
Recent Progress in Antibody Epitope Prediction


Abstract
1. Introduction
2. General Mechanism and Feature of Antibody–Antigen Recognition
3. Linear Epitope Prediction
4. Conformational Epitope Prediction
- Step 1: Determine all the surface residues in the protein antigen;
- Step 2: Search all possible unit patches within a 15 Å atom distance of residue r, map the pre-calculated propensity indices to the above unit patches, and calculate the propensity index avgr;
- Step 3: Calculate the clustering coefficient (ccr) for residue r using the Equation;
- Step 4: Summarize avgr and ccr as the antigenicity score for residue r;
- Step 5: Give the antigenicity score for each residue and highlight those residues with scores higher than a threshold.
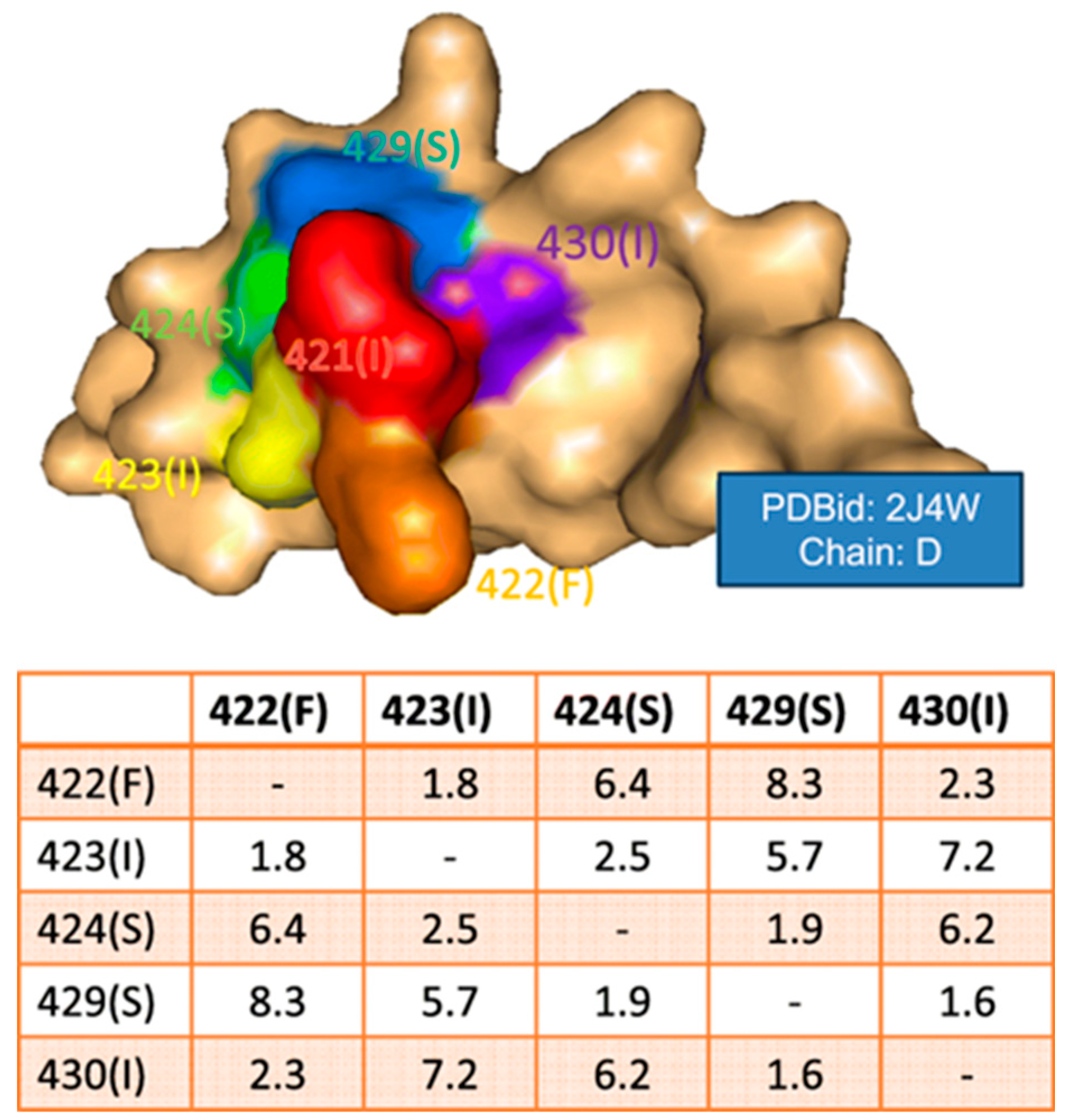
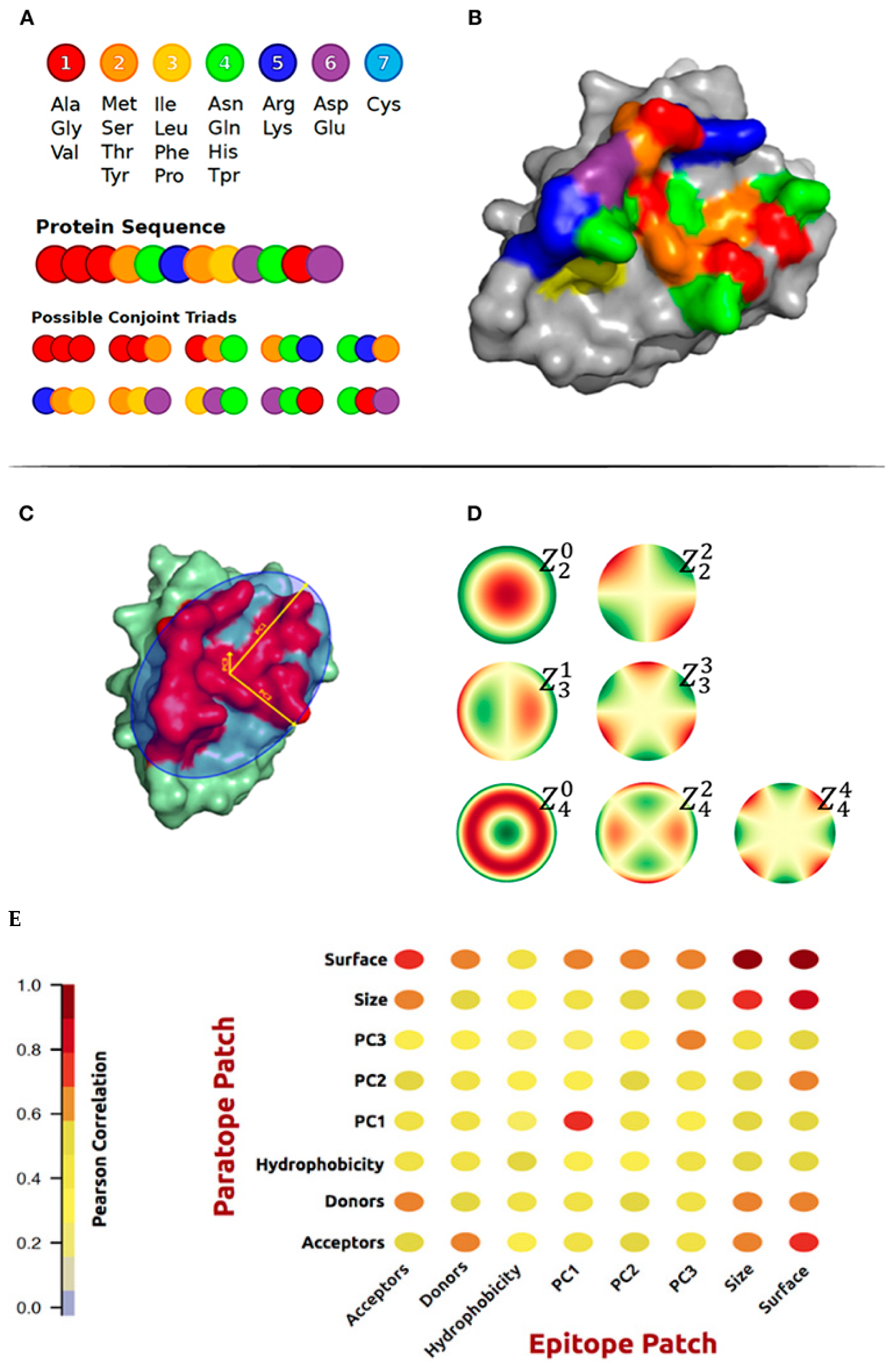
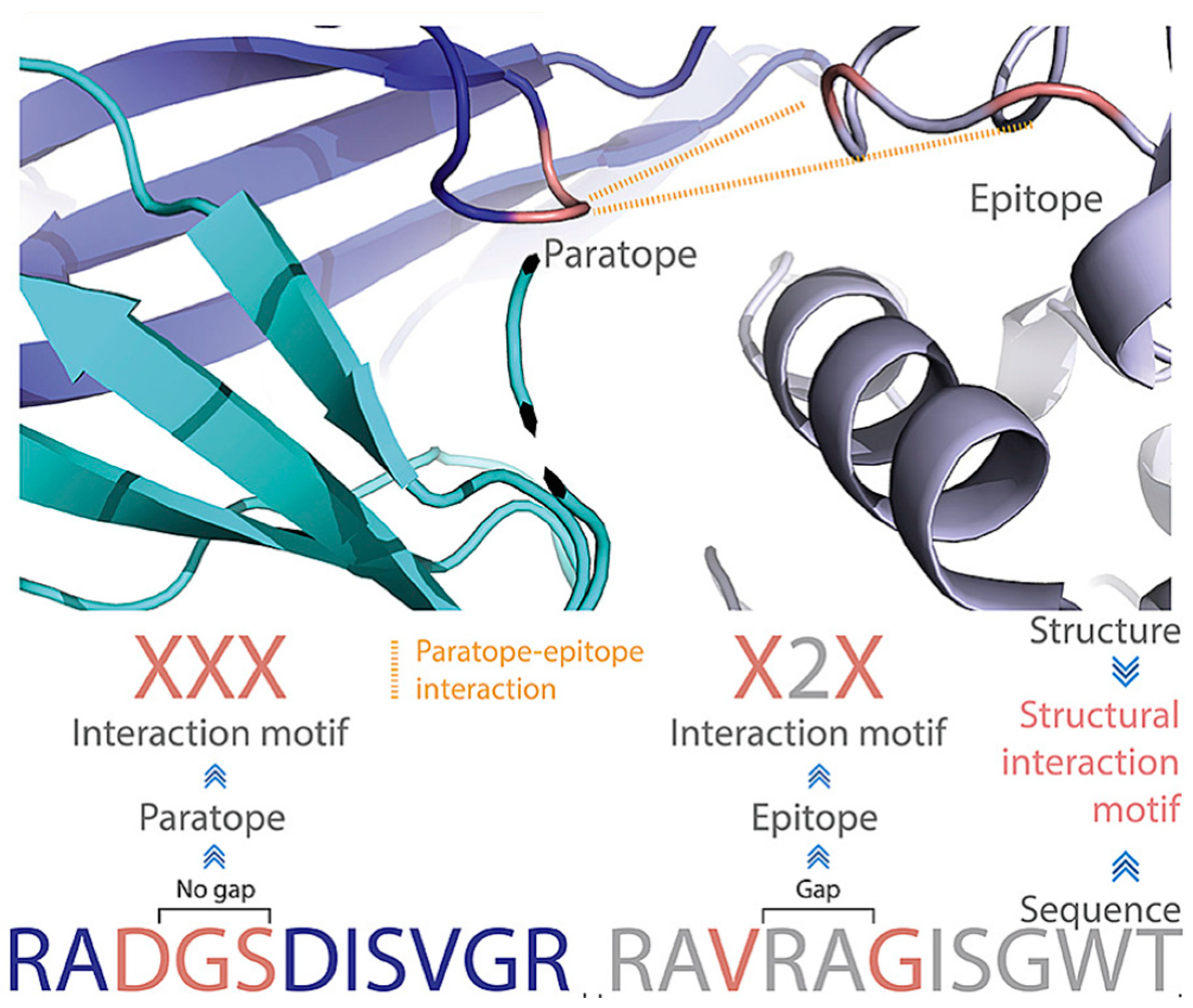
5. Epitope Prediction Based on Paratope–Epitope Interactions
6. Using Antibody–Antigen Dock to Predict Conformational Epitope
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Yang, S.; Cao, B.; Zhou, G.; Zhang, F.; Wang, Y.; Wang, R.; Zhu, L.; Meng, Y.; Hu, C.; et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J. Hematol. Oncol. 2021, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, Y.; Zhao, Q.; Zhu, Z.; Dimitrov, D.S. Human monoclonal antibodies targeting nonoverlapping epitopes on insulin-like growth factor II as a novel type of candidate cancer therapeutics. Mol. Cancer Ther. 2012, 11, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Z.; Han, B.-N.; Wei, R.; Yao, G.-Y.; Chen, Z.; Liu, J.; Poon, T.C.; Su, W.; Zhu, Z.; Dimitrov, D.S.; et al. N-terminal alpha-amino group modification of antibodies using a site-selective click chemistry method. MAbs 2018, 10, 712–719. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, J.; Zhou, G.; Ng, H.M.; Ang, I.L.; Ma, G.; Liu, Y.; Yang, S.; Zhang, F.; Miao, K.; et al. Targeting immune checkpoint B7-H3 antibody-chlorin e6 bioconjugates for spectroscopic photoacoustic imaging and photodynamic therapy. Chem. Commun. 2019, 55, 14255–14258. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Liu, M.; Wang, L.; Zhu, K.; Cai, M.; Chen, X.; Feng, Y.; Yang, S.; Fu, S.; Zhi, C.; et al. Remodelling of tumour microenvironment by microwave ablation potentiates immunotherapy of AXL-specific CAR T cells against non-small cell lung cancer. Nat. Commun. 2022, 13, 6203. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, F.; Yu, J.; Zhao, Q. Programmed death-ligand 1 expression on CD22-specific chimeric antigen receptor-modified T cells weakens antitumor potential. MedComm 2022, 3, e140. [Google Scholar] [CrossRef]
- Zhou, G.; Zhao, Q. Perspectives on therapeutic neutralizing antibodies against the Novel Coronavirus SARS-CoV-2. Int. J. Biol. Sci. 2020, 16, 1718–1723. [Google Scholar] [CrossRef]
- Zhao, Q. Bispecific Antibodies for Autoimmune and Inflammatory Diseases: Clinical Progress to Date. BioDrugs 2020, 34, 111–119. [Google Scholar] [CrossRef]
- Zhao, Q.; Ahmed, M.; Tassev, D.V.; Hasan, A.; Kuo, T.-Y.; Guo, H.-F.; O'Reilly, R.J.; Cheung, N.-K.V. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015, 29, 2238–2247. [Google Scholar] [CrossRef]
- Caoili, S.E.C. Hybrid Methods for B-Cell Epitope Prediction. In Immunoinformatics; De, R.K., Tomar, N., Eds.; Springer: New York, NY, USA, 2014; pp. 245–283. [Google Scholar] [CrossRef]
- Meloen, R.H.; Puijk, W.C.; Slootstra, J.W. Mimotopes: Realization of an unlikely concept. J. Mol. Recognit. 2000, 13, 352–359. [Google Scholar] [CrossRef]
- Deng, B.; Zhu, S.; Macklin, A.M.; Xu, J.; Lento, C.; Sljoka, A.; Wilson, D.J. Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. MAbs 2017, 9, 1327–1336. [Google Scholar] [CrossRef]
- Trkulja, C.L.; Jungholm, O.; Davidson, M.; Jardemark, K.; Marcus, M.M.; Hägglund, J.; Karlsson, A.; Karlsson, R.; Bruton, J.; Ivarsson, N.; et al. Rational antibody design for undruggable targets using kinetically controlled biomolecular probes. Sci. Adv. 2021, 7, eabe6397. [Google Scholar] [CrossRef]
- Tahir, S.; Bourquard, T.; Musnier, A.; Jullian, Y.; Corde, Y.; Omahdi, Z.; Mathias, L.; Reiter, E.; Crépieux, P.; Bruneau, G.; et al. Accurate determination of epitope for antibodies with unknown 3D structures. MAbs 2021, 13, 1961349. [Google Scholar] [CrossRef] [PubMed]
- Bourquard, T.; Musnier, A.; Puard, V.; Tahir, S.; Ayoub, M.A.; Jullian, Y.; Boulo, T.; Gallay, N.; Watier, H.; Bruneau, G.; et al. MAbTope: A Method for Improved Epitope Mapping. J. Immunol. 2018, 201, 3096–3105. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Teuwen, J.; Moriakov, N. Chapter 20—Convolutional neural networks. In Handbook of Medical Image Computing and Computer Assisted Intervention; Zhou, S.K., Rueckert, D., Fichtinger, G., Eds.; Academic Press: New York, NY, USA, 2020; pp. 481–501. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Bruna, J.; Zaremba, W.; Szlam, A.; LeCun, Y. Spectral Networks and Locally Connected Networks on Graphs. Comput. Sci. 2013. [Google Scholar] [CrossRef]
- Sakakibara, S.; Arimori, T.; Yamashita, K.; Jinzai, H.; Motooka, D.; Nakamura, S.; Li, S.; Takeda, K.; Katayama, J.; El Hussien, M.A.; et al. Clonal evolution and antigen recognition of anti-nuclear antibodies in acute systemic lupus erythematosus. Sci. Rep. 2017, 7, 16428. [Google Scholar] [CrossRef]
- Kappler, K.; Hennet, T. Emergence and significance of carbohydrate-specific antibodies. Genes. Immun. 2020, 21, 224–239. [Google Scholar] [CrossRef]
- Kunik, V.; Ofran, Y. The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Eng. Des. Sel. 2013, 26, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhu, D.; Zhu, J.W.; Nussinov, R.; Ma, B.Y. Local and global anatomy of antibody-protein antigen recognition. J. Mol. Recognit. 2018, 31, 14. [Google Scholar] [CrossRef]
- Kaur, H.; Sain, N.; Mohanty, D.; Salunke, D.M. Deciphering evolution of immune recognition in antibodies. BMC Struct. Biol. 2018, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; Wolfson, H.J.; Nussinov, R. Protein functional epitopes: Hot spots, dynamics and combinatorial libraries. Curr. Opin. Struct. Biol. 2001, 11, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Blackler, R.J.; Müller-Loennies, S.; Pokorny-Lehrer, B.; Legg, M.S.; Brade, L.; Brade, H.; Kosma, P.; Evans, S.V. Antigen binding by conformational selection in near-germline antibodies. J. Biol. Chem. 2022, 298, 101901. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; Kumar, S.; Tsai, C.J.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng. 1999, 12, 713–720. [Google Scholar] [CrossRef]
- Ma, B.Y.; Shatsky, M.; Wolfson, H.J.; Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. [Google Scholar] [CrossRef]
- Wei, G.H.; Xi, W.H.; Nussinov, R.; Ma, B.Y. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. [Google Scholar] [CrossRef]
- Fernandez-Quintero, M.L.; Kraml, J.; Georges, G.; Liedl, K.R. CDR-H3 loop ensemble in solution—Conformational selection upon antibody binding. mAbs 2019, 11, 1077–1088. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Loeffler, J.R.; Waibl, F.; Kamenik, A.S.; Hofer, F.; Liedl, K.R. Conformational selection of allergen-antibody complexes-surface plasticity of paratopes and epitopes. Protein Eng. Des. Sel. 2019, 32, 513–523. [Google Scholar] [CrossRef]
- Uversky, V.N.; Van Regenmortel, M.H.V. Mobility and disorder in antibody and antigen binding sites do not prevent immunochemical recognition. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, M.D.; Jandrlic, D.R.; Mitic, N.S. Epitope distribution in ordered and disordered protein regions. Part B—Ordered regions and disordered binding sites are targets of T- and B-cell immunity. J. Immunol. Methods 2014, 407, 90–107. [Google Scholar] [CrossRef] [PubMed]
- MacRaild, C.A.; Richards, J.S.; Anders, R.F.; Norton, R.S. Antibody Recognition of Disordered Antigens. Structure 2016, 24, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; Zhao, J.; Nussinov, R. Conformational selection in amyloid-based immunotherapy: Survey of crystal structures of antibody-amyloid complexes. Biochim. Biophys. Acta-Gen. Subj. 2016, 1860, 2672–2681. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, B.Y.; Nussinov, R. Compilation and Analysis of Enzymes, Engineered Antibodies, and Nanoparticles Designed to Interfere with Amyloid-beta Aggregation. Isr. J. Chem. 2017, 57, 622–633. [Google Scholar] [CrossRef]
- Schilz, J.; Binder, U.; Friedrich, L.; Gebauer, M.; Lutz, C.; Schlapschy, M.; Schiefner, A.; Skerra, A. Molecular recognition of structurally disordered Pro/Ala-rich sequences (PAS) by antibodies involves an Ala residue at the hot spot of the epitope. J. Mol. Biol. 2021, 433, 167113. [Google Scholar] [CrossRef]
- Biner, D.W.; Grosch, J.S.; Ortoleva, P.J. B-cell epitope discovery: The first protein flexibility-based algorithm-Zika virus conserved epitope demonstration. PLoS ONE 2023, 18, e0262321. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B. Mechanisms of recognition of amyloid-beta (Abeta) monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 2017, 292, 18325–18343. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef]
- Mollica, L.; Giachin, G. Recognition Mechanisms between a Nanobody and Disordered Epitopes of the Human Prion Protein: An Integrative Molecular Dynamics Study. J. Chem. Inf. Model. 2023, 63, 531–545. [Google Scholar] [CrossRef]
- Shrock, E.L.; Shrock, C.L.; Elledge, S.J. VirScan: High-throughput Profiling of Antiviral Antibody Epitopes. Bio-Protocol 2022, 12, e4464. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ma, M.; Hu, C.; Xu, Z.-W.; Wu, F.-L.; Wang, N.; Lai, D.-Y.; Li, Y.; Zhang, H.; Jiang, H.-W.; et al. Antibody Binding Epitope Mapping (AbMap) of Hundred Antibodies in a Single Run. Mol. Cell Proteom. 2021, 20, 100059. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.A.R.; Carnero, L.R.; Kuramoto, A.; Tang, F.H.F.; Gomes, C.H.; Pereira, N.B.; de Oliveira, L.C.; Garrini, R.; Monteiro, J.S.; Setubal, J.C.; et al. A refined genome phage display methodology delineates the human antibody response in patients with Chagas disease. iScience 2021, 24, 102540. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. BcePred: Prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. Lect. Notes Comput. Sci. 2004, 3239, 197–204. [Google Scholar]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins-Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [PubMed]
- Saha, I.; Maulik, U.; Bandyopadhyay, S.; Plewczynski, D. Fuzzy clustering of physicochemical and biochemical properties of amino acids. Amino Acids 2012, 43, 583–594. [Google Scholar] [CrossRef]
- Collatz, M.; Mock, F.; Barth, E.; Hölzer, M.; Sachse, K.; Marz, M. EpiDope: A deep neural network for linear B-cell epitope prediction. Bioinformatics 2021, 37, 1784. [Google Scholar] [CrossRef]
- Kozlova, E.; Viart, B.; de Avila, R.; Felicori, L.; Chavez-Olortegui, C. Classification epitopes in groups based on their protein family. BMC Bioinform. 2015, 19 (Suppl. S16), S7. [Google Scholar] [CrossRef]
- Ras-Carmona, A.; Lehmann, A.A.; Lehmann, P.V.; Reche, P.A. Prediction of B cell epitopes in proteins using a novel sequence similarity-based method. Sci. Rep. 2022, 12, 13739. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Ofran, Y.; Peters, B. Antibody specific epitope prediction—Emergence of a new paradigm. Curr. Opin. Virol. 2015, 11, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Najar, T.A.; Khare, S.; Pandey, R.; Gupta, S.K.; Varadarajan, R. Mapping Protein Binding Sites and Conformational Epitopes Using Cysteine Labeling and Yeast Surface Display. Structure 2017, 25, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, S.; Kelm, S.; Baker, T.S.; Shi, J.; Martin, A.C.R. B-cell epitopes: Discontinuity and conformational analysis. Mol. Immunol. 2019, 114, 643–650. [Google Scholar] [CrossRef]
- Hou, Q.; Stringer, B.; Waury, K.; Capel, H.; Haydarlou, R.; Xue, F.; Abeln, S.; Heringa, J.; Feenstra, K.A. SeRenDIP-CE: Sequence-based Interface Prediction for Conformational Epitopes. Bioinformatics 2021, 37, 3421–3427. [Google Scholar] [CrossRef]
- Andersen, P.H.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Sweredoski, M.J.; Baldi, P. PEPITO: Improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 2008, 24, 1459–1460. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Di Wu, D.; Xu, T.; Wang, X.; Xu, X.; Tao, L.; Li, Y.X.; Cao, Z.W. SEPPA: A computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 2009, 37, W612–W616. [Google Scholar] [CrossRef]
- Krawczyk, K.; Liu, X.F.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- Lo, Y.-T.; Shih, T.-C.; Pai, T.-W.; Ho, L.-P.; Wu, J.-L.; Chou, H.-Y. Conformational epitope matching and prediction based on protein surface spiral features. BMC Genom. 2021, 22, 116. [Google Scholar] [CrossRef]
- Hu, Y.J.; Lin, S.C.; Lin, Y.L.; Lin, K.H.; You, S.N. A meta-learning approach for B-cell conformational epitope prediction. BMC Bioinform. 2014, 15, 378. [Google Scholar] [CrossRef]
- Cia, G.; Pucci, F.; Rooman, M. Critical review of conformational B-cell epitope prediction methods. Brief. Bioinform. 2023, 24, bbac567. [Google Scholar] [CrossRef]
- Lu, S.; Li, Y.G.; Ma, Q.; Nan, X.F.; Zhang, S.T. A Structure-Based B-cell Epitope Prediction Model Through Combing Local and Global Features. Front. Immunol. 2022, 13, 890943. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, T.I.; Umerenkov, D.; Salnikov, M.; Strashnov, P.V.; Konstantinova, A.V.; Lebed, I.; Shcherbinin, D.N.; Asatryan, M.N.; Kardymon, O.L.; Ivanisenko, N.V. SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol. 2022, 13, 960985. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.M.; Myung, Y.; Ascher, D.B.; Pires, D.E.V. epitope3D: A machine learning method for conformational B-cell epitope prediction. Brief. Bioinform. 2022, 23, bbab423. [Google Scholar] [CrossRef] [PubMed]
- Robert, P.A.; Akbar, R.; Frank, R.; Pavlović, M.; Widrich, M.; Snapkov, I.; Slabodkin, A.; Chernigovskaya, M.; Scheffer, L.; Smorodina, E.; et al. Unconstrained generation of synthetic antibody-antigen structures to guide machine learning methodology for real-world antibody specificity prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liberis, E.; Velickovic, P.; Sormanni, P.; Vendruscolo, M.; Lio, P. Parapred: Antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 2018, 34, 2944–2950. [Google Scholar] [CrossRef]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef]
- Deac, A.; Velickovic, P.; Sormanni, P. Attentive Cross-Modal Paratope Prediction. J. Comput. Biol. 2019, 26, 536–545. [Google Scholar] [CrossRef]
- Pittala, S.; Bailey-Kellogg, C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics 2020, 36, 3996–4003. [Google Scholar] [CrossRef]
- Del Vecchio, A.; Deac, A.; Liò, P.; Veličković, P. Neural message passing for joint paratope-epitope prediction. arXiv 2021, arXiv:2106.00757. [Google Scholar]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Akbar, R.; Robert, P.A.; Pavlović, M.; Jeliazkov, J.R.; Snapkov, I.; Slabodkin, A.; Weber, C.R.; Scheffer, L.; Miho, E.; Haff, I.H.; et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021, 34, 108856. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.K.; Robinson, S.A.; Bujotzek, A.; Georges, G.; Lewis, A.P.; Shi, J.; Snowden, J.; Taddese, B.; Deane, C.M. Ab-Ligity: Identifying sequence-dissimilar antibodies that bind to the same epitope. MAbs 2021, 13, 1873478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, J. Mining for the antibody-antigen interacting associations that predict the B cell epitopes. BMC Struct. Biol. 2010, 10 (Suppl. S1), S6. [Google Scholar] [CrossRef]
- Wang, X.; Flannery, S.T.; Kihara, D. Protein Docking Model Evaluation by Graph Neural Networks. Front. Mol. Biosci. 2021, 8, 647915. [Google Scholar] [CrossRef]
- Ambrosetti, F.; Jimenez-Garcia, B.; Roel-Touris, J.; Bonvin, A. Modeling Antibody-Antigen Complexes by Information-Driven Docking. Structure 2020, 28, 119–129.e112. [Google Scholar] [CrossRef]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB: Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 2022, 38, 377–383. [Google Scholar] [CrossRef]
- Brooks, B.D.; Closmore, A.; Yang, J.; Holland, M.; Cairns, T.; Cohen, G.H.; Bailey-Kellogg, C. Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition. Molecules 2020, 25, 3659. [Google Scholar] [CrossRef]
- Hua, C.K.; Gacerez, A.T.; Sentman, C.L.; Ackerman, M.E.; Choi, Y.; Bailey-Kellogg, C. Computationally-driven identification of antibody epitopes. Elife 2017, 6, e29023. [Google Scholar] [CrossRef]
- Xu, Z.; Davila, A.; Wilamowski, J.; Teraguchi, S.; Standley, D.M. Improved Antibody-Specific Epitope Prediction Using AlphaFold and AbAdapt. Chembiochem 2022, 23, e202200303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method Name | Year | Methodology/Approach | Link |
|---|---|---|---|
| Bcepred | 2004 | prediction of linear B-cell epitopes, based on physicochemical properties | http://crdd.osdd.net/raghava/bcepred |
| ABCpred | 2006 | prediction of linear B-cell epitopes, based on recurrent neural network | http://crdd.osdd.net/raghava/abcpred |
| iBCE-EL | 2018 | prediction of linear B-cell epitopes, based on a fusion of randomized tree (ERT) and gradient boosting (GB) classifiers | http://thegleelab.org/iBCE-EL |
| EpiDope | 2021 | prediction of linear B-cell epitopes, based on bi-directional long short-term memory network (LSTM) | http://github.com/mcollatz/EpiDope |
| PECAN | 2020 | prediction of B-cell epitopes by paratope–epitope interactions, based on graph Convolution Attention Network and transfer learning | https://github.com/vamships/PECAN.git |
| EPMP | 2021 | prediction of B-cell epitopes by paratope–epitope interactions, based on separate neural message passing architectures | https://arxiv.org/abs/2106.00757 |
| Jespersen et al. | 2019 | prediction of B-cell epitopes by paratope–epitope specific interaction rules, based on geometric and physicochemical features, statistical and machine learning algorithms | https://doi.org/10.3389/fimmu.2019.00298 |
| Akbar et al. | 2021 | prediction of B-cell epitopes by paratope–epitope interactions, based on antibody–antigen interaction motifs | https://doi.org/10.1016/j.celrep.2021.108856 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Bai, G.; Sun, C.; Ma, B. Recent Progress in Antibody Epitope Prediction. Antibodies 2023, 12, 52. https://doi.org/10.3390/antib12030052
Zeng X, Bai G, Sun C, Ma B. Recent Progress in Antibody Epitope Prediction. Antibodies. 2023; 12(3):52. https://doi.org/10.3390/antib12030052
Chicago/Turabian StyleZeng, Xincheng, Ganggang Bai, Chuance Sun, and Buyong Ma. 2023. "Recent Progress in Antibody Epitope Prediction" Antibodies 12, no. 3: 52. https://doi.org/10.3390/antib12030052
APA StyleZeng, X., Bai, G., Sun, C., & Ma, B. (2023). Recent Progress in Antibody Epitope Prediction. Antibodies, 12(3), 52. https://doi.org/10.3390/antib12030052