
Profiling the Biophysical Developability Properties of Common IgG1 Fc Effector Silencing Variants
Abstract
:1. Introduction
2. Materials and Methods
2.1. Polyspecificity Reagent (PSR) Binding Assay
2.2. Affinity-Capture Self-Interaction Nanoparticle Spectroscopy (AC-SINS)
2.3. Hydrophobic Interaction Chromatography (HIC)
2.4. Size Exclusion Chromatography (SEC)
2.5. pH 3.5 Stress SEC
2.6. Thermal Melting (Tm) Measurements by Differential Scanning Fluorescence (DSF)
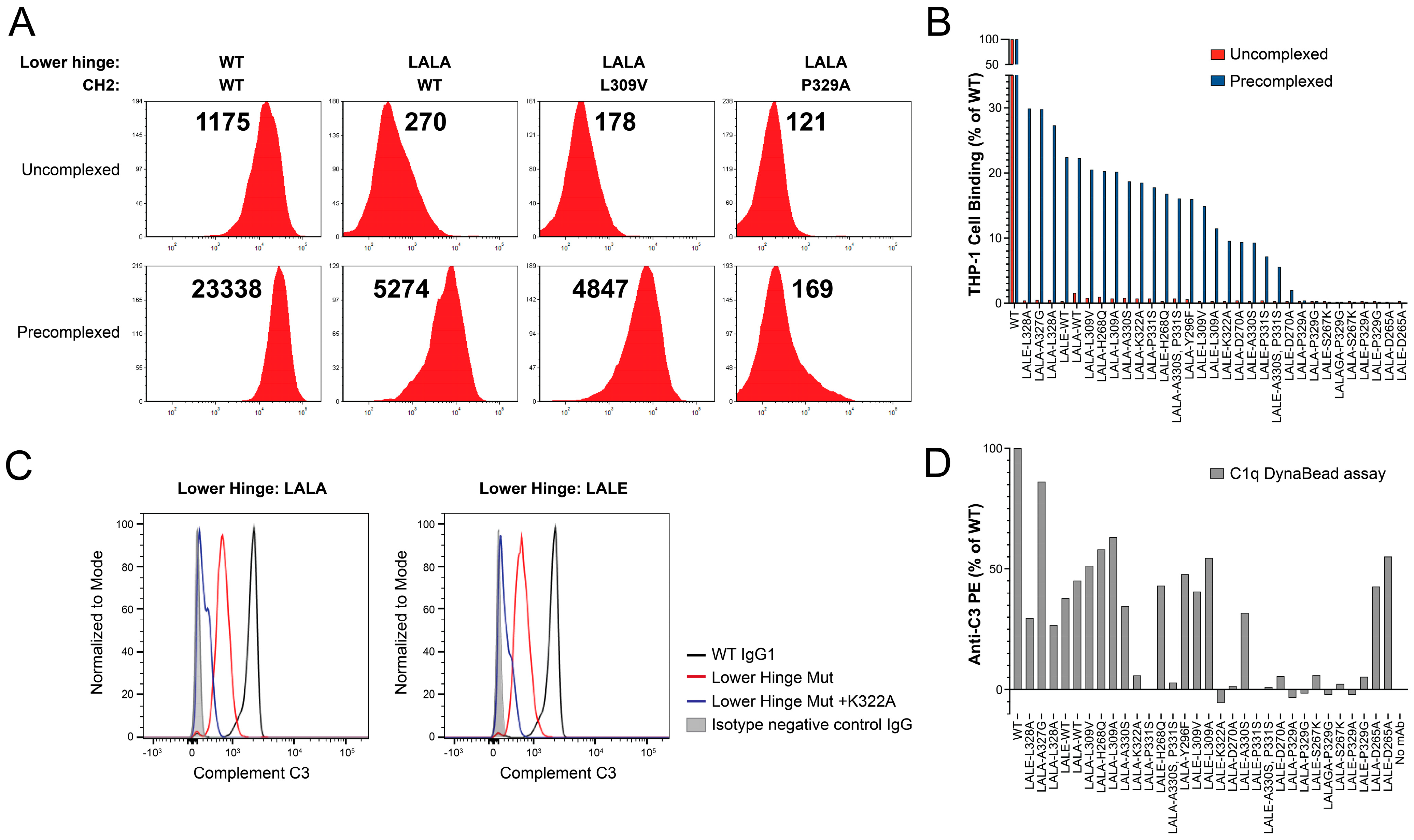
2.7. Profiling FcγR Present on Human Leukemia Monocytic Line THP-1
2.8. Assessing the Binding of Variant IgG1 Antibodies to THP-1 under Highly-Avid Conditions
2.9. Dynal Bead Assay for the Detection of Complement Activation by IgG
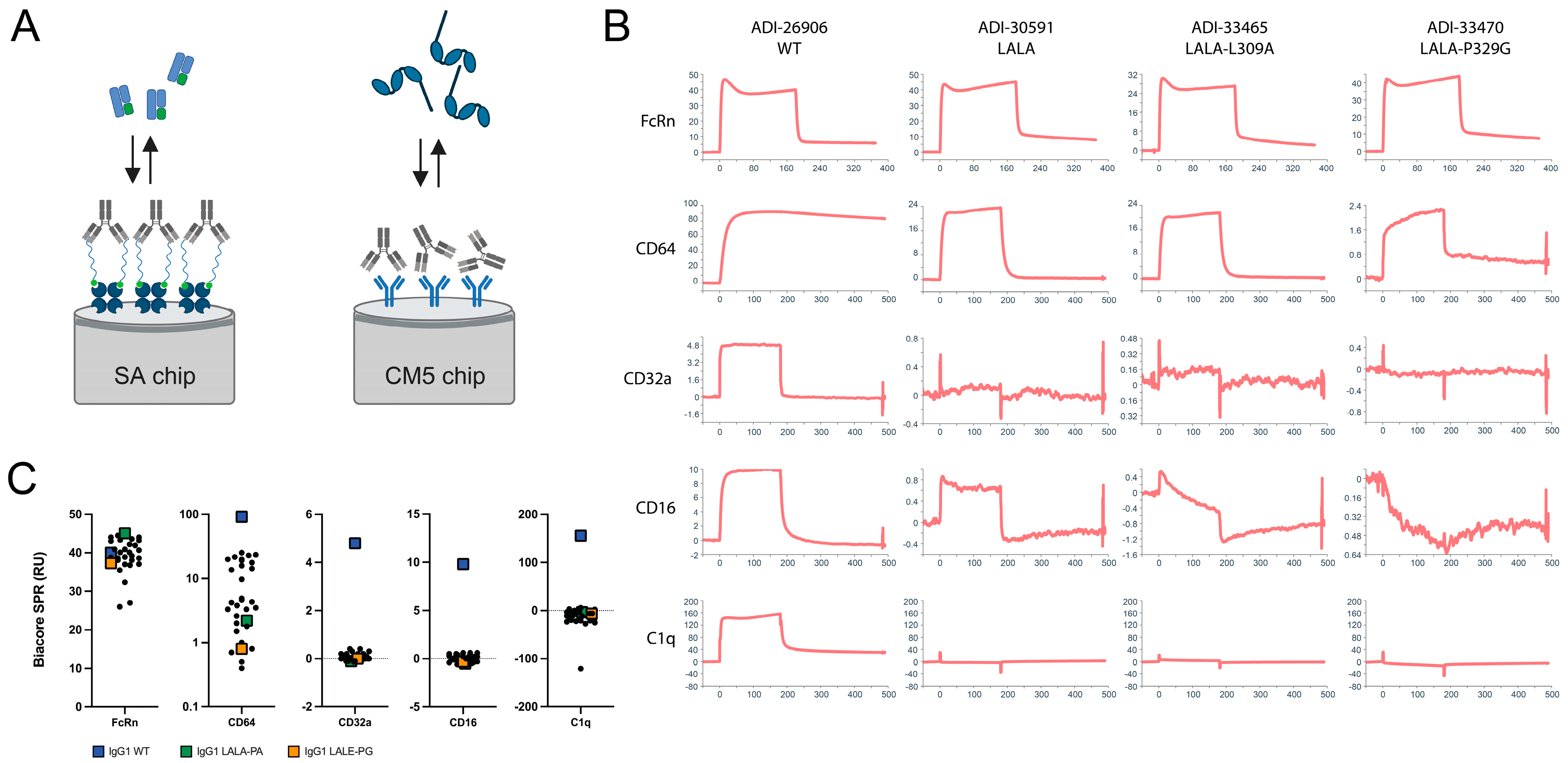
2.10. Biolayer Interferometry (BLI) Binding Asesssment of Variant Antibodies
2.11. Biacore Surface Plasmon Resonance (SPR) Binding Assessment of Variant Antibodies
3. Results
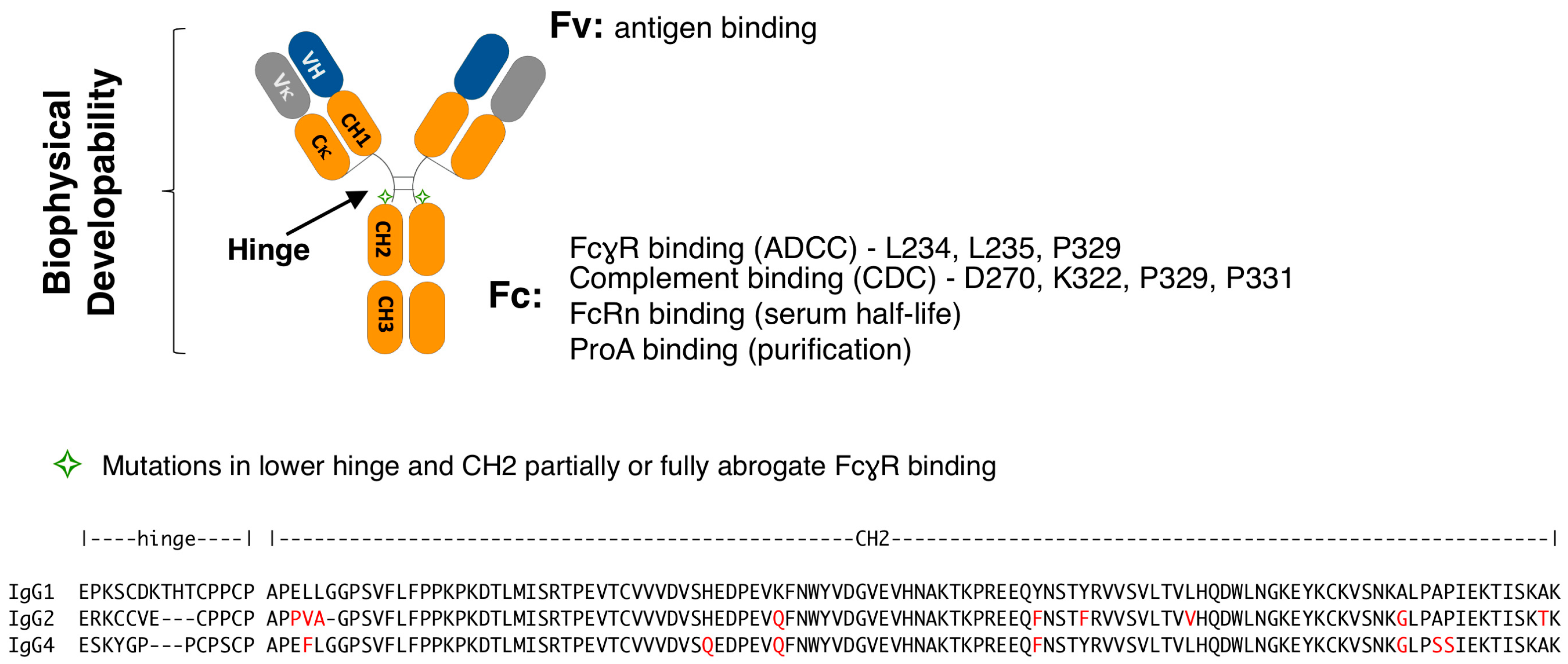
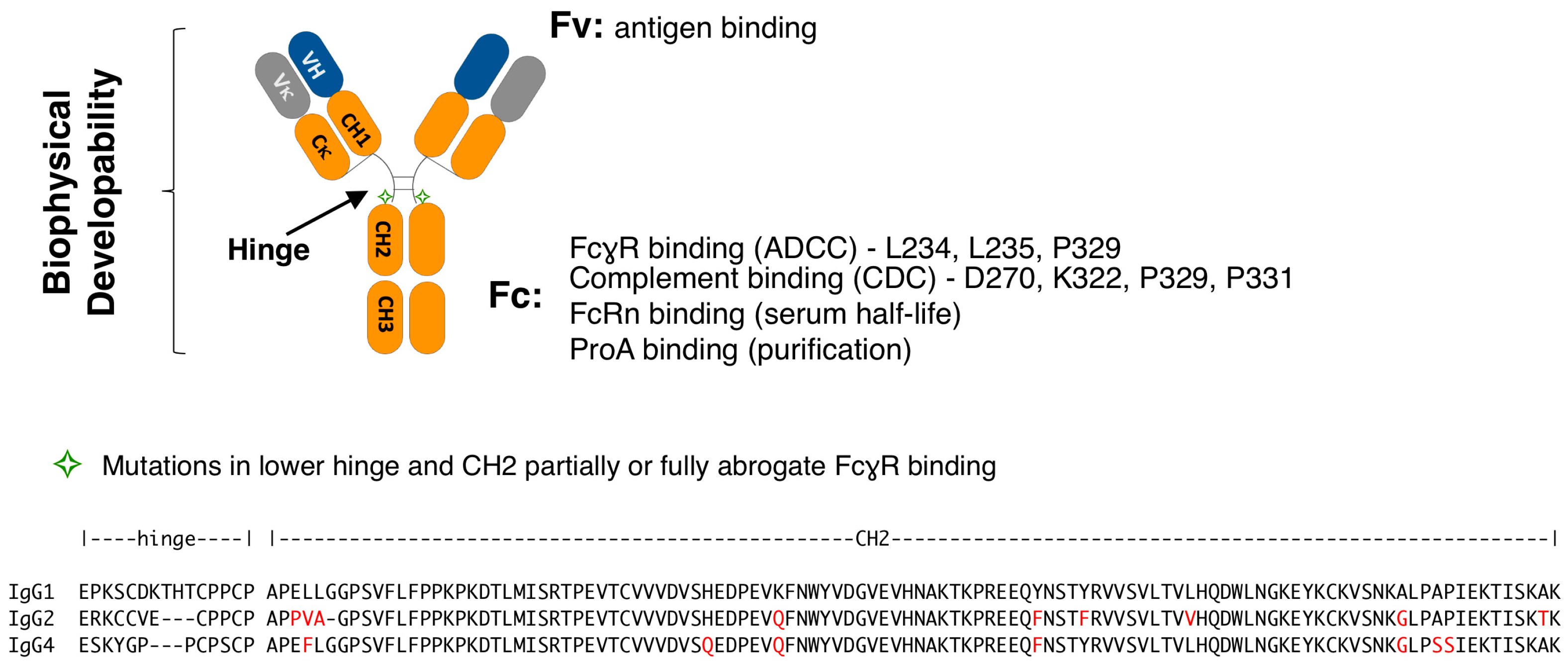
3.1. Lower Hinge Variants
3.2. Lower Hinge and CH2 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, P.J.; Rajpal, A. Designing antibodies as therapeutics. Cell 2022, 185, 2789–2805. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hoseini, S.S.; Xu, H.; Ponomarev, V.; Cheung, N.K. Silencing Fc Domains in T cell-Engaging Bispecific Antibodies Improves T-cell Trafficking and Antitumor Potency. Cancer Immunol. Res. 2019, 7, 2013–2024. [Google Scholar] [CrossRef]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Carter, P. Tunable antibodies. Nat. Biotechnol. 2005, 23, 556–557. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Vaughn, D.E.; Bjorkman, P.J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 1998, 6, 63–73. [Google Scholar] [CrossRef]
- Cooper, N.R. The classical complement pathway: Activation and regulation of the first complement component. Adv. Immunol. 1985, 37, 151–216. [Google Scholar] [CrossRef]
- Zarrineh, M.; Mashhadi, I.S.; Farhadpour, M.; Ghassempour, A. Mechanism of antibodies purification by protein A. Anal. Biochem. 2020, 609, 113909. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Salfeld, J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Guo, H.; Xu, J.; Qin, T.; Xu, L.; Zhang, J.; Guo, Q.; Zhang, D.; Qian, W.; Li, B.; et al. Acid-induced aggregation propensity of nivolumab is dependent on the Fc. MAbs 2016, 8, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tsumoto, K. Effects of subclass change on the structural stability of chimeric, humanized, and human antibodies under thermal stress. Protein Sci. 2013, 22, 1542–1551. [Google Scholar] [CrossRef]
- Ejima, D.; Tsumoto, K.; Fukada, H.; Yumioka, R.; Nagase, K.; Arakawa, T.; Philo, J.S. Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins 2007, 66, 954–962. [Google Scholar] [CrossRef]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Caaveiro, J.M.; Kiyoshi, M.; Tsumoto, K. Structural analysis of Fc/FcgammaR complexes: A blueprint for antibody design. Immunol. Rev. 2015, 268, 201–221. [Google Scholar] [CrossRef]
- Lu, J.; Ellsworth, J.L.; Hamacher, N.; Oak, S.W.; Sun, P.D. Crystal structure of Fcgamma receptor I and its implication in high affinity gamma-immunoglobulin binding. J. Biol. Chem. 2011, 286, 40608–40613. [Google Scholar] [CrossRef]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef]
- Maxwell, K.F.; Powell, M.S.; Hulett, M.D.; Barton, P.A.; McKenzie, I.F.; Garrett, T.P.; Hogarth, P.M. Crystal structure of the human leukocyte Fc receptor, Fc gammaRIIa. Nat. Struct. Biol. 1999, 6, 437–442. [Google Scholar] [CrossRef]
- Kiyoshi, M.; Caaveiro, J.M.; Kawai, T.; Tashiro, S.; Ide, T.; Asaoka, Y.; Hatayama, K.; Tsumoto, K. Structural basis for binding of human IgG1 to its high-affinity human receptor FcgammaRI. Nat. Commun. 2015, 6, 6866. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Gao, C.; Shirinian, L.; Wu, H.; Dall’Acqua, W.F. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jager, C.; Sondermann, P.; Brunker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Natsume, A.; Uehara, A.; Wakitani, M.; Iida, S.; Uchida, K.; Satoh, M.; Shitara, K. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J. Immunol. Methods 2005, 306, 151–160. [Google Scholar] [CrossRef]
- Edelman, G.M.; Cunningham, B.A.; Gall, W.E.; Gottlieb, P.D.; Rutishauser, U.; Waxdal, M.J. The covalent structure of an entire gammaG immunoglobulin molecule. Proc. Natl. Acad. Sci. USA 1969, 63, 78–85. [Google Scholar] [CrossRef]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef]
- Martin, W.L.; West, A.P., Jr.; Gan, L.; Bjorkman, P.J. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: Mechanism of pH-dependent binding. Mol. Cell 2001, 7, 867–877. [Google Scholar] [CrossRef]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef]
- Idusogie, E.E.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Wong, P.Y.; Ultsch, M.; Meng, Y.G.; Mulkerrin, M.G. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 2000, 164, 4178–4184. [Google Scholar] [CrossRef]
- Thommesen, J.E.; Michaelsen, T.E.; Loset, G.A.; Sandlie, I.; Brekke, O.H. Lysine 322 in the human IgG3 C(H)2 domain is crucial for antibody dependent complement activation. Mol. Immunol. 2000, 37, 995–1004. [Google Scholar] [CrossRef]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Winter, G.; Jones, P.T.; Pound, J.D.; Tanaka, T.; Walker, M.R.; Artymiuk, P.J.; Arata, Y.; Burton, D.R.; Jefferis, R.; et al. Human Fc gamma RI and Fc gamma RII interact with distinct but overlapping sites on human IgG. J. Immunol. 1991, 147, 2657–2662. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L. Compositions and Methods for the Treatment of Neuromyelitis Optica. US10654916B2, 2012. Available online: https://patents.google.com/patent/US10654916B2/en (accessed on 6 July 2023).
- Xu, Y.; Oomen, R.; Klein, M.H. Residue at position 331 in the IgG1 and IgG4 CH2 domains contributes to their differential ability to bind and activate complement. J. Biol. Chem. 1994, 269, 3469–3474. [Google Scholar] [CrossRef]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umana, P.; Mossner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine 2020, 52, 102625. [Google Scholar] [CrossRef] [PubMed]
- Armour, K.L.; Clark, M.R.; Hadley, A.G.; Williamson, L.M. Recombinant human IgG molecules lacking Fcgamma receptor I binding and monocyte triggering activities. Eur. J. Immunol. 1999, 29, 2613–2624. [Google Scholar] [CrossRef]
- Tam, S.H.; McCarthy, S.G.; Armstrong, A.A.; Somani, S.; Wu, S.J.; Liu, X.; Gervais, A.; Ernst, R.; Saro, D.; Decker, R.; et al. Functional, Biophysical, and Structural Characterization of Human IgG1 and IgG4 Fc Variants with Ablated Immune Functionality. Antibodies 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.; Parren, P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar] [CrossRef]
- Bolt, S.; Routledge, E.; Lloyd, I.; Chatenoud, L.; Pope, H.; Gorman, S.D.; Clark, M.; Waldmann, H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur. J. Immunol. 1993, 23, 403–411. [Google Scholar] [CrossRef]
- Leabman, M.K.; Meng, Y.G.; Kelley, R.F.; DeForge, L.E.; Cowan, K.J.; Iyer, S. Effects of altered FcgammaR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs 2013, 5, 896–903. [Google Scholar] [CrossRef]
- Jacobsen, F.W.; Stevenson, R.; Li, C.; Salimi-Moosavi, H.; Liu, L.; Wen, J.; Luo, Q.; Daris, K.; Buck, L.; Miller, S.; et al. Engineering an IgG Scaffold Lacking Effector Function with Optimized Developability. J. Biol. Chem. 2017, 292, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Ahonen, C.L.; Brown, M.E.; Zhou, L.; Welin, M.; Krauland, E.M.; Pejchal, R.; Widboom, P.F.; Battles, M.B. Structure-based engineering of a novel CD3epsilon-targeting antibody for reduced polyreactivity. MAbs 2023, 15, 2189974. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.L.; Geoghegan, J.C.; Feldman, J.; Jain, T.; Kauke, M.; Le, D.; Zhao, J.; Wittrup, K.D. Chaperone proteins as single component reagents to assess antibody nonspecificity. MAbs 2017, 9, 1036–1040. [Google Scholar] [CrossRef]
- Xu, Y.; Roach, W.; Sun, T.; Jain, T.; Prinz, B.; Yu, T.Y.; Torrey, J.; Thomas, J.; Bobrowicz, P.; Vasquez, M.; et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 2013, 26, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Shehata, L.; Maurer, D.P.; Wec, A.Z.; Lilov, A.; Champney, E.; Sun, T.; Archambault, K.; Burnina, I.; Lynaugh, H.; Zhi, X.; et al. Affinity Maturation Enhances Antibody Specificity but Compromises Conformational Stability. Cell Rep. 2019, 28, 3300–3308.e3304. [Google Scholar] [CrossRef]
- Chao, G.; Lau, W.L.; Hackel, B.J.; Sazinsky, S.L.; Lippow, S.M.; Wittrup, K.D. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006, 1, 755–768. [Google Scholar] [CrossRef]
- Sule, S.V.; Sukumar, M.; Weiss, W.F.t.; Marcelino-Cruz, A.M.; Sample, T.; Tessier, P.M. High-throughput analysis of concentration-dependent antibody self-association. Biophys. J. 2011, 101, 1749–1757. [Google Scholar] [CrossRef]
- Liu, Y.; Caffry, I.; Wu, J.; Geng, S.B.; Jain, T.; Sun, T.; Reid, F.; Cao, Y.; Estep, P.; Yu, Y.; et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs 2014, 6, 483–492. [Google Scholar] [CrossRef]
- Estep, P.; Caffry, I.; Yu, Y.; Sun, T.; Cao, Y.; Lynaugh, H.; Jain, T.; Vasquez, M.; Tessier, P.M.; Xu, Y. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. MAbs 2015, 7, 553–561. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Myszka, D.G. Improving biosensor analysis. J. Mol. Recognit. 1999, 12, 279–284. [Google Scholar] [CrossRef]
- Lund, J.; Takahashi, N.; Pound, J.D.; Goodall, M.; Jefferis, R. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J. Immunol. 1996, 157, 4963–4969. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y.; et al. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef]
- Kim, T.D.; Cho, S.E.; Yang, C.H.; Kim, J. Analysis of Fc gammaRIII and IgG Fc polymorphism reveals functional and evolutionary implications of protein-protein interaction. J. Mol. Evol. 2001, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Isoda, Y.; Yagi, H.; Satoh, T.; Shibata-Koyama, M.; Masuda, K.; Satoh, M.; Kato, K.; Iida, S. Importance of the Side Chain at Position 296 of Antibody Fc in Interactions with FcgammaRIIIa and Other Fcgamma Receptors. PLoS ONE 2015, 10, e0140120. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Y.; Li, M.; Shang, H.; Li, N.; Li, F.; Wang, W.; Wang, Y.; Jin, R.; Liu, S.; et al. A general Fc engineering platform for the next generation of antibody therapeutics. Theranostics 2021, 11, 1901–1917. [Google Scholar] [CrossRef]
- An, Z.; Forrest, G.; Moore, R.; Cukan, M.; Haytko, P.; Huang, L.; Vitelli, S.; Zhao, J.Z.; Lu, P.; Hua, J.; et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs 2009, 1, 572–579. [Google Scholar] [CrossRef]
- Hester, C.G.; Frank, M.M. Complement activation by IgG containing immune complexes regulates the interaction of C1q with its ligands. Mol. Immunol. 2019, 116, 117–130. [Google Scholar] [CrossRef]
- Jin, W.; Xing, Z.; Song, Y.; Huang, C.; Xu, X.; Ghose, S.; Li, Z.J. Protein aggregation and mitigation strategy in low pH viral inactivation for monoclonal antibody purification. MAbs 2019, 11, 1479–1491. [Google Scholar] [CrossRef]
- Durno, L.; Tounekti, O. Viral Inactivation: Low pH and Detergent. PDA J. Pharm. Sci. Technol. 2015, 69, 163–172. [Google Scholar] [CrossRef]
- Namisaki, H.; Saito, S.; Hiraishi, K.; Haba, T.; Tanaka, Y.; Yoshida, H.; Iida, S.; Takahashi, N. R409K mutation prevents acid-induced aggregation of human IgG4. PLoS ONE 2020, 15, e0229027. [Google Scholar] [CrossRef] [PubMed]
- Society, T.A. Therapeutic Monoclonal Antibodies Approved or in Review in the EU or US. 2023. Available online: https://www.antibodysociety.org/resources/approved-antibodies (accessed on 6 July 2023).
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.; Anderson, S.; Fry, J.; Julien, L.A.; Neville, D.; Qureshi, O.; Watts, G.; Hale, G. Fc-engineered antibodies with immune effector functions completely abolished. PLoS ONE 2021, 16, e0260954. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | Hinge Sequence | Isotype | PSR 1 (Score) | AC-SINS (Δmaxλ) | HIC RT (min) | SEC (%) | pH 3.5 Stress SEC (%) | Fc Tm (°C) | THP-1 (MFI) |
|---|---|---|---|---|---|---|---|---|---|
| N/A | PAPELLGG | WT IgG1 | 0.27 | 7.7 | 8.0 | 97.7 | 97.6 | 67.5 | 7136 |
| L234A/L235A (LALA) | PAPEAA2GG | IgG1 | 0.28 | 8.1 | 8.0 | 98.1 | 97.1 | 68.5 | 491 |
| L234F/L235E (LFLE) | PAPEFEGG | IgG1 | 0.28 | 6.9 | 8.0 | 97.5 | 98.1 | 66.0 | 369 |
| L234A/L235E (LALE) | PAPEAEGG | IgG1 | 0.28 | 8.4 | 8.0 | 98.4 | 97.9 | 66.0 | 192 |
| L235A/G237A (LAGA1) | PAPELAGA | IgG1 | 0.27 | 7.6 | 8.0 | 98.4 | 97.3 | 67.0 | 192 |
| L234A/G237A (LAGA2) | PAPEALGA | IgG1 | 0.28 | 7.4 | 8.0 | 98.5 | 98.5 | 67.0 | 184 |
| L234A/L235A/G237A (LALAGA) | PAPEAAGA | IgG1 | 0.29 | 7.6 | 8.0 | 98.2 | 96.7 | 67.5 | 150 |
| L234A/L235K (LALK) | PAPEAKGG | IgG1 | 0.28 | 7.7 | 8.0 | 97.5 | 81.3 | 70.5 | 140 |
| L234F/L235K (LFLK) | PAPEFKGG | IgG1 | 0.28 | 8.0 | 8.0 | 98.1 | 62.5 | 70.5 | 386 |
| L234E/L235K (LELK) | PAPEEKGG | IgG1 | 0.28 | 7.9 | 8.0 | 97.5 | 79.3 | 68.5 | 176 |
| N/A | PAPPVA-G | WT IgG2 | 0.38 | 10.0 | 8.0 | 96.1 | 97.8 | 68.0 | 565 |
| Biacore Kinetics | FACS Cell Binding | |||||||
|---|---|---|---|---|---|---|---|---|
| IgG1 Hinge × CH2 Mutations | FcRn (RU) | CD64 (RU) | CD32a (RU) | CD16 (RU) | C1q (RU) | THP-1 (MFI) | THP-1 CD3-BSA PC (MFI) | C1q Dynal FACS (MFI) |
| N/A | 40.0 | 92.0 | 4.8 | 9.8 | 155.7 | 11,755.2 | 23,338.7 | 3505.0 |
| LALA | 44.0 | 23.5 | 0.1 | 0.6 | −13.9 | 269.6 | 5274.1 | 1983.0 |
| LALA-D265A | 44.6 | 1.0 | 0.0 | −0.3 | −9.1 | 113.1 | 119.2 | 1948.0 |
| LALA-S267K | 43.0 | 1.5 | 0.0 | −0.6 | −8.7 | 119.0 | 127.5 | 797.0 |
| LALA-H268Q | 44.0 | 22.9 | 0.4 | 0.2 | −10.7 | 202.7 | 4799.2 | 2351.0 |
| LALA-D270A | 43.6 | 9.7 | 0.1 | −0.5 | −14.4 | 131.5 | 2274.4 | 769.0 |
| LALA-L309A | 27.0 | 21.6 | 0.2 | −0.5 | 3.0 | 168.3 | 4787.3 | 2519.0 |
| LALA-L309V | 43.3 | 25.2 | 0.1 | 0.2 | −2.3 | 177.8 | 4846.6 | 2143.0 |
| LALA-K322A | 38.1 | 19.9 | 0.1 | −0.4 | −20.9 | 171.8 | 4387.4 | 875.0 |
| LALA-A327G | 42.2 | 14.3 | 0.2 | 0.1 | 0.0 | 141.6 | 7006.4 | 3146.0 |
| LALA-L328A | 40.9 | 13.6 | 0.3 | 0.0 | −1.7 | 149.1 | 6424.2 | 1492.0 |
| LALA-P329A | 45.1 | 2.2 | −0.1 | −0.5 | −3.3 | 121.7 | 168.9 | 621.0 |
| LALA-P329G | 44.5 | 2.0 | −0.1 | −0.3 | −25.1 | 122.2 | 146.2 | 676.0 |
| LALA-Y296F | 41.9 | 19.1 | 0.2 | −0.1 | −3.2 | 150.9 | 3808.6 | 2077.0 |
| LALA-A330S | 43.2 | 19.9 | 0.3 | 0.2 | −2.9 | 175.3 | 4438.2 | 1703.0 |
| LALA-P331S | 40.7 | 17.7 | 0.0 | 0.0 | −22.1 | 167.5 | 4215.4 | 724.0 |
| LALA-A330S/P331S | 37.0 | 15.8 | 0.0 | −0.1 | −5.3 | 163.8 | 3840.6 | 813.0 |
| LALE | 38.9 | 3.8 | 0.1 | 0.4 | −27.3 | 126.6 | 5298.4 | 1787.0 |
| LALE-D265A | 36.8 | 0.5 | 0.0 | −0.4 | −16.4 | 125.3 | 115.4 | 2279.0 |
| LALE-S267K | 40.1 | 0.7 | −0.1 | −0.2 | −11.2 | 121.8 | 130.3 | 911.0 |
| LALE-H268Q | 39.2 | 4.3 | 0.1 | 0.6 | 5.7 | 125.7 | 4000.1 | 1934.0 |
| LALE-D270A | 40.1 | 1.8 | 0.0 | −0.1 | −19.6 | 119.7 | 545.8 | 885.0 |
| LALE-L309A | 26.0 | 4.2 | 0.1 | 0.5 | −11.7 | 124.1 | 2758.1 | 2312.0 |
| LALE-L309V | 40.9 | 4.9 | 0.1 | 0.6 | 2.3 | 122.7 | 3543.8 | 1840.0 |
| LALE-K322A | 32.4 | 4.6 | 0.0 | 0.0 | −4.9 | 120.7 | 2323.0 | 586.0 |
| LALE-L328A | 38.8 | 3.5 | 0.4 | 0.5 | −6.6 | 130.2 | 7040.7 | 1562.0 |
| LALE-P329G | 38.1 | 0.8 | −0.1 | −0.1 | −23.3 | 120.3 | 123.7 | 660.0 |
| LALE-P329A | 37.3 | 0.8 | 0.0 | −0.3 | −6.5 | 117.5 | 123.6 | 865.0 |
| LALE-A330S | 38.6 | 3.3 | 0.1 | 0.3 | −6.2 | 125.4 | 2259.5 | 1620.0 |
| LALE-P331S | 38.6 | 3.3 | 0.2 | 0.2 | −120.6 | 128.6 | 1762.6 | 725.0 |
| LALE-A330S/P331S | 35.5 | 2.6 | −0.1 | 0.0 | −21.0 | 123.0 | 1388.0 | 750.0 |
| LALAGA-P329G | 37.1 | 0.4 | −0.1 | −0.3 | −6.2 | 115.1 | 129.0 | 664.0 |
| IgG1 Hinge × CH2 Mutations | PSR (Score) | AC-SINS (Δmaxλ) | HIC RT (min) | SEC (%) | SEC Aggregate after 1 h at pH 3.5 (%) | Fc Tm (°C) |
|---|---|---|---|---|---|---|
| N/A | 0.33 | 8.1 | 8.5 | 97.6 | 0.7 | 67.5 |
| LALA | 0.37 | 9.1 | 8.4 | 93.8 | −0.2 | 66.5 |
| LALA-D265A | 0.40 | 6.9 | 8.4 | 96.1 | 9.6 | 63.0 |
| LALA-S267K | 0.39 | 14.6 | 8.4 | 94.9 | −0.4 | 67.0 |
| LALA-H268Q | 0.40 | 12.6 | 8.4 | 96.3 | −0.3 | 68.0 |
| LALA-D270A | 0.41 | 17.3 | 8.4 | 96.4 | 1.0 | 68.0 |
| LALA-L309A | 0.43 | 15.0 | 8.3 | 95.3 | 0.0 | 70.0 |
| LALA-L309V | 0.42 | 17.4 | 8.3 | 96.5 | 1.8 | 67.5 |
| LALA-K322A | 0.38 | 13.9 | 8.4 | 96.0 | 2.1 | 66.5 |
| LALA-A327G | 0.38 | 9.7 | 8.4 | 98.0 | −0.3 | 67.0 |
| LALA-L328A | 0.37 | 10.8 | 8.4 | 96.2 | 6.1 | 63.0 |
| LALA-P329A | 0.38 | 10.8 | 8.4 | 94.9 | −0.4 | 67.0 |
| LALA-P329G | 0.38 | 12.8 | 8.4 | 98.0 | 1.0 | 66.5 |
| LALA-Y296F | 0.40 | 12.6 | 8.4 | 96.1 | −0.1 | 67.5 |
| LALA-A330S | 0.37 | 11.9 | 8.4 | 96.3 | 1.0 | 67.0 |
| LALA-P331S | 0.39 | 14.7 | 8.4 | 96.3 | 3.2 | 62.5 |
| LALA-A330S/P331S | 0.37 | 13.7 | 8.4 | 92.8 | 3.1 | 64.0 |
| LALE | 0.37 | 11.2 | 8.4 | 96.6 | 0.5 | 66.5 |
| LALE-D265A | 0.38 | 9.3 | 8.4 | 96.0 | 11.3 | 62.0 |
| LALE-S267K | 0.36 | 8.0 | 8.4 | 97.8 | 3.3 | 66.5 |
| LALE-H268Q | 0.35 | 9.6 | 8.4 | 97.8 | −0.7 | 66.5 |
| LALE-D270A | 0.35 | 12.7 | 8.4 | 96.1 | 0.0 | 67.5 |
| LALE-L309A | 0.36 | 12.8 | 8.3 | 96.3 | 0.4 | 68.5 |
| LALE-L309V | 0.39 | 13.4 | 8.3 | 95.4 | 0.5 | 65.5 |
| LALE-K322A | 0.33 | 13.6 | 8.4 | 95.7 | 0.1 | 65.0 |
| LALE-L328A | 0.36 | 8.4 | 8.4 | 94.1 | 8.6 | 62.0 |
| LALE-P329G | 0.36 | 11.3 | 8.4 | 97.4 | 0.7 | 66.0 |
| LALE-P329A | 0.35 | 10.5 | 8.4 | 96.5 | 0.8 | 65.0 |
| LALE-A330S | 0.35 | 13.5 | 8.4 | 96.9 | 0.8 | 65.5 |
| LALE-P331S | 0.35 | 12.8 | 8.4 | 93.9 | 1.6 | 60.5 |
| LALE-A330S/P331S | 0.35 | 12.9 | 8.4 | 94.5 | 3.1 | 61.0 |
| LALAGA-P329G | 0.38 | 16.4 | 8.4 | 96.8 | 0.6 | 65.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pejchal, R.; Cooper, A.B.; Brown, M.E.; Vásquez, M.; Krauland, E.M. Profiling the Biophysical Developability Properties of Common IgG1 Fc Effector Silencing Variants. Antibodies 2023, 12, 54. https://doi.org/10.3390/antib12030054
Pejchal R, Cooper AB, Brown ME, Vásquez M, Krauland EM. Profiling the Biophysical Developability Properties of Common IgG1 Fc Effector Silencing Variants. Antibodies. 2023; 12(3):54. https://doi.org/10.3390/antib12030054
Chicago/Turabian StylePejchal, Robert, Anthony B. Cooper, Michael E. Brown, Maximiliano Vásquez, and Eric M. Krauland. 2023. "Profiling the Biophysical Developability Properties of Common IgG1 Fc Effector Silencing Variants" Antibodies 12, no. 3: 54. https://doi.org/10.3390/antib12030054
APA StylePejchal, R., Cooper, A. B., Brown, M. E., Vásquez, M., & Krauland, E. M. (2023). Profiling the Biophysical Developability Properties of Common IgG1 Fc Effector Silencing Variants. Antibodies, 12(3), 54. https://doi.org/10.3390/antib12030054