Structure and Dynamics Guiding Design of Antibody Therapeutics and Vaccines

 , , , ,
, , , , 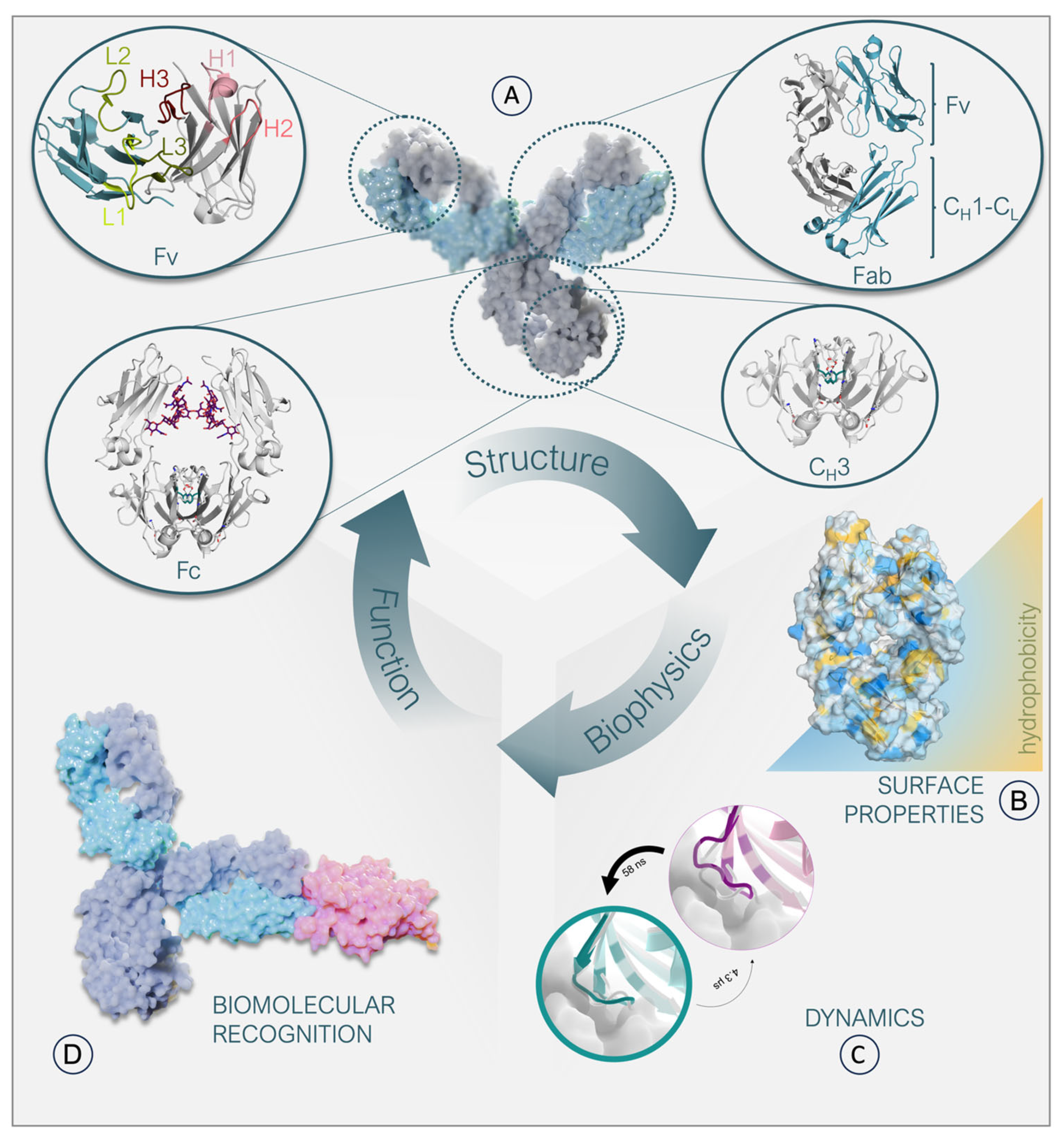{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Antibody Structure Characteristics
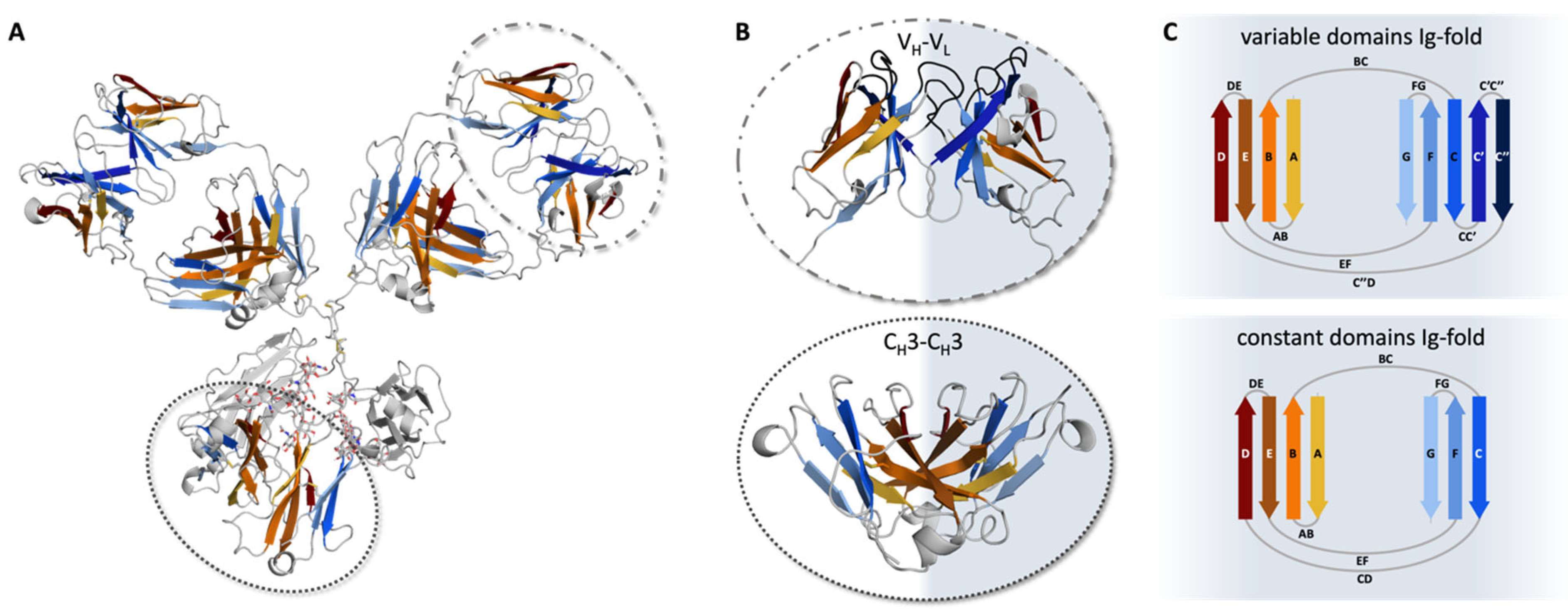
2. The Ig-Fold
3. Antibody Structure Prediction
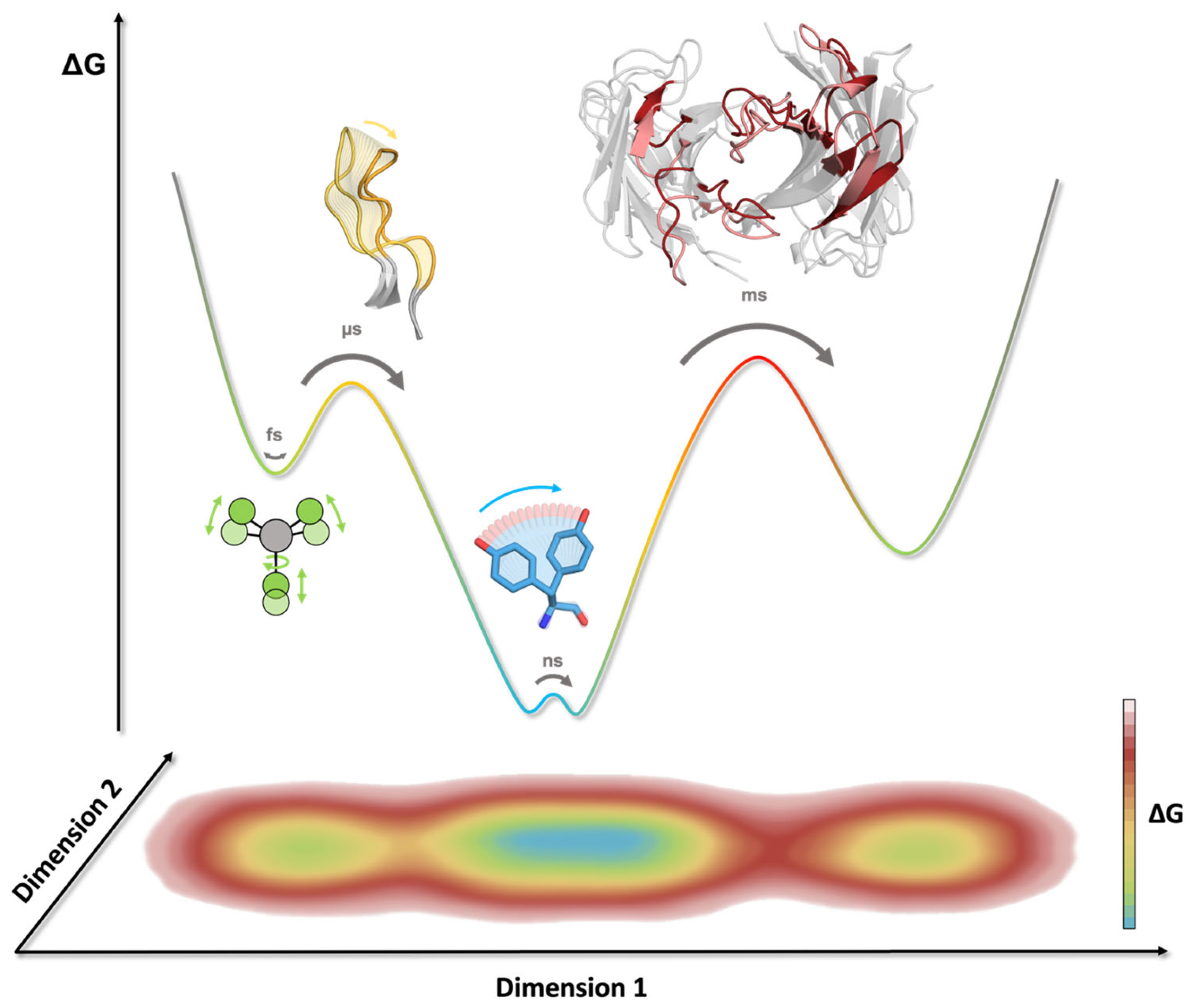
4. Antibody Dynamics
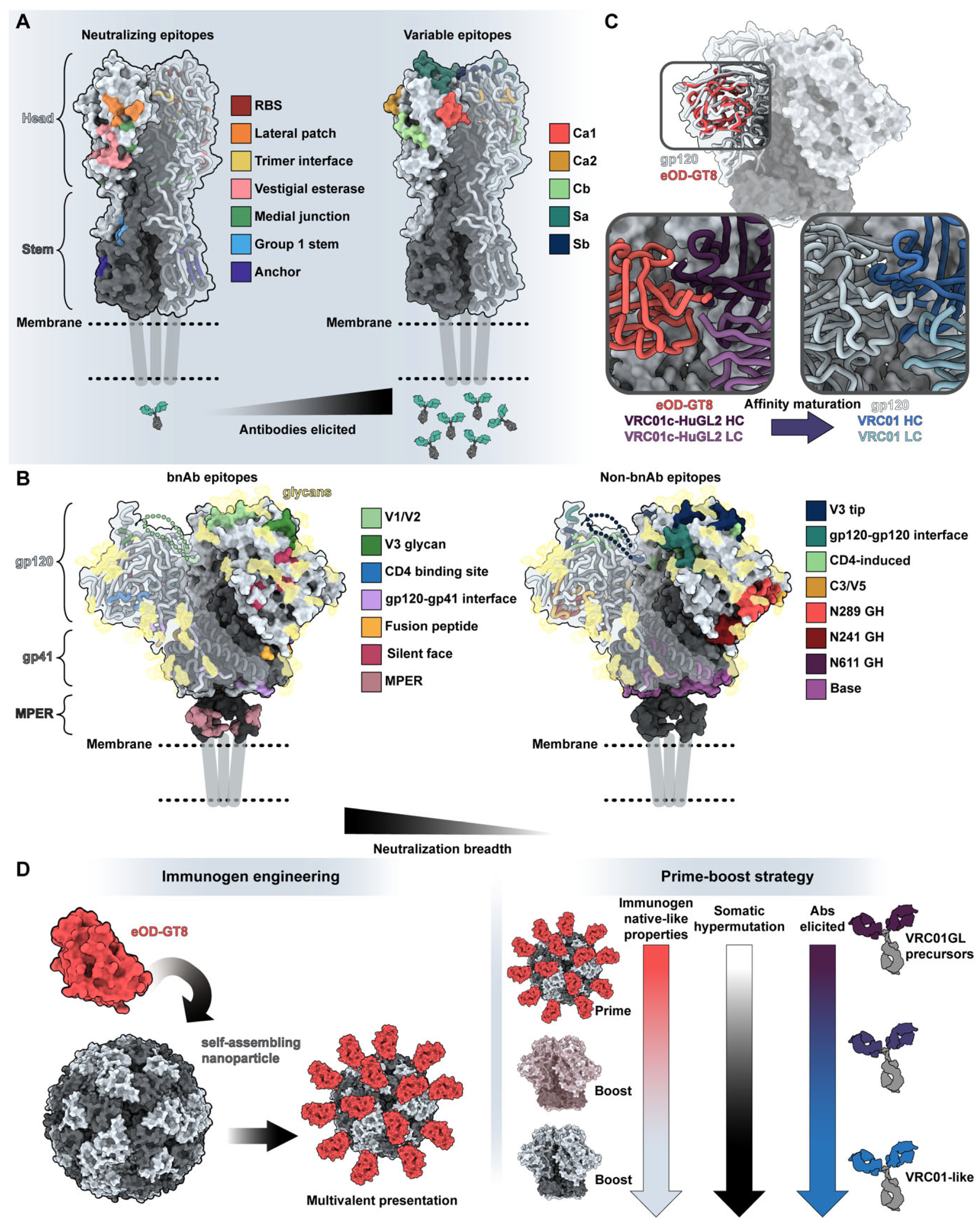
5. Antibody-Antigen Recognition and Structure Guided Vaccine Design

6. State-of-the-Art Experimental Structure Determination
7. Antibody Engineering—Design of Special Formats/Interface Characterization
7.1. The Role and Organization of Constant Domains
7.2. Engineering Techniques Led to New Antibody Formats
7.3. Humanization
7.4. Affinity Maturation
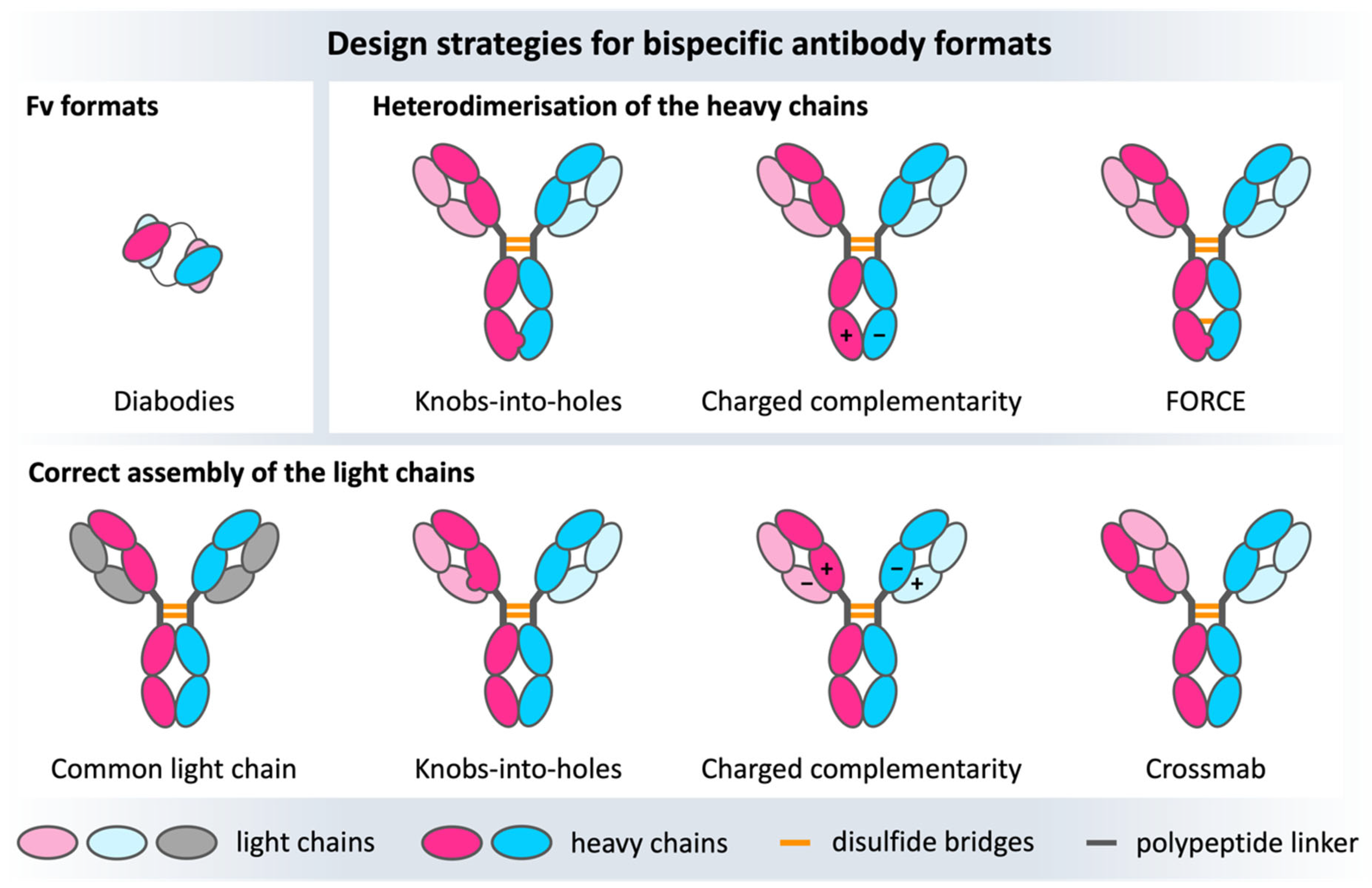
7.5. Bispecific Antibodies
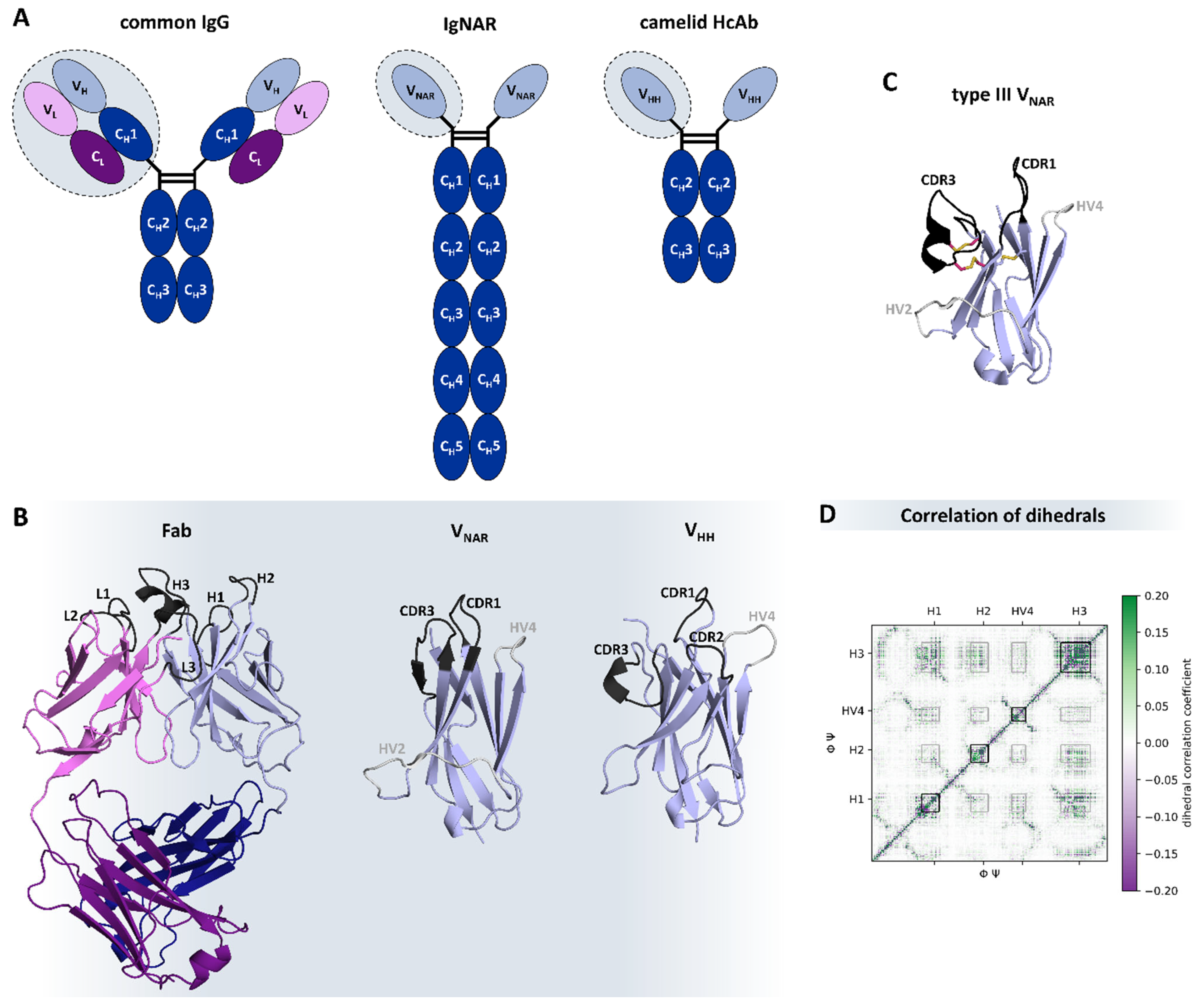
7.6. Small Size Antibody Formats
8. Special Formats from Natural Sources

9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2023. mAbs 2023, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to Watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.; Ju, T.W.; Wang, Y.; Prabakaran, P.; Dimitrov, D.S. Interactions of IgG1 CH2 and CH3 Domains with FcRn. Front. Immunol. 2014, 5, 146. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Kroell, K.B.; Hofer, F.; Riccabona, J.R.; Liedl, K.R. Mutation of Framework Residue H71 Results in Different Antibody Paratope States in Solution. Front. Immunol. 2021, 12, 243. [Google Scholar] [CrossRef] [PubMed]
- Foote, J.; Winter, G. Antibody Framework Residues Affecting the Conformation of the Hypervariable Loops. J. Mol. Biol. 1992, 224, 487–499. [Google Scholar] [CrossRef]
- Krauss, J.; Arndt, M.A.E.; Zhu, Z.; Newton, D.L.; Vu, B.K.; Choudhry, V.; Darbha, R.; Ji, X.; Courtenay-Luck, N.S.; Deonarain, M.P.; et al. Impact of Antibody Framework Residue VH-71 on the Stability of a Humanised Anti-MUC1 scFv and Derived Immunoenzyme. Br. J. Cancer 2004, 90, 1863–1870. [Google Scholar] [CrossRef]
- Tramontano, A.; Chothia, C.; Lesk, A.M. Framework Residue 71 Is a Major Determinant of the Position and Conformation of the Second Hypervariable Region in the VH Domains of Immunoglobulins. J. Mol. Biol. 1990, 215, 175–182. [Google Scholar] [CrossRef]
- Xiang, J.; Sha, Y.; Jia, Z.; Prasad, L.; Delbaere, L.T.J. Framework Residues 71 and 93 of the Chimeric B72.3 Antibody Are Major Determinants of the Conformation of Heavy-Chain Hypervariable Loops. J. Mol. Biol. 1995, 253, 385–390. [Google Scholar] [CrossRef]
- Rappazzo, C.G.; Fernández-Quintero, M.L.; Mayer, A.; Wu, N.C.; Greiff, V.; Guthmiller, J.J. Defining and Studying B Cell Receptor and TCR Interactions. J. Immunol. 2023, 211, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Hoerschinger, V.J.; Lamp, L.M.; Bujotzek, A.; Georges, G.; Liedl, K.R. VH -VL Interdomain Dynamics Observed by Computer Simulations and NMR. Proteins 2020, 88, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Kroell, K.B.; Heiss, M.C.; Loeffler, J.R.; Quoika, P.K.; Waibl, F.; Bujotzek, A.; Moessner, E.; Georges, G.; Liedl, K.R. Surprisingly Fast Interface and Elbow Angle Dynamics of Antigen-Binding Fragments. Front. Mol. Biosci. 2020, 7, 609088. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, S.A.; Chothia, C. Immunoglobulin Superfamily Proteins in Caenorhabditis Elegans. J. Mol. Biol. 2000, 296, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Youkharibache, P. Topological and Structural Plasticity of the Single Ig Fold and the Double Ig Fold Present in CD19. Biomolecules 2021, 11, 1290. [Google Scholar] [CrossRef] [PubMed]
- Lesk, A.M.; Chothia, C. Evolution of Proteins Formed by Beta-Sheets. II. The Core of the Immunoglobulin Domains. J. Mol. Biol. 1982, 160, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An Efficient Route to Human Bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic Structure of an Intact IgG1 Monoclonal Antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L. Second Antibody Modeling Assessment (AMA-II). Proteins Struct. Funct. Bioinform. 2014, 82, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Jung, J.; Xiang, Z.; Honig, B. Protein Structure Prediction. Curr. Opin. Chem. Biol. 2001, 5, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Khetan, R.; Curtis, R.; Deane, C.M.; Hadsund, J.T.; Kar, U.; Krawczyk, K.; Kuroda, D.; Robinson, S.A.; Sormanni, P.; Tsumoto, K.; et al. Current Advances in Biopharmaceutical Informatics: Guidelines, Impact and Challenges in the Computational Developability Assessment of Antibody Therapeutics. mAbs 2022, 14, 2020082. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody Structure, Instability, and Formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Kraml, J.; Georges, G.; Liedl, K.R. CDR-H3 Loop Ensemble in Solution—Conformational Selection upon Antibody Binding. mAbs 2019, 11, 1077–1088. [Google Scholar] [CrossRef]
- Weitzner, B.D.; Dunbrack, R.L.J.; Gray, J.J. The Origin of CDR H3 Structural Diversity. Structure 2015, 23, 302–311. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. Canonical Structures for the Hypervariable Regions of Immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- North, B.; Lehmann, A.; Dunbrack, R.L.J. A New Clustering of Antibody CDR Loop Conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Morea, V.; Lesk, A.M.; Tramontano, A. Antibody Modeling: Implications for Engineering and Design. Methods 2000, 20, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Kuroda, D.; Marze, N.; Xu, J.; Gray, J.J. Blind Prediction Performance of RosettaAntibody 3.0: Grafting, Relaxation, Kinematic Loop Modeling, and Full CDR Optimization. Proteins 2014, 82, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Bujotzek, A.; Dunbar, J.; Lipsmeier, F.; Schäfer, W.; Antes, I.; Deane, C.M.; Georges, G. Prediction of VH-VL Domain Orientation for Antibody Variable Domain Modeling. Proteins 2015, 83, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Abhinandan, K.R.; Martin, A.C.R. Analysis and Prediction of VH/VL Packing in Antibodies. Protein Eng. Des. Sel. PEDS 2010, 23, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Marze, N.A.; Lyskov, S.; Gray, J.J. Improved Prediction of Antibody VL-VH Orientation. Protein Eng. Des. Sel. PEDS 2016, 29, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Marks, C.; Deane, C.M. Antibody H3 Structure Prediction. Comput. Struct. Biotechnol. J. 2017, 15, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Third Generation Antibody Discovery Methods: In Silico Rational Design. Chem. Soc. Rev. 2018, 47, 9137–9157. [Google Scholar] [CrossRef]
- Wilman, W.; Wróbel, S.; Bielska, W.; Deszynski, P.; Dudzic, P.; Jaszczyszyn, I.; Kaniewski, J.; Młokosiewicz, J.; Rouyan, A.; Satława, T.; et al. Machine-Designed Biotherapeutics: Opportunities, Feasibility and Advantages of Deep Learning in Computational Antibody Discovery. Brief. Bioinform. 2022, 23, bbac267. [Google Scholar] [CrossRef]
- Hummer, A.M.; Abanades, B.; Deane, C.M. Advances in Computational Structure-Based Antibody Design. Curr. Opin. Struct. Biol. 2022, 74, 102379. [Google Scholar] [CrossRef]
- Norman, R.A.; Ambrosetti, F.; Bonvin, A.M.J.J.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational Approaches to Therapeutic Antibody Design: Established Methods and Emerging Trends. Brief. Bioinform. 2020, 21, 1549–1567. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L.J.; Gray, J.J. Modeling and Docking of Antibody Structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.; Dunbar, J.; Georges, G.; Shi, J.; Deane, C.M. ABodyBuilder: Automated Antibody Structure Prediction with Data-Driven Accuracy Estimation. mAbs 2016, 8, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Sulam, J.; Gray, J.J. Antibody Structure Prediction Using Interpretable Deep Learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Chu, L.-S.; Mahajan, S.P.; Gray, J.J. Fast, Accurate Antibody Structure Prediction from Deep Learning on Massive Set of Natural Antibodies. Nat. Commun. 2023, 14, 2389. [Google Scholar] [CrossRef]
- Abanades, B.; Georges, G.; Bujotzek, A.; Deane, C.M. ABlooper: Fast Accurate Antibody CDR Loop Structure Prediction with Accuracy Estimation. Bioinformatics 2022, 38, 1877–1880. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Georges, G.; Varga, J.M.; Liedl, K.R. Ensembles in Solution as a New Paradigm for Antibody Structure Prediction and Design. mAbs 2021, 13, 1923122. [Google Scholar] [CrossRef]
- Nishigami, H.; Kamiya, N.; Nakamura, H. Revisiting Antibody Modeling Assessment for CDR-H3 Loop. Protein Eng. Des. Sel. PEDS 2016, 29, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Kokot, J.; Waibl, F.; Fischer, A.-L.M.; Quoika, P.K.; Deane, C.M.; Liedl, K.R. Challenges in Antibody Structure Prediction. mAbs 2023, 15, 2175319. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.; Luo, J.; Obmolova, G.; Malia, T.J.; Sweet, R.; Stanfield, R.L.; Kodangattil, S.; Almagro, J.C.; Gilliland, G.L. Antibody Modeling Assessment II. Structures and Models. Proteins 2014, 82, 1563–1582. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Beavers, M.P.; Hernandez-Guzman, F.; Maier, J.; Shaulsky, J.; Butenhof, K.; Labute, P.; Thorsteinson, N.; Kelly, K.; Teplyakov, A.; et al. Antibody Modeling Assessment. Proteins 2011, 79, 3050–3066. [Google Scholar] [CrossRef] [PubMed]
- Kern, D. From Structure to Mechanism: Skiing the Energy Landscape. Nat. Methods 2021, 18, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic Personalities of Proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Petsko, G.A. Molecular Dynamics Simulations in Biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Landsteiner, K.; Van Der Scheer, J. On the specificity of serological reactions with simple chemical compounds (inhibition reactions). J. Exp. Med. 1931, 54, 295–305. [Google Scholar] [CrossRef]
- Pauling, L. A Theory of the Structure and Process of Formation of Antibodies *. J. Am. Chem. Soc. 1940, 62, 2643–2657. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Kroell, K.B.; Bacher, L.M.; Loeffler, J.R.; Quoika, P.K.; Georges, G.; Bujotzek, A.; Kettenberger, H.; Liedl, K.R. Germline-Dependent Antibody Paratope States and Pairing Specific VH-VL Interface Dynamics. Front. Immunol. 2021, 12, 675655. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Pomarici, N.D.; Math, B.A.; Kroell, K.B.; Waibl, F.; Bujotzek, A.; Georges, G.; Liedl, K.R. Antibodies Exhibit Multiple Paratope States Influencing VH–VL Domain Orientations. Commun. Biol. 2020, 3, 589. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Heiss, M.C.; Pomarici, N.D.; Math, B.A.; Liedl, K.R. Antibody CDR Loops as Ensembles in Solution vs. Canonical Clusters from X-Ray Structures. mAbs 2020, 12, 1744328. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Vangone, A.; Loeffler, J.R.; Seidler, C.A.; Georges, G.; Liedl, K.R. Paratope States in Solution Improve Structure Prediction and Docking. Structure 2022, 30, 430–440.e3. [Google Scholar] [CrossRef] [PubMed]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An Online Tool for Systematic Identification of Antigen-Binding Regions in Antibodies Based on Sequence or Structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, R.M.; Martin, A.C.R.; Thornton, J.M. Antibody-Antigen Interactions: Contact Analysis and Binding Site Topography. J. Mol. Biol. 1996, 262, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Waibl, F.; Fernández-Quintero, M.L.; Kamenik, A.S.; Kraml, J.; Hofer, F.; Kettenberger, H.; Georges, G.; Liedl, K.R. Conformational Ensembles of Antibodies Determine Their Hydrophobicity. Biophys. J. 2021, 120, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Waibl, F.; Fernández-Quintero, M.L.; Wedl, F.S.; Kettenberger, H.; Georges, G.; Liedl, K.R. Comparison of Hydrophobicity Scales for Predicting Biophysical Properties of Antibodies. Front. Mol. Biosci. 2022, 9, 960194. [Google Scholar] [CrossRef]
- Waibl, F.; Pomarici, N.D.; Hoerschinger, V.J.; Loeffler, J.R.; Deane, C.M.; Georges, G.; Kettenberger, H.; Fernández-Quintero, M.L.; Liedl, K.R. PEP-Patch: Electrostatics in Protein-Protein Recognition, Specificity and Antibody Developability. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dunbar, J.; Fuchs, A.; Shi, J.; Deane, C.M. ABangle: Characterising the VH-VL Orientation in Antibodies. Protein Eng. Des. Sel. 2013, 26, 611–620. [Google Scholar] [CrossRef]
- Hoerschinger, V.J.; Fernández-Quintero, M.L.; Waibl, F.; Kraml, J.; Bujotzek, A.; Georges, G.; Liedl, K.R. OCD.Py-Characterizing Immunoglobulin Inter-Domain Orientations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Zemla, A.; Wilson, I.A.; Rupp, B. Antibody Elbow Angles Are Influenced by Their Light Chain Class. J. Mol. Biol. 2006, 357, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Quoika, P.K.; Wedl, F.S.; Seidler, C.A.; Kroell, K.B.; Loeffler, J.R.; Pomarici, N.D.; Hoerschinger, V.J.; Bujotzek, A.; Georges, G.; et al. Comparing Antibody Interfaces to Inform Rational Design of New Antibody Formats. Front. Mol. Biosci. 2022, 9, 812750. [Google Scholar]
- Sotriffer, C.A.; Liedl, K.R.; Linthicum, D.S.; Rode, B.M.; Varga, J.M. Ligand-Induced Domain Movement in an Antibody Fab: Molecular Dynamics Studies Confirm the Unique Domain Movement Observed Experimentally for Fab NC6.8 upon Complexation and Reveal Its Segmental flexibility. J. Mol. Biol. 1998, 278, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Kroell, K.B.; Grunewald, L.J.; Fischer, A.-L.M.; Riccabona, J.R.; Liedl, K.R. CDR Loop Interactions Can Determine Heavy and Light Chain Pairing Preferences in Bispecific Antibodies. mAbs 2022, 14, 2024118. [Google Scholar] [CrossRef] [PubMed]
- Bujotzek, A.; Lipsmeier, F.; Harris, S.F.; Benz, J.; Kuglstatter, A.; Georges, G. VH-VL Orientation Prediction for Antibody Humanization Candidate Selection: A Case Study. mAbs 2016, 8, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. Scaffolding to Build a Rational Vaccine Design Strategy. Proc. Natl. Acad. Sci. USA 2010, 107, 17859–17860. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Wilson, I.A. Innovations in Structure-Based Antigen Design and Immune Monitoring for next Generation Vaccines. Curr. Opin. Immunol. 2020, 65, 50–56. [Google Scholar] [CrossRef]
- Kulp, D.W.; Schief, W.R. Advances in Structure-Based Vaccine Design. Curr. Opin. Virol. 2013, 3, 322–331. [Google Scholar] [CrossRef]
- Krammer, F. The Human Antibody Response to Influenza A Virus Infection and Vaccination. Nat. Rev. Immunol. 2019, 19, 383–397. [Google Scholar] [CrossRef]
- Kirkpatrick, E.; Qiu, X.; Wilson, P.C.; Bahl, J.; Krammer, F. The Influenza Virus Hemagglutinin Head Evolves Faster than the Stalk Domain. Sci. Rep. 2018, 8, 10432. [Google Scholar] [CrossRef]
- Nachbagauer, R.; Feser, J.; Naficy, A.; Bernstein, D.I.; Guptill, J.; Walter, E.B.; Berlanda-Scorza, F.; Stadlbauer, D.; Wilson, P.C.; Aydillo, T.; et al. A Chimeric Hemagglutinin-Based Universal Influenza Virus Vaccine Approach Induces Broad and Long-Lasting Immunity in a Randomized, Placebo-Controlled Phase I Trial. Nat. Med. 2021, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Guptill, J.; Naficy, A.; Nachbagauer, R.; Berlanda-Scorza, F.; Feser, J.; Wilson, P.C.; Solórzano, A.; Van der Wielen, M.; Walter, E.B.; et al. Immunogenicity of Chimeric Haemagglutinin-Based, Universal Influenza Virus Vaccine Candidates: Interim Results of a Randomised, Placebo-Controlled, Phase 1 Clinical Trial. Lancet Infect. Dis. 2020, 20, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.-J.; Kanekiyo, M.; Kong, W.-P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-Stem Nanoparticles Generate Heterosubtypic Influenza Protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Wei, C.-J.; Yassine, H.M.; McTamney, P.M.; Boyington, J.C.; Whittle, J.R.R.; Rao, S.S.; Kong, W.-P.; Wang, L.; Nabel, G.J. Self-Assembling Influenza Nanoparticle Vaccines Elicit Broadly Neutralizing H1N1 Antibodies. Nature 2013, 499, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Boyoglu-Barnum, S.; Ellis, D.; Gillespie, R.A.; Hutchinson, G.B.; Park, Y.-J.; Moin, S.M.; Acton, O.; Ravichandran, R.; Murphy, M.; Pettie, D.; et al. Elicitation of Broadly Protective Immunity to Influenza by Multivalent Hemagglutinin Nanoparticle Vaccines. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kanekiyo, M.; Joyce, M.G.; Gillespie, R.A.; Gallagher, J.R.; Andrews, S.F.; Yassine, H.M.; Wheatley, A.K.; Fisher, B.E.; Ambrozak, D.R.; Creanga, A.; et al. Mosaic Nanoparticle Display of Diverse Influenza Virus Hemagglutinins Elicits Broad B Cell Responses. Nat. Immunol. 2019, 20, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Steichen, J.M.; Lin, Y.-C.; Havenar-Daughton, C.; Pecetta, S.; Ozorowski, G.; Willis, J.R.; Toy, L.; Sok, D.; Liguori, A.; Kratochvil, S.; et al. A Generalized HIV Vaccine Design Strategy for Priming of Broadly Neutralizing Antibody Responses. Science 2019, 366, eaax4380. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.R.; Berndsen, Z.T.; Ma, K.M.; Steichen, J.M.; Schiffner, T.; Landais, E.; Liguori, A.; Kalyuzhniy, O.; Allen, J.D.; Baboo, S.; et al. Human Immunoglobulin Repertoire Analysis Guides Design of Vaccine Priming Immunogens Targeting HIV V2-Apex Broadly Neutralizing Antibody Precursors. Immunity 2022, 55, 2149–2167.e9. [Google Scholar] [CrossRef]
- Melzi, E.; Willis, J.R.; Ma, K.M.; Lin, Y.-C.; Kratochvil, S.; Berndsen, Z.T.; Landais, E.A.; Kalyuzhniy, O.; Nair, U.; Warner, J.; et al. Membrane-Bound mRNA Immunogens Lower the Threshold to Activate HIV Env V2 Apex-Directed Broadly Neutralizing B Cell Precursors in Humanized Mice. Immunity 2022, 55, 2168–2186.e6. [Google Scholar] [CrossRef]
- Leggat, D.J.; Cohen, K.W.; Willis, J.R.; Fulp, W.J.; deCamp, A.C.; Kalyuzhniy, O.; Cottrell, C.A.; Menis, S.; Finak, G.; Ballweber-Fleming, L.; et al. Vaccination Induces HIV Broadly Neutralizing Antibody Precursors in Humans. Science 2022, 378, eadd6502. [Google Scholar] [CrossRef]
- Guthmiller, J.J.; Han, J.; Utset, H.A.; Li, L.; Lan, L.Y.-L.; Henry, C.; Stamper, C.T.; McMahon, M.; O’Dell, G.; Fernández-Quintero, M.L.; et al. Broadly Neutralizing Antibodies Target a Haemagglutinin Anchor Epitope. Nature 2022, 602, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.R.; Zhang, R.; Khurana, S.; King, L.R.; Manischewitz, J.; Golding, H.; Dormitzer, P.R.; Haynes, B.F.; Walter, E.B.; Moody, M.A.; et al. Broadly Neutralizing Human Antibody That Recognizes the Receptor-Binding Pocket of Influenza Virus Hemagglutinin. Proc. Natl. Acad. Sci. USA 2011, 108, 14216–14221. [Google Scholar] [CrossRef]
- Bangaru, S.; Lang, S.; Schotsaert, M.; Vanderven, H.A.; Zhu, X.; Kose, N.; Bombardi, R.; Finn, J.A.; Kent, S.J.; Gilchuk, P.; et al. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell 2019, 177, 1136–1152.e18. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.-A.; Friesen, R.H.E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Rowan-Nash, A.D.; Korry, B.J.; Mylonakis, E.; Belenky, P. Cross-Domain and Viral Interactions in the Microbiome. Microbiol. Mol. Biol. Rev. MMBR 2019, 83, e00044-18. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Wilson, I.A. Influenza Hemagglutinin Structures and Antibody Recognition. Cold Spring Harb. Perspect. Med. 2020, 10, a038778. [Google Scholar] [CrossRef]
- Antanasijevic, A.; Ueda, G.; Brouwer, P.J.M.; Copps, J.; Huang, D.; Allen, J.D.; Cottrell, C.A.; Yasmeen, A.; Sewall, L.M.; Bontjer, I.; et al. Structural and Functional Evaluation of de Novo-Designed, Two-Component Nanoparticle Carriers for HIV Env Trimer Immunogens. PLoS Pathog. 2020, 16, e1008665. [Google Scholar] [CrossRef] [PubMed]
- Ozorowski, G.; Pallesen, J.; de Val, N.; Lyumkis, D.; Cottrell, C.A.; Torres, J.L.; Copps, J.; Stanfield, R.L.; Cupo, A.; Pugach, P.; et al. Open and Closed Structures Reveal Allostery and Pliability in the HIV-1 Envelope Spike. Nature 2017, 547, 360–363. [Google Scholar] [CrossRef]
- Barnes, C.O.; Schoofs, T.; Gnanapragasam, P.N.P.; Golijanin, J.; Huey-Tubman, K.E.; Gruell, H.; Schommers, P.; Suh-Toma, N.; Lee, Y.E.; Cetrulo Lorenzi, J.C.; et al. A Naturally Arising Broad and Potent CD4-Binding Site Antibody with Low Somatic Mutation. Sci. Adv. 2022, 8, eabp8155. [Google Scholar] [CrossRef]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.-B.; Fu, B.Z.; et al. Somatic Mutations of the Immunoglobulin Framework Are Generally Required for Broad and Potent HIV-1 Neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef]
- Scharf, L.; Wang, H.; Gao, H.; Chen, S.; McDowall, A.W.; Bjorkman, P.J. Broadly Neutralizing Antibody 8ANC195 Recognizes Closed and Open States of HIV-1 Env. Cell 2015, 162, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Cottrell, C.A.; Ozorowski, G.; van Gils, M.J.; Kumar, S.; Wu, N.C.; Sarkar, A.; Torres, J.L.; de Val, N.; Copps, J.; et al. Conformational Plasticity in the HIV-1 Fusion Peptide Facilitates Recognition by Broadly Neutralizing Antibodies. Cell Host Microbe 2019, 25, 873–883.e5. [Google Scholar] [CrossRef]
- Schoofs, T.; Barnes, C.O.; Suh-Toma, N.; Golijanin, J.; Schommers, P.; Gruell, H.; West, A.P.J.; Bach, F.; Lee, Y.E.; Nogueira, L.; et al. Broad and Potent Neutralizing Antibodies Recognize the Silent Face of the HIV Envelope. Immunity 2019, 50, 1513–1529.e9. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and Potent Neutralization of HIV-1 by a Gp41-Specific Human Antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Rantalainen, K.; Berndsen, Z.T.; Antanasijevic, A.; Schiffner, T.; Zhang, X.; Lee, W.-H.; Torres, J.L.; Zhang, L.; Irimia, A.; Copps, J.; et al. HIV-1 Envelope and MPER Antibody Structures in Lipid Assemblies. Cell Rep. 2020, 31, 107583. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.G.; Kulp, D.W.; Havenar-Daughton, C.; Sarkar, A.; Briney, B.; Sok, D.; Sesterhenn, F.; Ereño-Orbea, J.; Kalyuzhniy, O.; Deresa, I.; et al. HIV-1 Broadly Neutralizing Antibody Precursor B Cells Revealed by Germline-Targeting Immunogen. Science 2016, 351, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.-Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meining, W.; Cushman, M.; Haase, I.; Fischer, M.; Bacher, A.; Ladenstein, R. A Structure-Based Model of the Reaction Catalyzed by Lumazine Synthase from Aquifex Aeolicus. J. Mol. Biol. 2003, 328, 167–182. [Google Scholar] [CrossRef]
- Bennett, N.R.; Zwick, D.B.; Courtney, A.H.; Kiessling, L.L. Multivalent Antigens for Promoting B and T Cell Activation. ACS Chem. Biol. 2015, 10, 1817–1824. [Google Scholar] [CrossRef]
- De Gregorio, E.; Rappuoli, R. From Empiricism to Rational Design: A Personal Perspective of the Evolution of Vaccine Development. Nat. Rev. Immunol. 2014, 14, 505–514. [Google Scholar] [CrossRef]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse Vaccinology 2.0: Human Immunology Instructs Vaccine Antigen Design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Wilson, I.A. Structural Characterization of Viral Epitopes Recognized by Broadly Cross-Reactive Antibodies. Curr. Top. Microbiol. Immunol. 2015, 386, 323–341. [Google Scholar]
- Wilson, I.A.; Stanfield, R.L. Antibody-Antigen Interactions: New Structures and New Conformational Changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A Computer-Based Archival File for Macromolecular Structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein Crystallography for Aspiring Crystallographers or How to Avoid Pitfalls and Traps in Macromolecular Structure Determination. FEBS J. 2013, 280, 5705–5736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Yang, F.; Smith-Gill, S.J.; Mariuzza, R.A. X-Ray Snapshots of the Maturation of an Antibody Response to a Protein Antigen. Nat. Struct. Biol. 2003, 10, 482–488. [Google Scholar] [CrossRef]
- Purslow, J.A.; Khatiwada, B.; Bayro, M.J.; Venditti, V. NMR Methods for Structural Characterization of Protein-Protein Complexes. Front. Mol. Biosci. 2020, 7, 9. [Google Scholar] [CrossRef]
- Rosen, O.; Anglister, J. Epitope Mapping of Antibody-Antigen Complexes by Nuclear Magnetic Resonance Spectroscopy. Methods Mol. Biol. 2009, 524, 37–57. [Google Scholar]
- Bardelli, M.; Livoti, E.; Simonelli, L.; Pedotti, M.; Moraes, A.; Valente, A.P.; Varani, L. Epitope Mapping by Solution NMR Spectroscopy. J. Mol. Recognition JMR 2015, 28, 393–400. [Google Scholar] [CrossRef]
- Sapienza, P.J.; Lee, A.L. Using NMR to Study Fast Dynamics in Proteins: Methods and Applications. Curr. Opin. Pharmacol. 2010, 10, 723–730. [Google Scholar] [CrossRef]
- Krishnan, V.; Rupp, B. Macromolecular Structure Determination: Comparison of X-ray Crystallography and NMR Spectroscopy. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Shoemaker, S.C.; Ando, N. X-Rays in the Cryo-Electron Microscopy Era: Structural Biology’s Dynamic Future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; McMullan, G.; Scheres, S.H.W. How Cryo-EM Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. Single-Particle Cryo-EM at Crystallographic Resolution. Cell 2015, 161, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Turner, H.L.; Nogal, B.; Cottrell, C.A.; Oyen, D.; Pauthner, M.; Bastidas, R.; Nedellec, R.; McCoy, L.E.; Wilson, I.A.; et al. Electron-Microscopy-Based Epitope Mapping Defines Specificities of Polyclonal Antibodies Elicited during HIV-1 BG505 Envelope Trimer Immunization. Immunity 2018, 49, 288–300.e8. [Google Scholar] [CrossRef] [PubMed]
- Antanasijevic, A.; Bowman, C.A.; Kirchdoerfer, R.N.; Cottrell, C.A.; Ozorowski, G.; Upadhyay, A.A.; Cirelli, K.M.; Carnathan, D.G.; Enemuo, C.A.; Sewall, L.M.; et al. From Structure to Sequence: Antibody Discovery Using cryoEM. Sci. Adv. 2022, 8, eabk2039. [Google Scholar] [CrossRef] [PubMed]
- Janda, A.; Bowen, A.; Greenspan, N.S.; Casadevall, A. Ig Constant Region Effects on Variable Region Structure and Function. Front. Microbiol. 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic Generation of Antibody Diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.J.; Shikhman, A.R.; Glass, D.D.; Kangisser, D.; Cunningham, M.W.; Greenspan, N.S. Role of Heavy Chain Constant Domains in Antibody-Antigen Interaction. Apparent Specificity Differences among Streptococcal IgG Antibodies Expressing Identical Variable Domains. J. Immunol. 1993, 150, 2231–2242. [Google Scholar] [CrossRef]
- Cooper, L.J.; Robertson, D.; Granzow, R.; Greenspan, N.S. Variable Domain-Identical Antibodies Exhibit IgG Subclass-Related Differences in Affinity and Kinetic Constants as Determined by Surface Plasmon Resonance. Mol. Immunol. 1994, 31, 577–584. [Google Scholar] [CrossRef]
- Huber, R.; Deisenhofer, J.; Colman, P.M.; Matsushima, M.; Palm, W. Crystallographic Structure Studies of an IgG Molecule and an Fc Fragment. Nature 1976, 264, 415–420. [Google Scholar] [CrossRef]
- Pritsch, O.; Magnac, C.; Dumas, G.; Bouvet, J.P.; Alzari, P.; Dighiero, G. Can Isotype Switch Modulate Antigen-Binding Affinity and Influence Clonal Selection? Eur. J. Immunol. 2000, 30, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Sheriff, S.; Jeffrey, P.D.; Bajorath, J. Comparison of CH1 Domains in Different Classes of Murine Antibodies. J. Mol. Biol. 1996, 263, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Normansell, D.E. Human Immunoglobulin Subclasses. Diagn. Clin. Immunol. 1987, 5, 115–128. [Google Scholar] [PubMed]
- Torres, M.; Casadevall, A. The Immunoglobulin Constant Region Contributes to Affinity and Specificity. Trends Immunol. 2008, 29, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.F.; Barclay, A.N. The Immunoglobulin Superfamily—Domains for Cell Surface Recognition. Annu. Rev. Immunol. 1988, 6, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.A.; Kota, P.; Demarest, S.J.; Caravella, J.A.; Dokholyan, N.V. Highly Covarying Residues Have a Functional Role in Antibody Constant Domains. Proteins Struct. Funct. Bioinform. 2013, 81, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Pomarici, N.D.; Fernández-Quintero, M.L.; Quoika, P.K.; Waibl, F.; Bujotzek, A.; Georges, G.; Liedl, K.R. Bispecific Antibodies—Effects of Point Mutations on CH3-CH3 Interface Stability. Protein Eng. Des. Sel. 2022, 35, gzac012. [Google Scholar] [CrossRef]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural Analysis of Human IgG-Fc Glycoforms Reveals a Correlation Between Glycosylation and Structural Integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Radaev, S.; Motyka, S.; Fridman, W.-H.; Sautes-Fridman, C.; Sun, P.D. The Structure of a Human Type III Fcγ Receptor in Complex with Fc*. J. Biol. Chem. 2001, 276, 16469–16477. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Zhang, C. Hybridoma Technology for the Generation of Monoclonal Antibodies. In Antibody Methods and Protocols; Proetzel, G., Ebersbach, H., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 117–135. [Google Scholar] [CrossRef]
- ul Haque Saeed, A.F.; ul Haque Saeed, A.F. Advances in Monoclonal Antibodies Production and Cancer Therapy. MOJ Immunol. 2016, 3, 99. [Google Scholar] [CrossRef]
- Vaisman-Mentesh, A.; Rosenstein, S.; Yavzori, M.; Dror, Y.; Fudim, E.; Ungar, B.; Kopylov, U.; Picard, O.; Kigel, A.; Ben-Horin, S.; et al. Molecular Landscape of Anti-Drug Antibodies Reveals the Mechanism of the Immune Response Following Treatment With TNFα Antagonists. Front. Immunol. 2019, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
- Mosch, R.; Guchelaar, H.-J. Immunogenicity of Monoclonal Antibodies and the Potential Use of HLA Haplotypes to Predict Vulnerable Patients. Front. Immunol. 2022, 13, 885672. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.H.; Yang, X.-D. Therapeutic Potential of ABX-EGF: A Fully Human Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Cancer Treatment. Semin. Oncol. 2002, 29 (Suppl. S4), 47–50. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development Trends for Human Monoclonal Antibody Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.-C.; Mashayekh, R.; Wymore, K.; McCabe, J.G.; et al. Antigen-Specific Human Antibodies from Mice Comprising Four Distinct Genetic Modifications. Nature 1994, 368, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Green, L.L. Antibody Engineering via Genetic Engineering of the Mouse: XenoMouse Strains Are a Vehicle for the Facile Generation of Therapeutic Human Monoclonal Antibodies. J. Immunol. Methods 1999, 231, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.D.; Carmack, C.E.; Schramm, S.R.; Mashayekh, R.; Higgins, K.M.; Kuo, C.C.; Woodhouse, C.; Kay, R.M.; Lonberg, N. A Transgenic Mouse That Expresses a Diversity of Human Sequence Heavy and Light Chain Immunoglobulins. Nucleic Acids Res. 1992, 20, 6287–6295. [Google Scholar] [CrossRef]
- Hwang, W.Y.K.; Foote, J. Immunogenicity of Engineered Antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef]
- Shankar, G.; Shores, E.; Wagner, C.; Mire-Sluis, A. Scientific and Regulatory Considerations on the Immunogenicity of Biologics. Trends Biotechnol. 2006, 24, 274–280. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric Human Antibody Molecules: Mouse Antigen-Binding Domains with Human Constant Region Domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the Complementarity-Determining Regions in a Human Antibody with Those from a Mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Bedouelle, H.; Rees, A.R.; Crennell, S.J.; Lefranc, M.-P.; Thullier, P. Germline Humanization of a Non-Human Primate Antibody That Neutralizes the Anthrax Toxin, by in Vitro and in Silico Engineering. J. Mol. Biol. 2008, 384, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Robert, R.; Streltsov, V.A.; Newman, J.; Pearce, L.A.; Wark, K.L.; Dolezal, O. Germline Humanization of a Murine Aβ Antibody and Crystal Structure of the Humanized Recombinant Fab Fragment. Protein Sci. 2010, 19, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.-K.V.; Guo, H.; Hu, J.; Tassev, D.V.; Cheung, I.Y. Humanizing Murine IgG3 Anti-GD2 Antibody m3F8 Substantially Improves Antibody-Dependent Cell-Mediated Cytotoxicity While Retaining Targeting in Vivo. OncoImmunology 2012, 1, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hong, H.J. Humanization by CDR Grafting and Specificity-Determining Residue Grafting. In Antibody Engineering: Methods and Protocols, 2nd ed.; Chames, P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 237–245. [Google Scholar] [CrossRef]
- Makabe, K.; Nakanishi, T.; Tsumoto, K.; Tanaka, Y.; Kondo, H.; Umetsu, M.; Sone, Y.; Asano, R.; Kumagai, I. Thermodynamic Consequences of Mutations in Vernier Zone Residues of a Humanized Anti-Human Epidermal Growth Factor Receptor Murine Antibody, 528*. J. Biol. Chem. 2008, 283, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Doria-Rose, N.A.; Joyce, M.G. Strategies to Guide the Antibody Affinity Maturation Process. Curr. Opin. Virol. 2015, 11, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Ducancel, F.; Muller, B.H. Molecular Engineering of Antibodies for Therapeutic and Diagnostic Purposes. mAbs 2012, 4, 445–457. [Google Scholar] [CrossRef]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed Evolution of Antibody Fragments with Monovalent Femtomolar Antigen-Binding Affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef]
- Rajpal, A.; Beyaz, N.; Haber, L.; Cappuccilli, G.; Yee, H.; Bhatt, R.R.; Takeuchi, T.; Lerner, R.A.; Crea, R. A General Method for Greatly Improving the Affinity of Antibodies by Using Combinatorial Libraries. Proc. Natl. Acad. Sci. USA 2005, 102, 8466–8471. [Google Scholar] [CrossRef]
- Lippow, S.M.; Wittrup, K.D.; Tidor, B. Computational Design of Antibody Affinity Improvement beyond in Vivo Maturation. Nat. Biotechnol. 2007, 25, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Boriack-Sjodin, P.A.; Eldredge, J.; Fitch, C.; Friedman, B.; Hanf, K.J.M.; Jarpe, M.; Liparoto, S.F.; Li, Y.; Lugovskoy, A.; et al. Affinity Enhancement of an in Vivo Matured Therapeutic Antibody Using Structure-Based Computational Design. Protein Sci. 2006, 15, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Tamura, H.; Caaveiro, J.M.M.; Tsumoto, K. Computer-Guided Library Generation Applied to the Optimization of Single-Domain Antibodies. Protein Eng. Des. Sel. 2019, 32, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Okuno, Y.; Ohta, M. Structure-Based Affinity Maturation of Antibody Based on Double-Point Mutations. In Computer-Aided Antibody Design; Tsumoto, K., Kuroda, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; pp. 323–331. [Google Scholar] [CrossRef]
- Sulea, T.; Deprez, C.; Corbeil, C.R.; Purisima, E.O. Optimizing Antibody–Antigen Binding Affinities with the ADAPT Platform. In Computer-Aided Antibody Design; Tsumoto, K., Kuroda, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; pp. 361–374. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Loeffler, J.R.; Bacher, L.M.; Waibl, F.; Seidler, C.A.; Liedl, K.R. Local and Global Rigidification Upon Antibody Affinity Maturation. Front. Mol. Biosci. 2020, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Seidler, C.A.; Quoika, P.K.; Liedl, K.R. Shark Antibody Variable Domains Rigidify Upon Affinity Maturation—Understanding the Potential of Shark Immunoglobulins as Therapeutics. Front. Mol. Biosci. 2021, 8, 639166. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E. Einfluss Der Configuration Auf Die Wirkung Der Enzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Braden, B.C.; Dall’Acqua, W.; Eisenstein, E.; Fields, B.A.; Goldbaum, F.A.; Malchiodi, E.L.; Mariuzza, R.A.; Schwarz, F.P.; Ysern, X.; Poljak, R.J. Protein Motion and Lock and Key Complementarity in Antigen-Antibody Reactions. Pharm. Acta Helv. 1995, 69, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Kumar, S.; Tsai, C.-J.; Nussinov, R. Folding Funnels and Binding Mechanisms. Protein Eng. Des. Sel. 1999, 12, 713–720. [Google Scholar] [CrossRef]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced Fit, Conformational Selection and Independent Dynamic Segments: An Extended View of Binding Events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef]
- Wang, W.; Ye, W.; Yu, Q.; Jiang, C.; Zhang, J.; Luo, R.; Chen, H.-F. Conformational Selection and Induced Fit in Specific Antibody and Antigen Recognition: SPE7 as a Case Study. J. Phys. Chem. B 2013, 117, 4912–4923. [Google Scholar] [CrossRef]
- Klein, C.; Sustmann, C.; Thomas, M.; Stubenrauch, K.; Croasdale, R.; Schanzer, J.; Brinkmann, U.; Kettenberger, H.; Regula, J.T.; Schaefer, W. Progress in Overcoming the Chain Association Issue in Bispecific Heterodimeric IgG Antibodies. mAbs 2012, 4, 653–663. [Google Scholar] [CrossRef]
- Krah, S.; Sellmann, C.; Rhiel, L.; Schröter, C.; Dickgiesser, S.; Beck, J.; Zielonka, S.; Toleikis, L.; Hock, B.; Kolmar, H.; et al. Engineering Bispecific Antibodies with Defined Chain Pairing. New Biotechnol. 2017, 39, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Krah, S.; Kolmar, H.; Becker, S.; Zielonka, S. Engineering IgG-Like Bispecific Antibodies—An Overview. Antibodies 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Seok, S.-H.; Kim, Y.-J.; Seo, M.-D.; Kim, Y.-S. Crystal Structures of Immunoglobulin Fc Heterodimers Reveal the Molecular Basis for Heterodimer Formation. Mol. Immunol. 2015, 65, 377–383. [Google Scholar] [CrossRef] [PubMed]
- De Nardis, C.; Hendriks, L.J.A.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.H.; de Kruif, J. A New Approach for Generating Bispecific Antibodies Based on a Common Light Chain Format and the Stable Architecture of Human Immunoglobulin G1. J. Biol. Chem. 2017, 292, 14706–14717. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing Antibody Fc Heterodimer Formation through Electrostatic Steering Effects. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. “Knobs-into-Holes” Engineering of Antibody CH3 Domains for Heavy Chain Heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Ultsch, M.; Lee, J.; Tong, R.; Takeda, K.; Spiess, C.; Eigenbrot, C.; Scheer, J.M. Antiparallel Conformation of Knob and Hole Aglycosylated Half-Antibody Homodimers Is Mediated by a CH2-CH3 Hydrophobic Interaction. J. Mol. Biol. 2014, 426, 1947–1957. [Google Scholar] [CrossRef]
- Dengl, S.; Mayer, K.; Bormann, F.; Duerr, H.; Hoffmann, E.; Nussbaum, B.; Tischler, M.; Wagner, M.; Kuglstatter, A.; Leibrock, L.; et al. Format Chain Exchange (FORCE) for High-Throughput Generation of Bispecific Antibodies in Combinatorial Binder-Format Matrices. Nat. Commun. 2020, 11, 4974. [Google Scholar] [CrossRef]
- Krah, S.; Schröter, C.; Eller, C.; Rhiel, L.; Rasche, N.; Beck, J.; Sellmann, C.; Günther, R.; Toleikis, L.; Hock, B.; et al. Generation of Human Bispecific Common Light Chain Antibodies by Combining Animal Immunization and Yeast Display. Protein Eng. Des. Sel. 2017, 30, 291–301. [Google Scholar] [CrossRef]
- Van Blarcom, T.; Lindquist, K.; Melton, Z.; Cheung, W.L.; Wagstrom, C.; McDonough, D.; Valle Oseguera, C.; Ding, S.; Rossi, A.; Potluri, S.; et al. Productive Common Light Chain Libraries Yield Diverse Panels of High Affinity Bispecific Antibodies. mAbs 2018, 10, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a Bispecific Antibody with a Common Light Chain: Identification and Optimization of an Anti-CD3 Epsilon and Anti-GPC3 Bispecific Antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, M.; Sellmann, C.; Maresch, D.; Halbig, C.; Becker, S.; Toleikis, L.; Hock, B.; Rüker, F. Novel CH1:CL Interfaces That Enhance Correct Light Chain Pairing in Heterodimeric Bispecific Antibodies. Protein Eng. Des. Sel. 2017, 30, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin Domain Crossover as a Generic Approach for the Production of Bispecific IgG Antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Yan, H.; Zhang, Y.; Mernaugh, R.L.; Zeng, X. Engineering Peptide Linkers for scFv Immunosensors. Anal. Chem. 2008, 80, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Yusakul, G.; Sakamoto, S.; Pongkitwitoon, B.; Tanaka, H.; Morimoto, S. Effect of Linker Length between Variable Domains of Single Chain Variable Fragment Antibody against Daidzin on Its Reactivity. Biosci. Biotechnol. Biochem. 2016, 80, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small Bivalent and Bispecific Antibody Fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed]
- Kwon, N.-Y.; Kim, Y.; Lee, J.-O. Structural Diversity and Flexibility of Diabodies. Methods 2019, 154, 136–142. [Google Scholar] [CrossRef]
- Math, B.A.; Waibl, F.; Lamp, L.M.; Fernández-Quintero, M.L.; Liedl, K.R. Cross-linking Disulfide Bonds Govern Solution Structures of Diabodies. Proteins 2023, 91, 1316–1328. [Google Scholar] [CrossRef]
- Harmsen, M.; de Haard, H. Properties, Production, and Applications of Camelid Single-Domain Antibody Fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal Structure of a Shark Single-Domain Antibody V Region in Complex with Lysozyme. Science 2004, 305, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.; Flajnik, M.F. Antibody Repertoire Development in Cartilaginous Fish. Dev. Comp. Immunol. 2006, 30, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Matz, H.; Dooley, H. Shark IgNAR-Derived Binding Domains as Potential Diagnostic and Therapeutic Agents. Dev. Comp. Immunol. 2019, 90, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, F.; Corbeil, C.R.; Purisima, E.O.; Sulea, T. Coevolved Canonical Loops Conformations of Single-Domain Antibodies: A Tale of Three Pockets Playing Musical Chairs. Front. Immunol. 2022, 13, 884132. [Google Scholar] [CrossRef] [PubMed]
- Sehlin, D.; Stocki, P.; Gustavsson, T.; Hultqvist, G.; Walsh, F.S.; Rutkowski, J.L.; Syvänen, S. Brain Delivery of Biologics Using a Cross-Species Reactive Transferrin Receptor 1 VNAR Shuttle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13272–13283. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.; Stocki, P.; Sinclair, E.H.; Gauhar, A.; Fletcher, E.J.R.; Krawczun-Rygmaczewska, A.; Duty, S.; Walsh, F.S.; Doherty, P.; Rutkowski, J.L. A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease. Pharmaceutics 2022, 14, 1335. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S.; Atarhouch, T.; Saldanha, J.; Barbosa, J.A.R.G.; Hamers, R. Sequence and Structure of VH Domain from Naturally Occurring Camel Heavy Chain Immunoglobulins Lacking Light Chains. Protein Eng. Des. Sel. 1994, 7, 1129–1135. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular Basis for the Preferential Cleft Recognition by Dromedary Heavy-Chain Antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef]
- Zavrtanik, U.; Lukan, J.; Loris, R.; Lah, J.; Hadži, S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J. Mol. Biol. 2018, 430, 4369–4386. [Google Scholar] [CrossRef]
- Muyldermans, S.; Cambillau, C.; Wyns, L. Recognition of Antigens by Single-Domain Antibody Fragments: The Superfluous Luxury of Paired Domains. Trends Biochem. Sci. 2001, 26, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Govaert, J.; Pellis, M.; Deschacht, N.; Vincke, C.; Conrath, K.; Muyldermans, S.; Saerens, D. Dual Beneficial Effect of Interloop Disulfide Bond for Single Domain Antibody Fragments*. J. Biol. Chem. 2011, 287, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Löhr, T.; Sormanni, P.; Vendruscolo, M. Conformational Entropy as a Potential Liability of Computationally Designed Antibodies. Biomolecules 2022, 12, 718. [Google Scholar] [CrossRef] [PubMed]
- Ewert, S.; Cambillau, C.; Conrath, K.; Plückthun, A. Biophysical Properties of Camelid V(HH) Domains Compared to Those of Human V(H)3 Domains. Biochemistry 2002, 41, 3628–3636. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.; Zhao, Y.; Malia, T.J.; Obmolova, G.; Gilliland, G.L. IgG2 Fc Structure and the Dynamic Features of the IgG CH2-CH3 Interface. Mol. Immunol. 2013, 56, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Fischer, A.-L.M.; Kokot, J.; Waibl, F.; Seidler, C.A.; Liedl, K.R. The Influence of Antibody Humanization on Shark Variable Domain (VNAR) Binding Site Ensembles. Front. Immunol. 2022, 13, 953917. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; Seidler, C.A.; Liedl, K.R. T-Cell Receptor Variable β Domains Rigidify During Affinity Maturation. Sci. Rep. 2020, 10, 4472. [Google Scholar] [CrossRef]
- Kovalenko, O.; Olland, A.; Piché-Nicholas, N.; Godbole, A.; King, D.; Svenson, K.; Calabro, V.; Müller, M.; Barelle, C.; Somers, W.; et al. Atypical Antigen Recognition Mode of a Shark IgNAR Variable Domain Characterized by Humanization and Structural Analysis. J. Biol. Chem. 2013, 288, 17408–17419. [Google Scholar] [CrossRef]
- Brazeau, M.D.; Friedman, M. The Origin and Early Phylogenetic History of Jawed Vertebrates. Nature 2015, 520, 490–497. [Google Scholar] [CrossRef]
- Flajnik, M.F. A Cold-Blooded View of Adaptive Immunity. Nat. Rev. Immunol. 2018, 18, 438–453. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel Heavy-Chain Antibodies: Diverse Germline V(H)H and Specific Mechanisms Enlarge the Antigen-Binding Repertoire. EMBO J. 2000, 19, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.A. The History of Old World Camelids in the Light of Molecular Genetics. Trop. Anim. Health Prod. 2016, 48, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Flajnik, M.F.; Deschacht, N.; Muyldermans, S. A Case of Convergence: Why Did a Simple Alternative to Canonical Antibodies Arise in Sharks and Camels? PLoS Biol. 2011, 9, e1001120. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.K.; Su, C.; Muyldermans, S.; van der Loo, W. Heavy-Chain Antibodies in Camelidae; a Case of Evolutionary Innovation. Immunogenetics 2002, 54, 39–47. [Google Scholar] [PubMed]
- Klarenbeek, A.; Mazouari, K.E.; Desmyter, A.; Blanchetot, C.; Hultberg, A.; de Jonge, N.; Roovers, R.C.; Cambillau, C.; Spinelli, S.; Del-Favero, J.; et al. Camelid Ig V Genes Reveal Significant Human Homology Not Seen in Therapeutic Target Genes, Providing for a Powerful Therapeutic Antibody Platform. mAbs 2015, 7, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Rossotti, M.A.; Bélanger, K.; Henry, K.A.; Tanha, J. Immunogenicity and Humanization of Single-Domain Antibodies. FEBS J. 2022, 289, 4304–4327. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Dooley, H.; Verdino, P.; Flajnik, M.F.; Wilson, I.A. Maturation of Shark Single-Domain (IgNAR) Antibodies: Evidence for Induced-Fit Binding. J. Mol. Biol. 2007, 367, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Hoey, R.J.; Eom, H.; Horn, J.R. Structure and Development of Single Domain Antibodies as Modules for Therapeutics and Diagnostics. Exp. Biol. Med. 2019, 244, 1568–1576. [Google Scholar] [CrossRef]
- Sulea, T. Humanization of Camelid Single-Domain Antibodies. Methods Mol. Biol. 2022, 2446, 299–312. [Google Scholar]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as Therapeutics: Big Opportunities for Small Antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Steven, J.; Müller, M.R.; Carvalho, M.F.; Ubah, O.C.; Kovaleva, M.; Donohoe, G.; Baddeley, T.; Cornock, D.; Saunders, K.; Porter, A.J.; et al. In Vitro Maturation of a Humanized Shark VNAR Domain to Improve Its Biophysical Properties to Facilitate Clinical Development. Front. Immunol. 2017, 8, 1361. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Nakakido, M.; Mori, C.; Kuroda, D.; Caaveiro, J.M.M.; Tsumoto, K. Molecular Basis for Thermal Stability and Affinity in a VHH: Contribution of the Framework Region and Its Influence in the Conformation of the CDR3. Protein Sci. A Publ. Protein Soc. 2022, 31, e4450. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintero, M.L.; DeRose, E.F.; Gabel, S.A.; Mueller, G.A.; Liedl, K.R. Nanobody Paratope Ensembles in Solution Characterized by MD Simulations and NMR. Int. J. Mol. Sci. 2022, 23, 5419. [Google Scholar] [CrossRef] [PubMed]
- Regep, C.; Georges, G.; Shi, J.; Popovic, B.; Deane, C.M. The H3 Loop of Antibodies Shows Unique Structural Characteristics. Proteins 2017, 85, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Abanades, B.; Wong, W.K.; Boyles, F.; Georges, G.; Bujotzek, A.; Deane, C.M. ImmuneBuilder: Deep-Learning Models for Predicting the Structures of Immune Proteins. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Halfon, M.; Schneidman-Duhovny, D. NanoNet: Rapid and Accurate End-to-End Nanobody Modeling by Deep Learning. Front. Immunol. 2022, 13, 958584. [Google Scholar] [CrossRef] [PubMed]
- West, B.R.; Wec, A.Z.; Moyer, C.L.; Fusco, M.L.; Ilinykh, P.A.; Huang, K.; Wirchnianski, A.S.; James, R.M.; Herbert, A.S.; Hui, S.; et al. Structural Basis of Broad Ebolavirus Neutralization by a Human Survivor Antibody. Nat. Struct. Mol. Biol. 2019, 26, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Tereshko, V.; Uysal, S.; Margalef, K.; Kossiakoff, A.A.; Koide, S. Exploring the Capacity of Minimalist Protein Interfaces: Interface Energetics and Affinity Maturation to Picomolar KD of a Single-Domain Antibody with a Flat Paratope. J. Mol. Biol. 2007, 373, 941–953. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Quintero, M.L.; Pomarici, N.D.; Fischer, A.-L.M.; Hoerschinger, V.J.; Kroell, K.B.; Riccabona, J.R.; Kamenik, A.S.; Loeffler, J.R.; Ferguson, J.A.; Perrett, H.R.; et al. Structure and Dynamics Guiding Design of Antibody Therapeutics and Vaccines. Antibodies 2023, 12, 67. https://doi.org/10.3390/antib12040067
Fernández-Quintero ML, Pomarici ND, Fischer A-LM, Hoerschinger VJ, Kroell KB, Riccabona JR, Kamenik AS, Loeffler JR, Ferguson JA, Perrett HR, et al. Structure and Dynamics Guiding Design of Antibody Therapeutics and Vaccines. Antibodies. 2023; 12(4):67. https://doi.org/10.3390/antib12040067
Chicago/Turabian StyleFernández-Quintero, Monica L., Nancy D. Pomarici, Anna-Lena M. Fischer, Valentin J. Hoerschinger, Katharina B. Kroell, Jakob R. Riccabona, Anna S. Kamenik, Johannes R. Loeffler, James A. Ferguson, Hailee R. Perrett, and et al. 2023. "Structure and Dynamics Guiding Design of Antibody Therapeutics and Vaccines" Antibodies 12, no. 4: 67. https://doi.org/10.3390/antib12040067
APA StyleFernández-Quintero, M. L., Pomarici, N. D., Fischer, A.-L. M., Hoerschinger, V. J., Kroell, K. B., Riccabona, J. R., Kamenik, A. S., Loeffler, J. R., Ferguson, J. A., Perrett, H. R., Liedl, K. R., Han, J., & Ward, A. B. (2023). Structure and Dynamics Guiding Design of Antibody Therapeutics and Vaccines. Antibodies, 12(4), 67. https://doi.org/10.3390/antib12040067