway (1) above is the only pathway to result in iso-aspartate, it is logical to conclude that N102 within ADC-A/mAb intermediate solely deamidates through the formation of succinimide intermediate.
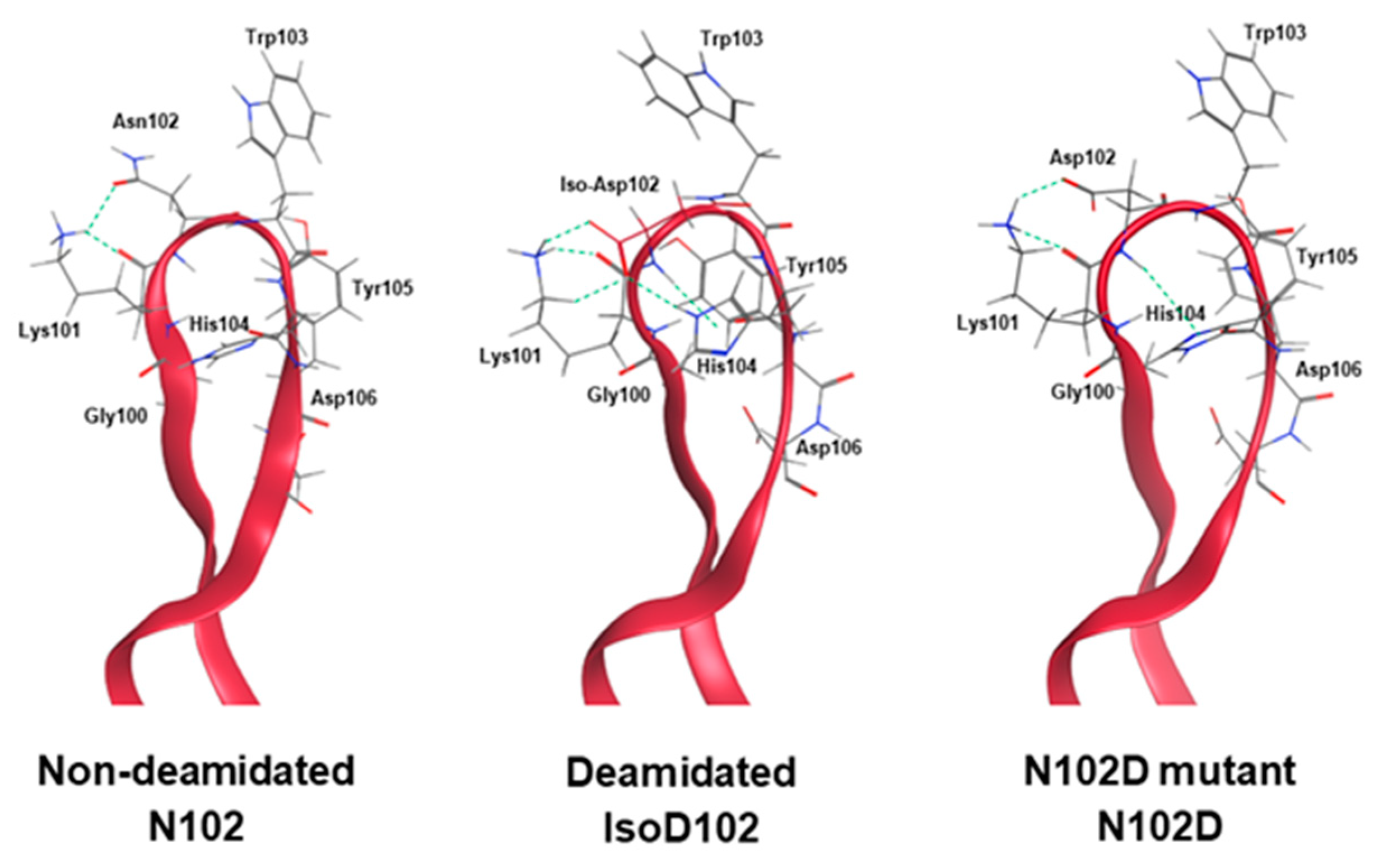
Results within
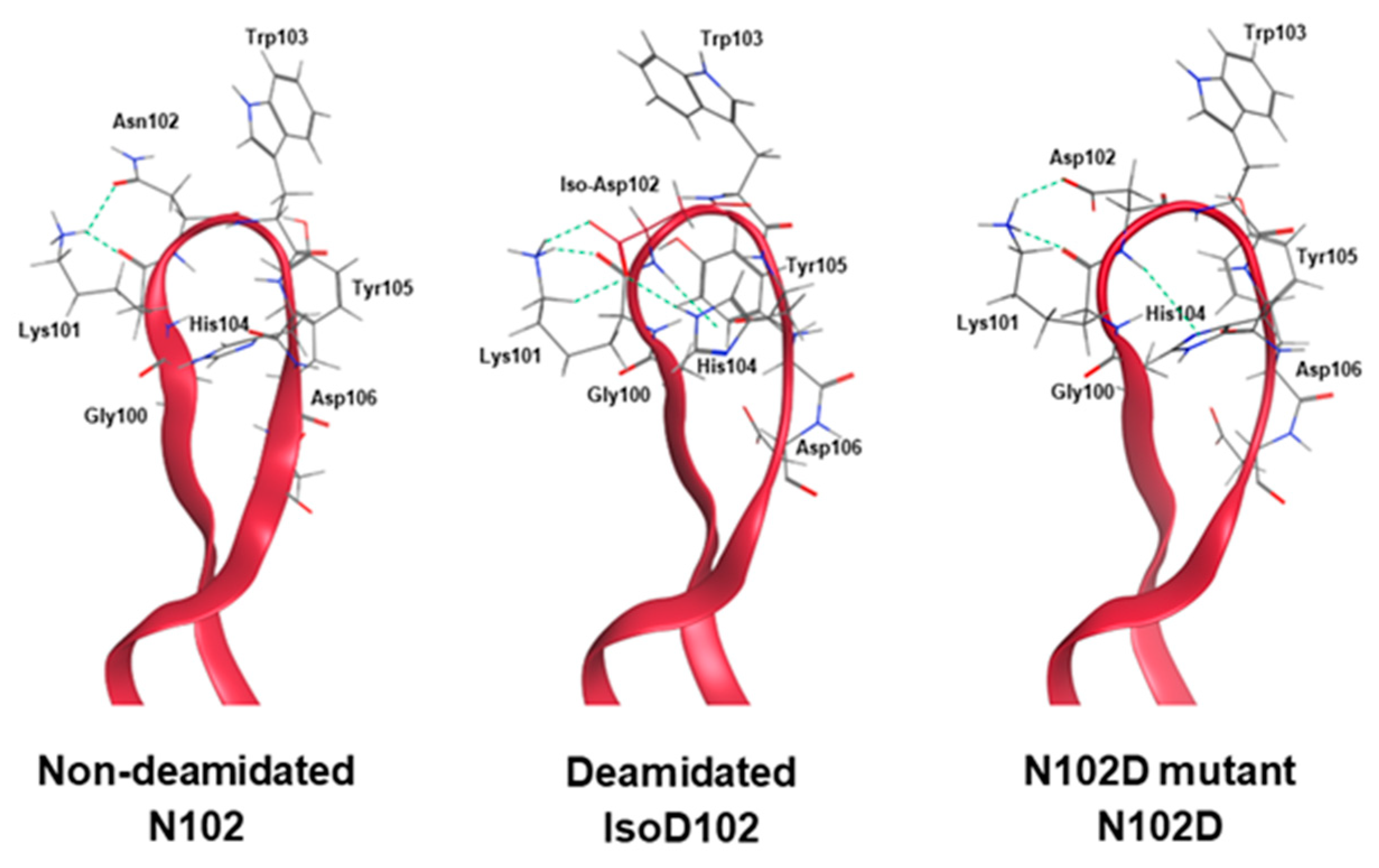
Figure 5A demonstrate that the NW motif itself can deamidate into a typical mixture of iso-aspartate and aspartate, at least when the motif is contained within a simple peptide and the deamidation demonstrates solvent dependencies. However, deamidation of N102–W103 continued to convert solely into iso-aspartate within ADC-A and mAb intermediate, independent of the solvent pH. Thus, we conclude that this phenomenon is not inherent to the NW motif itself, but instead is induced by the unique higher-order structure surrounding N102–W103 within the ADC-A/mAb intermediate. Structural modeling confirmed that the conversion of N102 to isoD102 is thermodynamically favored over D102 in this antibody, as it results in additional hydrogen bonding with the adjacent K101 as opposed to D102 (
Figure 11). In addition, the conversion to isoD102 appears to have a greater impact on the micro-environment structure compared to D102, as evident by both the modeling in
Figure 11 and HDX data in
Figure 9 versus
Figure 10.
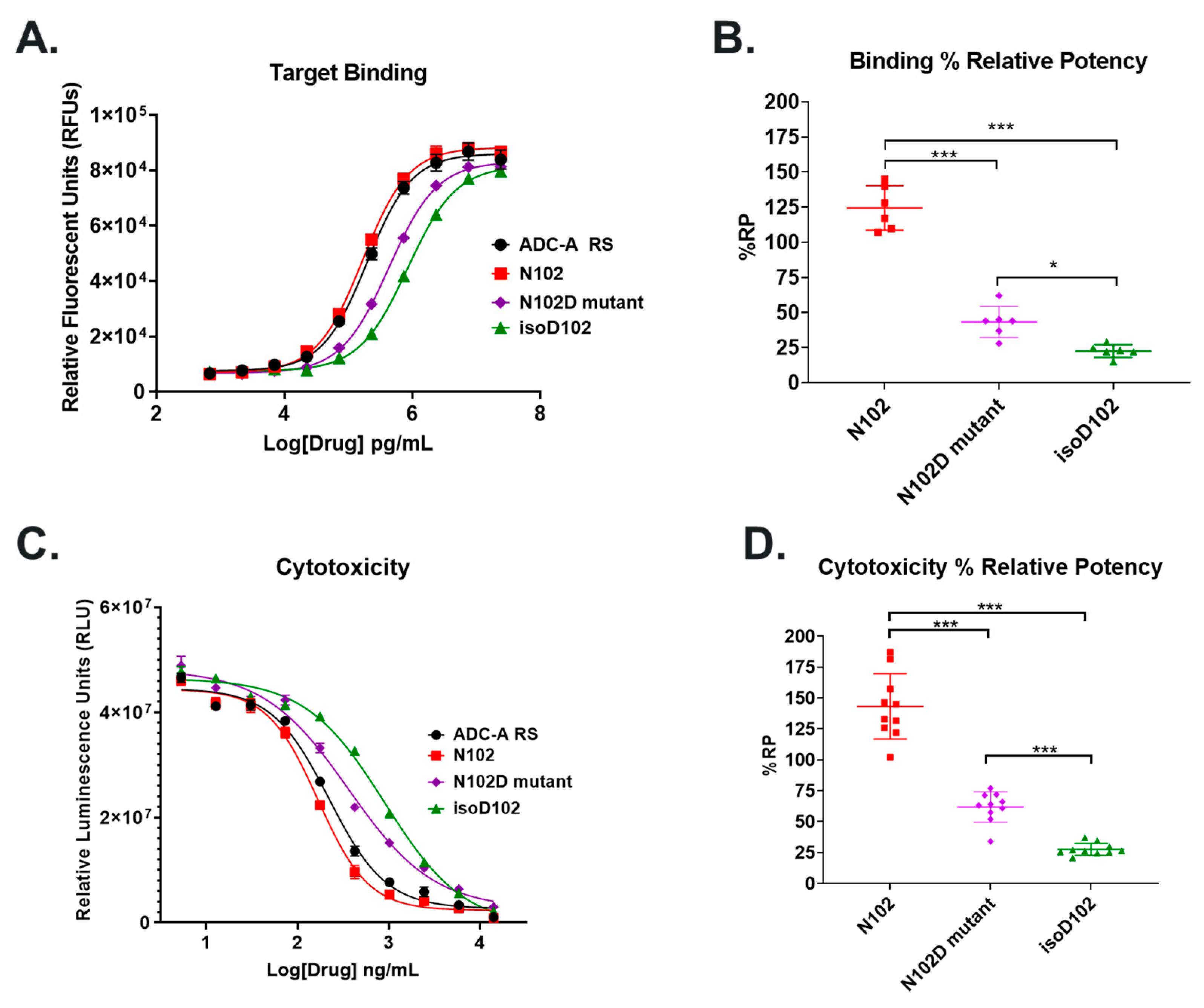
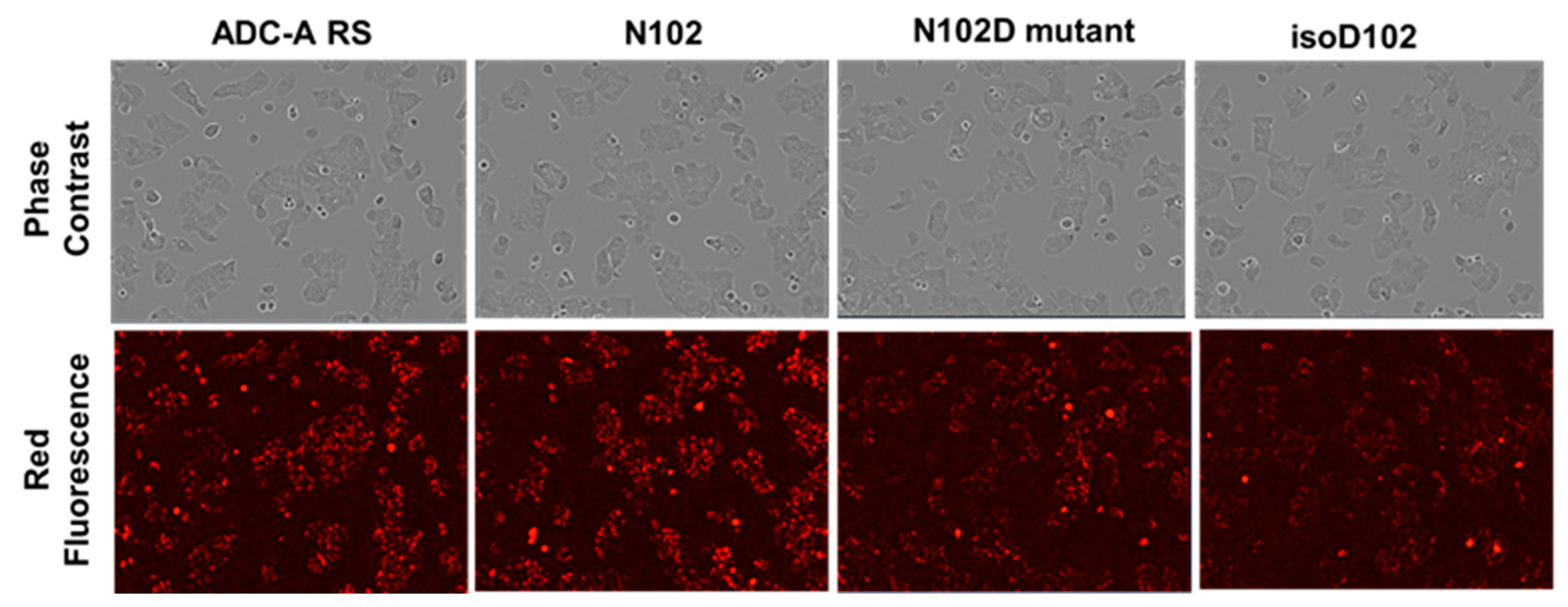
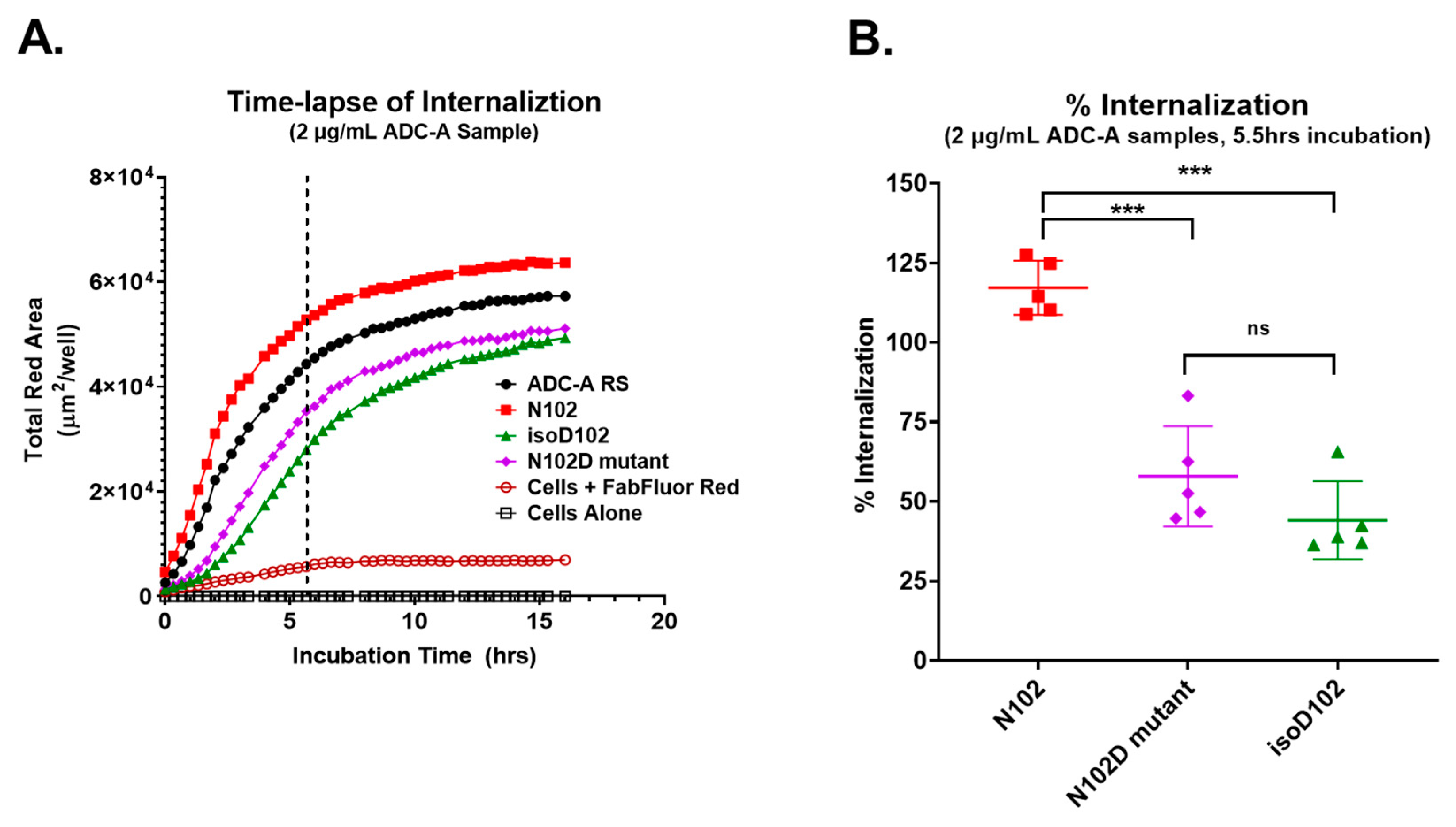
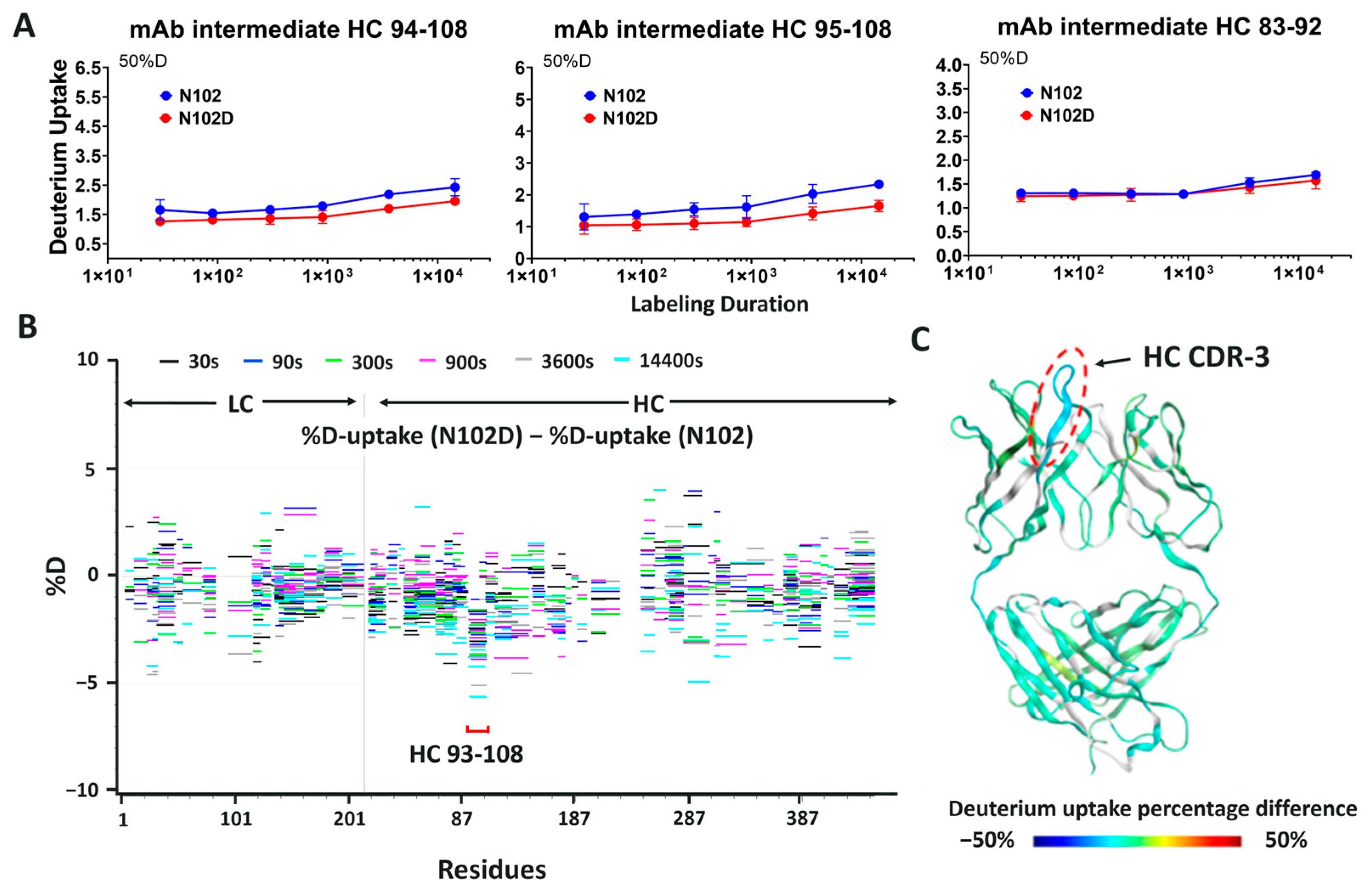
Both isoD102 and the N102D mutant impacted the biological activity of ADC-A in vitro. This impact stems from a reduced antigen binding affinity, which consequentially reduces ADC-A’s rate of cellular internalization, delivery of payload, and ultimately cytotoxicity. The reduced biological activities of isoD102 were more severe than the N102D mutant. In fact, the reduction in isoD102′s bioactivity is even greater than the apparent reductions in
Figure 6,
Figure 7 and
Figure 8, as the isoD102 used in these experiments (i.e., from CEX pre-peak 1) likely possessed deamidation at only one Fab arm of ADC-A, whereas the N102D mutant possessed the intended mutation at both Fab arms. IsoD102 ADC-A from the CEX pre-peak 2, which likely possesses deamidation at both Fab arms, could not be used for biological characterization due to limitations in material amounts.
The HDX data in
Figure 9 and
Figure 10 show that isoD102 induces greater changes to local structure than N102D, which generally explains why isoD102 impacted biological activity more than the N102D mutant. MOE modeling provides some insight into the specific structural changes that may be occurring under isoD102 and the N102D mutant. Specifically, the conversion of N102 to isoD102 alters the orientation of the residues W103, H104, and Y105 more severely than conversion to N102D (
Figure 11), which correlates with the severity of the reduction in biological activity in vitro. Altogether, these results demonstrate that N102 within the ADC-A/mAb intermediate solely deamidates into iso-aspartate because it is thermodynamically favored, which in turn induces greater changes to the local CDR-3 structure, and, thus, impacts biological activities more severely.
The impact of N102 deamidation on ADC-A’s in vivo activities has not been studied yet. However, we hypothesize that the profound reduction in ADC-A’s in vitro biological activities reported here will translate to reduced potency and/or efficacy for inhibiting tumor cell growth in vivo. N102 deamidation could also impact ADC-A’s pharmacokinetic properties, as cellular internalization and subsequent degradation of an ADC is not only integral to its mechanism of action, but is a major route to its metabolism, too [
29]. Thus, reduced on-target cellular internalization could translate to increased systemic circulation of ADC-A in vivo, which may in turn increase the chances of off-target toxicities. Future studies are needed to fully assess whether N102 deamidation impacts these in vivo activities, and to test whether the differential impact between isoD102 and N102D mutant on in vitro activity is recapitulated in vivo.
Likewise, future studies are needed to determine the impact of N102 deamidation on the stability of ADC-A and its mAb intermediate as a drug product. While we show that accelerated stability under thermal stress increases N102 deamidation (
Figure 1,
Figure 2 and
Figure 5), we have not yet studied how N102 deamidation feeds back to influence the overall stability of ADC-A and its mAb intermediate under accelerated and/or normal manufacturing and storage conditions. Previous studies report that asparagine deamidation can affect antibody colloidal stability and increase susceptibility to aggregation, especially under acidic conditions [
30,
31]. Considering these reports, it is possible that N102 deamidation will decrease the overall stability of ADC-A and its mAb intermediate, and that this instability will be more severe when N102 is converted to isoD102 versus N102D. Future studies are needed to detail this impact.
In summary, this is the first report on an atypical increase in asparagine deamidation in the non-canonical NW motif within an ADC and its mAb intermediate, which converts the asparagine residue solely into iso-aspartate. The observed NW motif is located within a site critical for antigen binding, which consequently impacts all other biological activities required for ADC-A’s mechanism of action. Interestingly, the conversion of the asparagine residue into iso-aspartate impacts biological activities more severely than conversion into aspartate, which correlates with HDX and modeling data showing more severe structural changes when the asparagine is converted to iso-aspartate. The unique susceptibility of the otherwise stable, non-canonical NW motif within the ADC-A/mAb intermediate is likely due to a distinct higher-order structure and specific susceptibilities in the N102 micro-environment brought on by the hinge region, including the inserted cysteines and/or conjugation of payload [
21,
22]. Future studies are needed to fully elucidate the mechanism of this NW deamidation, as well as to evaluate its impact on in vivo activity and influence on drug product stability. Overall, these findings help in assessing sequence liabilities, as well as ADC designs and constructs, when engineering and manufacturing antibody-drug conjugates and other mAb-based biotherapeutics.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}